

Кристаллография, 2020, T. 65, № 3, стр. 449-456
Особенности кристаллической упаковки потенциально мезоморфных производных азобензола R1–C6H4–N=N–C6H4–R2 (R1, R2 = CH3COO, C2H5O; CH2=C(CH3)COO, C2H5; C6H13COO, C2H5O)
Л. Г. Кузьмина 1, *, И. И. Константинов 2, **, М. А. Навасардян 1
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
2 Институт нефтехимического синтеза им. А.В. Топчиева РАН
Москва, Россия
* E-mail: kuzmina@igic.ras.ru
** E-mail: konst@ipc.ac.ru
Поступила в редакцию 13.08.2019
После доработки 28.08.2019
Принята к публикации 02.10.2019
Аннотация
Методами дифференциальной сканирующей калориметрии и рентгеноструктурного анализа изучены структура и термические свойства производных азобензола R1–C6H4–N=N–C6H4–R2 (R1/R2 = CH3COO/C2H5O (I), CH2=C(CH3)COO/C2H5 (II), C6H13COO/C2H5O (III). Соединения I и III при плавлении образуют мезофазу (нематик), соединение II мезофазу не образует. Соединение III характеризуется двумя высокотемпературными фазовыми переходами типа кристалл–кристалл IIIIII–IIIII–IIII. Структурно изучена низкотемпературная кристаллическая модификация, не являющаяся предшественником мезофазы. В кристаллической упаковке I обнаружены взаимодействия C−H···π, и наблюдается ее разделение на чередующиеся плотно и рыхло упакованные области. Найденные слабые направленные взаимодействия, связывающие молекулы в зигзагообразные цепочки, являются основой структурированности мезофазы I. Обсуждается возможный вид структурированности мезофазы III.
ВВЕДЕНИЕ
Настоящая работа является продолжением исследований методами дифференциальной сканирующей калориметрии (ДСК) и рентгеноструктурного анализа (РСА) потенциально мезоморфных органических соединений в гомологических рядах. Мезоморфизм проявляют дихотомные соединения, т.е. такие соединения, молекулы которых содержат по крайней мере два фрагмента, контрастирующих химически и структурно. Молекулы простейших органических мезоморфов состоят из линейных единиц. Их жесткий фрагмент (чаще всего ненасыщенный, несущий функциональные группы) соединен с насыщенным конформационно подвижным протяженным фрагментом (чаще всего это алифатическая цепочка). Образуемый дихотомными молекулами структурированный расплав, т.е. мезофаза, – это агрегатное состояние, промежуточное между жидким и кристаллическим, оно может возникать при плавлении кристалла и/или охлаждении изотропного расплава. Мезоморфные (или жидкокристаллические) соединения были открыты около 130 лет назад [1], однако интерес к их исследованию до сих пор не ослабевает. За истекшее время созданы многочисленные теории мезофазы и фазовых переходов с ее участием, основанные на ряде модельных представлений [2–11]. Но ни одна из них не позволяет хотя бы приблизительно рассчитать энтальпию фазовых переходов или предсказать число таких переходов, а также не дает ответа на многие вопросы. Например, почему некоторые представители одного и того же гомологического ряда потенциально мезоморфных соединений образуют мезофазу при плавлении кристалла (энантиотропный мезоморфизм), другие – только при охлаждении изотропного расплава (монотропный мезоморфизм), а третьи вообще мезоморфизма не обнаруживают [12]. Предыдущие исследования [13–21] показывают, что такого рода различия могут объясняться особенностями кристаллической упаковки. С целью выявления таких особенностей и характера их влияния на термическое поведение вещества в настоящей работе проведено исследование методами ДСК и РСА трех производных азобензола:
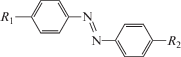
Все молекулы приведенных соединений содержат жесткий ароматический фрагмент, но только одна из них (III) имеет достаточно длинную насыщенную цепь и является дихотомной. Поэтому проявление мезоморфизма веществом III вполне ожидаемo. Что касается двух других молекул (I и II), то имеющиеся в них алифатические фрагменты очень короткие. Поэтому предсказать существование или отсутствие мезоморфизма в них априори затруднительно. Кроме того, интересно понять, какое соотношение размеров контрастирующих фрагментов молекулы будет достаточным, чтобы соединениe можно было считать дихотомным и потенциально мезоморфным.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез соединений. Сочетая 4-этил- или 4-этилоксианилин с фенолом, по методике [22] получали 4-этил- или 4-этокси-4'-гидроксиазобензол. В результате взаимодействия ОН-группы с соответствующим ацилхлоридом получали 4-этокси-4'-ацетилоксиазобензол (I), 4-этил-4'-метакрилоилоксиазобензол (II) или 4-этокси-4'-гептаноилоксиазобензол (III). Полученные продукты очищали путем многократной перекристаллизации из изопропанола до постоянной температуры изотропизации (Тизо). Оптические исследования выполняли с помощью микроскопа Leitz Ortholux Pol-BK, оснащенного нагревательным столиком “Mettler FP82HT” и центральным процессором FP90.
ДСК-исследование. Температуры фазовых переходов и соответствующие им значения теплоты измеряли на калориметре Mettler DSC 828e при скорости сканирования 5 град/мин. Результаты измерения образцов, полученных кристаллизацией из раствора, приведены в табл. 1.
Таблица 1.
Параметры фазовых переходов по данным ДСК
| Соединение | R1 | R2 | Т, °С (ΔН, кДж/моль) |
|---|---|---|---|
| I | C2H5–O | CH3COO | Cr 122.5 (25.5) N 137.4 (4.5) Iso |
| II | C2H5 | CH2=C(CH3)COO | Cr 58 (24) Iso |
| III | C2H5–O | C6H13COO | CrIII 57.3(3.18) CrII 61.4(21.66) CrI 66.3(15.88) N 118.7(1.67) Iso |
Рентгеноструктурый анализ. Оранжевые кристаллы соединений выращивали из раствора бензол-ацетонитрил. Кристаллы I и III получили в виде тонких пластин, а кристаллы II имели форму иголок. Монокристалл каждого соединения исследовали на CCD-дифрактометре Smart-6K в потоке охлажденного азота (излучение MoKα, λ = = 0.71073 Å, графитовый монохроматор, ω-сканирование). Обработку экспериментальных отражений проводили по программе SAINT [23]. Структура расшифрована прямыми методами и уточнена МНК в анизотропном приближении смещений всех атомов, кроме атомов водорода. Положения атомов водорода рассчитаны геометрически и включены в дальнейшее уточнение методом наименьших квадратов по модели наездника. Кристаллографические параметры, а также параметры расшифровки и уточнения структур приведены в табл. 2. Все расчеты выполняли по программам Olex2 [24].
Таблица 2.
Кристаллографические характеристики, данные эксперимента и результаты уточнения структур I, II, III
| Соединение | I | II | III |
|---|---|---|---|
| CCDC | 1545983 | 1545984 | 1545985 |
| Формула | C16H16N2O3 | C18H18N2O2 | C21H26N2O3 |
| М, г/моль | 284.31 | 294.34 | 354.44 |
| Пр. гр., Z | P$\bar {1}$, 4 | P21/c, 4 | P$\bar {1}$, 2 |
| a, b, c, Å | 8.521(2), 9.707(2), 17.867(3) | 13.2316(8), 5.6305(3), 1.551(1) | 7.7065(5), 8.5439(6), 16.112(1) |
| α, β, γ, град | 79.450(3), 84.315(3), 88.410(3) | 90, 101.207(4), 90 | 97.055(5), 103.151(5), 108.634(3) |
| V, Å3 | 1445.7(5) | 1574.9(2) | 956.6(1) |
| Dx, г/см3 | 1.306 | 1.241 | 1.230 |
| μ, см–1 | 0.091 | 0.082 | 0.082 |
| Т, K | 150 | 120 | 120 |
| Размеры кристалла, мм | 0.36 × 0.22 × 0.10 | 0.52 × 0.10 ×0.06 | 0.54 × 0.18 × 0.03 |
| F(000) | 600 | 624.0 | 380.0 |
| Число отражений: измер./независ., Rуср | 11 630/5613, 0.0349 | 10 613/3609, 0.1084 | 5967/3717, 0.0445 |
| Число параметров | 383 | 271 | 339 |
| S | 0.977 | 0.986 | 0.986 |
| R1/wR2 (все отражения) | 0.0935/0.1342 | 0.1732/0.1735 | 0.1229/0.2136 |
| R1/wR2 [I > 2σ(I)] | 0.0483/0.1129 | 0.0753/0.1482 | 0.0737/0.1877 |
| Δρmin/Δρmax, э/Å–3 | –0.24/0.27 | –0.25/0.25 | –0.46/0.32 |
| Программы | SAINT [22], Olex-2 [23] | ||
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Элементарная ячейка I содержит две кристаллографически независимые молекулы, а кристаллы II и III – по одной. Строение формульных единиц кристаллов показано на рис. 1. В молекулах I азобензольный фрагмент почти плоский. Двугранный угол между бензольными кольцами С1…С6 (плоскость А) и С7…С12 (плоскость В) в двух молекулах составляет 13.5° и 8.8°, двугранный угол между фрагментом С1–N1–N2–C7 (плоскость С) и плоскостями колец (A/C, а также B/С) составляют 9.1° и 4.7°, а также 6.5° и 2.3° для независимых молекул соответственно. Карбоксильная группа повернута относительно плоскости А на большие углы – 56.4° и 61.8°, тогда как этокси-заместитель лежит в плоскости кольца В (двугранные углы составляют 3.6° и 8.2°). Этокси-группа включена в систему сопряжения центрального ненасыщенного фрагмента молекулы, а карбоксильный заместитель из нее выведен.
Рис. 1.
Строение молекул I, II и III; эллипсоиды тепловых смещений атомов приведены на уровне вероятности 50%.
В молекуле II азабензольный фрагмент уплощен в большей степени. При таком же обозначении плоскостей двугранные углы A/B, A/C, B/C составляют 4.0°, 3.0° и 4.3° соответственно. Карбоксильная группа повернута относительно плоскости А на 74.3°. В молекуле III двугранные углы A/B, A/C, B/C составляют 15.4°, 8.6° и 6.7° соответственно и указывают на практическую планарность этого фрагмента. В отличие от I и II в молекуле III в общую систему сопряжения вовлечен карбоксилатный фрагмент О1–С15–О2. Соответствующий двугранный угол равен всего 18.5°. Этокси-заместитель второго бензольного кольца также находится с ним в сопряжении (соответствующий угол 3.9°). Таким образом, каркас атомов молекулы III почти плоский.
По данным ДСК вещество II не проявляет мезоморфизма. Это означает, что особенности его кристаллической упаковки не типичны для жидкокристаллических соединений. Согласно исследованиям [13–21] наблюдаются следующие особенности: разделение кристаллической упаковки на чередующиеся алифатические и ароматические области; очень рыхлая упаковка алифатических областей; плотная упаковка ароматических областей как за счет большого количества ненаправленных дисперсионных взаимодействий, так и за счет слабых направленных взаимодействий (вторичных связей) между функциональными группами соседних молекул.
Чередование рыхлых и плотных областей в кристаллической упаковке является следствием дихотомной природы молекул мезоморфных соединений. Алифатические цепoчки имеют высокую конформационную подвижность, тогда как остальная часть молекулы достаточно жесткая. Это означает, что эффективный размер алифатических атомов больше, чем атомов жестких фрагментов, т.е. для них требуется большее свободное пространство в процессе образования зародышей и дальнейшего роста кристалла. Анализ большого количества кристаллических упаковок мезоморфных соединений свидетельствует о том, что формирование кристаллической упаковки с чередующейся плотностью может достигаться двумя путями:
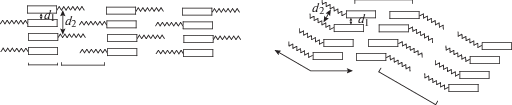
На схеме прямоугольник – жесткий фрагмент молекулы, зигзаг – подвижная цепочка. Слева показан способ расположения молекул “голова-к-хвосту”, а справа – “голова-к-голове”. В первом случае увеличение расстояния между подвижными цепочками соседних молекул возможно как следствие их расположения по разные стороны от жестких ядер соседних молекул, уложенных в колонки. Увеличение расстояния между соседними подвижными цепочками во втором случае происходит за счет перегиба молекул и их взаимного сдвига в параллельных плоскостях. Молекулы в колонках объединены слабыми направленными взаимодействиями (водородными связями, взаимодействиями С–Н···О [25], π–π, С–Н···π [26–28] и другими).
Плавление кристалла с такой “разделенной” упаковкой происходит аномально. Оно начинается с рыхлых алифатических областей, что приводит к разрушению кристалла и образованию не изотропного расплава, а геля (т.е. мезофазы), в котором сохраняются структурированные ассоциаты. Структуру ассоциатов поддерживают вторичные связи. При дальнейшем повышении температуры вторичные связи разной природы поочередно разрушаются, и система переходит вначале к менее структурированному расплаву (мезофазе иного типа), а затем к изотропной жидкости. Разрушение слабых направленных взаимодействий характеризуется соответствующим тепловым эффектом на кривой ДСК, т.е. оно сопряжено с фазовыми переходами. Количество фазовых переходов с участием мезофазы зависит от количества типов вторичных связей и соотношения значений их энергии.
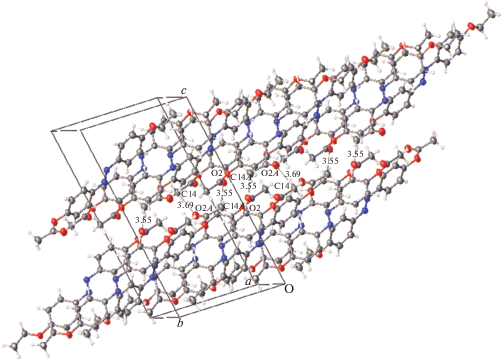
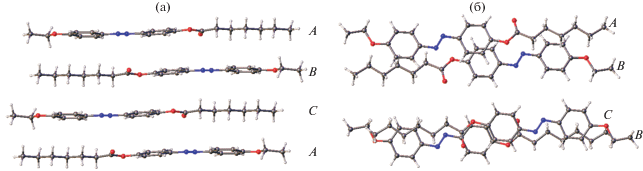
Строение молекулы II показано на рис. 2, кристаллическая упаковка II – на рис. 3. В этой упаковке выделяются чередующиеся молекулярные слои, которые “соприкасаются” ацетатными фрагментами. Анализ межслоевых расстояний с участием атомов этих групп (О···О, О···С, С···С О···Н, Н···Н) показывает, что лишь небольшое их число может быть рассмотрено как ван-дер-ваальсовы контакты, а остальные превышают суммы ван-дер-ваальсовых радиусов (∼2.8, 3.2, 3.6, 2.4 Å соответственно). Поэтому рыхлый слой как предпосылка начала плавления существует и в этой упаковке. Однако только рыхлых слоев недостаточно для формирования мезофазы. Необходимо существование в системе вторичных связей. Такие взаимодействия в кристалле I действительно существуют (рис. 4). Это взаимодействия С–Н···π, связывающие молекулы в зигзагообразные цепочки. Как обычно бывает при взаимодействиях такого рода, атом водорода проектируется не в центр ароматического кольца, а сдвинут к одной из его связей.
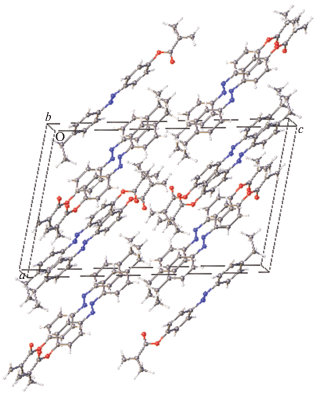
Рис. 3.
Фрагмент кристаллической упаковки I, состоящий из двух слоев; приведены кратчайшие межслоевые расстояния в Å.
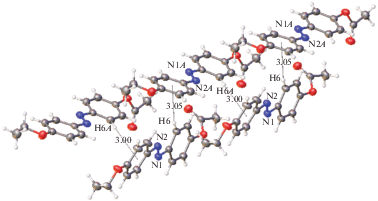
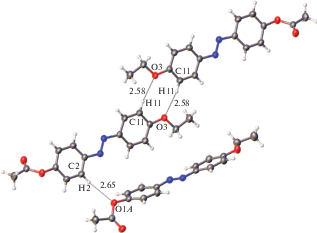
Кроме того, в данной упаковке обнаруживаются очень слабые водородные связи двух типов С–Н···О (рис. 5). Молекула образует центросимметричный димер за счет пары взаимодействий О3···Н11–С11. Эта же молекула соединена со второй независимой молекулой (атомы обозначены буквой А) водородной связью С2–Н2···О1А. Во всех случаях во взаимодействиях участвует достаточно “кислый” водород, поскольку в орто-положении для связанного с ним углерода находится электроноакцепторный заместитель (или азо-группа, или алкилокси-заместитель). Отметим, что эти очень слабые водородные связи едва ли могут оказывать какое-то влияние на вид образующейся мезофазы, поскольку она возникает при довольно высокой температуре (122°С). Естественно предположить, что очень слабые водородные связи С–Н···О скорее всего разрушаются до достижения таких температур, если учесть, что даже более прочные водородные связи О–Н···О, существующие в воде, разрушаются при 100°С. Таким образом, в области температур существования мезофазы в ней имеются вторичные связи только одного типа (взаимодействия С–Н···π), которые обусловливают структурированность системы.
Несколько более сложный случай представляет соединение III. Проблема в том, что методом РСА исследована структура кристалла, который не является предшественником мезофазы, образующейся, согласно данным ДСК, в результате серии фазовых переходов, а именно IIIIII–IIIII–IIII–N–Iso (N – нематик, Iso – изотроп, IIIIII, IIIII, IIII – разные кристаллические модификации соединения III). Исследование методом РСА структуры IIIIII показало, что кристаллическая упаковка в структуре IIIIII не разделена на рыхлые алифатические и плотные ароматические области, поэтому она не может быть прямым источником мезофазы. Однако ее изучение позволяет выявить возможности, которыми располагает данная молекула для образования разных вторичных связей.
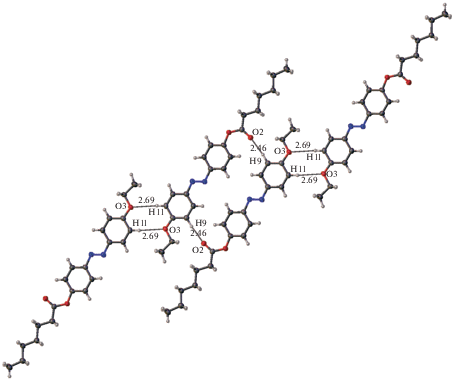
В кристалле III найдена система слабых водородных связей С–Н···О (рис. 6). Имеются контакты двух типов. Первый – это пара водородных связей между связанными центром инверсии молекулами: O3···H11–C11. Группа С–Н этого фрагмента находится в орто-положении относительно электроноакцепторного этокси-заместителя бензольного кольца, т.е. ее водород достаточно “кислый”, чтобы быть вовлеченным в водородную связь. Геометрические параметры этого взаимодействия (расстояния О···Н 2.69 Å, углы H11···O3–C10(С13) ∼127(115)° и C11–H11···O3 ~175°) оптимальны для водородной связи. Два таких трансляционно связанных димера образуют другой связанный центром симметрии димер за счет пары взаимодействий O2···H9–C9. Атом Н9 также расположен в орто-положении относительно акцепторного заместителя и является “кислым”. Хотя расстояние Н9···О2 (2.46 Å) заметно короче, угловые параметры (угол при атоме Н9 151° и угол H9···O2–C15 146°) менее благоприятны для образования водородной связи. Тем не менее хорошо известно, что угловая зависимость водородных связей (особенно слабых) не слишком сильно выражена, поскольку эти взаимодействия в основном имеют электростатическую природу.
Кроме описанных вторичных связей молекулы в данном кристалле образуют еще одну систему слабых взаимодействий. Уплощенные молекулы III упакованы в стопки (рис. 7). В этих стопках может осуществляться π–π-стэкинг, хотя это взаимодействие не самое эффективное вследствие существенного взаимного сдвига π-сопряженных систем соседних молекул (рис. 7б). Расстояние между π-системами, равное 3.41 Å в стопке А/B и 3.76 Å в стопке В/C, отвечает указанному взаимодействию.
Рис. 7.
Стопки молекул в кристалле III (а) и взаимное расположение молекул А и В, а также В и С (б).
При температуре ∼57°С в кристалле реализуется фазовый переход во вторую кристаллическую модификацию с энтальпией 3.18 кДж/моль. При наблюдении за этим процессом под микроскопом никаких видимых изменений в кристалле не обнаружено, т.е. данный фазовый переход не сопровождается разрушением кристалла. При этом переходе вполне возможно сохранение в кристалле IIIII слабых направленных взаимодействий обоих типов. Однако следующий фазовый переход при ∼61°C характеризуется высоким значением энтальпии (∼21.66 кДж/моль) и сопровождается превращением кристалла в мелкокристаллический порошок IIII. По-видимому, в момент перехода структура не разрушается радикально до аморфного состояния, а какие-то элементы упаковки предыдущей модификации сохраняются, и скорее всего это фрагменты стопочной структуры, образованной вследствие π–π-стэкинг-взаимодействий.
Дальнейшее повышение температуры до ∼66°C сопровождается очередным фазовым переходом в мезофазу с низким уровнем структурированности (нематик). Температурная область существования образовавшейся нематической фазы (от 66 до ∼119°С) доходит до довольно высоких значений. Это дает основание считать, что структурированность данной мезофазы обусловлена достаточно сильными стэкин-взаимодействиями, а не очень слабыми водородными связями.
Работа выполнена при финансовой поддержке Российского научного фонда (грант № 16-13-10273). Образцы синтезированы и охарактеризованы И.И. Константиновым в рамках государственной программы ИНХС РАН.
Список литературы
Lehman O. // Z. Phys. Chem. 1889. V. 4. P. 462.
Maier W., Saupe A. // Z. Naturforsch. 1960. B. 15a. S. 287.
Humphrie R.L., Jame P.G., Luckhurs G.R. // J. Chem. Soc. Faraday Trans. II. 1972. V. 68. P. 1031.
De Gennes P.G. // Mol. Cryst. Liq. Cryst. 1973. V. 21. P. 49.
Mc Millan W.L. // Phys. Rev. A. 1973. V. 8. P. 1921.
Wulf A. // Phys. Rev. A. 1975. V. 11. P. 365.
Cotter M.A. // Mol. Cryst. Liq. Cryst. 1983. V. 97. P. 29.
Osipov M.A. Molecular Theories of Liquid Crystals. Sections 2. Ch. III. V. 1 // Handbook of Liquid Crystals. / Ed. Demus D. et al. Weinheim: Wiley–VCH, 1998. P. 40.
Vertoge G., Jeu W.H. Thermotropic Liquid Crystals, Fundamentals. Berlin: Springer-Verlag, 1988. 324 p.
Gennes P.G. de, Prost J. The Physics of Liquid Crystals. New York: Oxford University Press, 1995. 616 p.
Singh S. // Phys. Rep. 2000. V. 324. № 2–4. P. 107.
Кузьмина Л.Г., Константинов И.И., Беззубов С.И. // Химия высоких энергий. 2016. Т. 50. № 6. С. 478.
Кочетов А.Н., Кузьмина Л.Г., Чураков А.В. и др. // Кристаллография. 2006. Т. 51. № 1. С. 59.
Кузьмина Л.Г., Кучерепа Н.С., Пестов С.М. и др. // Кристаллография. 2009. Т. 54. № 5. С. 908.
Кузьмина Л.Г., Пестов С.М., Кочетов А.Н. и др. // Кристаллография. 2010. Т. 55. № 5. С. 835.
Кузьмина Л.Г., Кучерепа Н.С. // Кристаллография. 2011. Т. 56. № 2. С. 267.
Кузьмина Л.Г., Кучерепа Н.С., Чураков А.В. // Кристаллография. 2012. Т. 56. № 2. С. 212.
Kuz’mina L.G., Navasardyan M.A., Churakov A.V., Howard J.A.K. // Mol. Cryst. Liq. Cryst. 2016. V. 638. P. 60.
Константинов И.И., Чураков А.В., Кузьмина Л.Г. // Кристаллография. 2013. Т. 58. № 1. С. 89.
Kuz’mina L.G., Konstantinov I.I., Lermontova E.Kh. // Mol. Cryst. Liq. Cryst. 2014. V. 588. P. 1.
Kuz’mina L.G., Konstantinov I.I., Churakov A.V., Navasardyan M.A. // Acta Cryst. E. 2017. V. 73. P. 1052.
Vogel A.I. Practical Organic Chemistry. 3rd Ed. London: Longman, 1966. 622 p.
Bruker (2008). APEX2, SADABS and SAINT. Bruker AXS Inc. Madison, Wiskonsin, USA.
Dolomanov O.V., Bourhis L.J., Gildea R. J. et al. // J. Appl. Cryst. 2009. V. 42. P. 339.
Desiraju G.R., Syeiner T. The Weak Hydrogen Bond in Structural Chemistry and Biology. Oxford: Oxford University Press, 1999.
Nangia A. // Cryst. Eng. Comun. 2002. V. 4. № 17. P. 93.
Muller-Dethlefs K., Hobza P. // Chem. Rev. 2000. V. 100. P. 143.
Janiak Ch. // J. Chem. Soc. Dalton Trans. 2000. P. 3885.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография