

Кристаллография, 2021, T. 66, № 5, стр. 776-781
Пространственная структура термостабильной трансаминазы D-аминокислот из археи Methanocaldococcus jannaschii DSM 2661
К. М. Бойко 1, *, А. Ю. Николаева 2, А. К. Бакунова 1, Т. Н. Стеханова 1, Т. В. Ракитина 2, В. О. Попов 1, 2, Е. Ю. Безсуднова 1
1 Институт биохимии им. А.Н. Баха РАН, Федеральный исследовательский центр “Фундаментальные основы биотехнологии” РАН
Москва, Россия
2 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
* E-mail: boiko_konstantin@inbi.ras.ru
Поступила в редакцию 14.07.2020
После доработки 14.08.2020
Принята к публикации 14.08.2020
Аннотация
Пиридоксаль-5'-фосфат-зависимые трансаминазы (PLP) катализируют стереоспецифический перенос аминогруппы с аминокислоты или амина на кетон или кетокислоту. Трансаминазы участвуют в метаболизме аминокислот у всех организмов; на основе этого суперсемейства ферментов активно разрабатываются биокатализаторы стереоселективного аминирования органических соединений для тонкого органического синтеза. Представлены краткая биохимическая характеристика и кристаллическая структура термостабильной трансаминазы I типа укладки PLP-связывающего домена из термофильной археи Methanocaldococcus jannaschii DSM 2661, полученная с разрешением 1.8 Å. Описаны строение функционального димера фермента и организация его активного центра, проведено сравнение с ближайшими гомологами.
ВВЕДЕНИЕ
Пиридоксаль-5'-фосфат-зависимые трансаминазы (PLP) (TА, аминотрансферазы, EC 2.6.1.Х) катализируют обратимый стереоселективный перенос аминогруппы с аминосубстрата (амин/аминокислота) на кетосубстрат (кетон/кетокислота/альдегид) с образованием хирального амина/аминокислоты и нового кетосоединения [1]. В клетках всех организмов ТА являются ключевыми ферментами метаболизма аминокислот. В биотехнологии ТА применяются для стереоселективного аминирования органических соединений, на основе природных ТА разрабатываются биокатализаторы для синтеза оптически активных аминов и неприродных аминокислот [2–6].
Принятая на сегодня классификация ТА составлена по множественному выравниванию последовательностей в базе данных семейств белковых доменов PFAM и объединяет, ориентируясь на субстратную специфичность, ТА в классы и подклассы (семейства) по паре реагирующих субстратов: донору аминогруппы/акцептору аминогруппы (механизм трансаминирования приведен ниже). Имеются шесть основных классов ТА [1]. Кроме того, все ТА разделяются по типу укладки PLP-связывающего домена на ферменты I или IV типа укладки [7, 8].
Любой фермент характеризуется оптимальным природным субстратом (или набором субстратов). Для всех ТА такими субстратами являются α-аминокислоты и их кетоаналоги, однако некоторые ферменты в определенных условиях активны с отличными от природных субстратов соединениями, т.е. проявляют разнородную (промискуитетную) активность (promiscuous activity). Такая активность представляет интерес для целей биотехнологии, так как позволяет использовать ферменты в синтезе неприродных соединений [9].
Трансаминазы III класса относятся к I типу PLP-укладки и являются примером ферментов с разнородной активностью, т.е. проявляют активность не только с α-, β-, γ- и ε-аминокислотами и их кетоаналогами, но и с (S)-ароматическими и алифатическими аминами, а также с альдегидами и кетонами [10, 11]. Трансаминазы III класса, катализирующие аминирование (S)-8-амино-7-оксононаноата (КАРА) с образованием 7,8-диаминопеларгоновой кислоты (DAPA), составляют подсемейство DAPA TA и в клетках участвуют в синтезе биотина [12]. Большинство DAPA ТА узкоспецифичны, используют S-аденозил-L-метионин (SAM) в качестве аминодонора. Среди ТА III класса – это одни из самых “медленных” ферментов [13, 14]. В [15] была охарактеризована DAPA TA из Psychrobacter cryohalolentis, проявляющая активность не только к природным SAM, DAPA и KAPA, но и к (S)-(-)-1-фенилэтиламину (S-PEA) и ряду альдегидов и кетонов. Для проявления такой разнородной субстратной специфичности в активном центре фермента присутствует ряд аминокислотных замен, а также повышенная мобильность отдельных остатков по сравнению с аминокислотными остатками в активном центре канонической DAPA TA из Escherichia coli.
Исследованная в настоящей работе термостабильная DAPA ТА из археи Methanocaldococcus jannaschii DSM 2661 (BioA, EC 2.6.1.62) гомологична описанной ранее DAPA ТА из P. cryohololentis. Охарактеризована неприродная активность BioA в реакции с S-PEA и его энантиомером R-PEA с кетосубстратом изобутаналем, получена пространственная структура фермента с разрешением 1.8 Å и проведены ее детальный анализ и сравнение со структурами гомологичных ТА. Полученные данные дополняют многообразие структур ТА I типа укладки, а также могут быть полезны при разработке новых термостабильных биокатализаторов, проявляющих максимальную активность и стабильность при температурах 50°С и выше, а также стабильных в присутствии органических растворителей, улучшающих растворимость органических субстратов в биотехнологических процессах.
МАТЕРИАЛЫ И МЕТОДЫ
Клонирования, экспрессия и очистка BioA. Ген BioA был получен полимеразной цепной реакцией (ПЦР) с использованием геномной ДНК M. jannaschii DSM 2661 [16] в качестве матрицы. Продукт ПЦР лигировали в плазмиду pET-21b (Novagen, Germany) и модифицировали согласно методике, описанной в [17], для получения целевого белка, на N-конце слитого с His-тагом и сайтом узнавания TEV протеазы. Трансформированные клетки E. coli BL21Star (Stratagene, USA) выращивали в среде LB/ампицилин при 37°C до А600 = 0.8, экспрессию индуцировали 1 мМ изопропил-β-тиогалактопиранозида. После инкубирования при 25°C клетки собирали центрифугированием, далее ресуспендировали в 50 мМ Трис-HCl-буфере, pH 7.5, содержащем 200 мМ NaCl, 20 мМ имидазола, 10% глицерина, 2 мМ β-меркаптоэтанола, 0.1% Тритона X-100 и 1 мМ фенилметилсульфонилфторида, и разрушали ультразвуком. Грубый экстракт центрифугировали при 10 000 g в течение 25 мин. Супернатант наносили на колонку HisTrap HP (GE Healthcare, USA), уравновешенную 50 мМ Трис-HCl-буфером, pH 7.5, содержащим 500 мМ NaCl, 20 мМ имидазола и 0.1% Тритона X-100. BioA с 6-His-тагом элюировали линейным градиентом имидазола (20–500 мМ) в том же буфере без Тритона X-100. Фракцию с целевым ферментом инкубировали с десятикратным молярным избытком PLP при 4°C в течение 12 ч, переносили в буфер 50 мМ Трис-HCl, pH 7.9, содержащий 100 мМ NaCl, 1 мМ ЭДТА, 1 мМ β-меркаптоэтанола и TEV протеазу (1 мг на 10 мг белка). Раствор инкубировали при комнатной температуре в течение 2 ч, диализировали против 50 мМ Трис-HCl-буфера, pH 7.5, содержащего 500 мМ NaCl, 20 мМ имидазола, и наносили на колонку HisTrap HP. Проскок собирали, концентрировали и наносили на колонку Superdex 200 10/300 GL (GE Healthcare), уравновешенную 50 мМ Трис-HCl-буфером, pH 7.5, содержащим 50 мМ NaCl и 15 мкМ PLP. Очищенный BioA в холоформе хранили при –20°C с добавлением 50% глицерина. Чистоту белка подтверждали при помощи ДДС-ПААГ электрофореза. Концентрацию белка определяли методом Бредфорда.
Определение активности BioA и стационарных параметров реакции. Удельную активность BioA в реакции с R-PEA и S-PEA с изобутаналем в качестве аминоакцептора рассчитывали из начального линейного участка зависимости накопления продукта от времени. За единицу ферментативной активности U принимали количество BioA, катализирующее образование 1 мкМ ацетофенона в минуту. За образованием ацетофенона следили по увеличению оптической плотности при 245 нм (∆ε = 12 мМ–1 см–1) на спектрофотометре Evolution 300 UV-Vis (Thermo Scientific, USA). Стандартную реакцию проводили при 65°C в 100 мМ бикарбонатном буфере, pH 10, с добавлением 90 мкМ PLP при концентрации каждого субстрата 10 мМ.
Для получения кинетических параметров варьировали концентрацию одного субстрата при постоянной концентрации второго (концентрация для изобутаналя составляла 12 мМ, для R-PEA – 10 мМ). Измерения проводили при 35°C в 75 мМ натрий-фосфатном буфере, pH 8, с добавлением 90 мкМ PLP. Кинетические параметры получали, обрабатывая экспериментальные данные согласно уравнению Михаэлиса–Ментен, используя Origin 7.5 (Origin Lab, USA).
Кристаллизацию BioA проводили методом диффузии в парах, как описано в [18]. Финальные условия кристаллизации включали 0.2 M MgCl2; 0.1 M Tris, pH 8.5; 19% PEG 3350, кристаллизация велась при температуре +20°С. Кристаллы палочкообразной формы размером ∼100 × 100 × 50 мкм росли в течение семи дней.
Сбор и обработка дифракционных данных. Решение и уточнение структуры. Перед сбором дифракционных данных кристаллы вылавливали петлей и переносили в криораствор, содержащий кроме компонентов, входящих в противораствор, 25% глицерина. После чего кристалл в петле замораживали в парах азота. Дифракционные наборы собирали при температуре 100 K на станции ID29 синхротронного источника ESRF (Франция). Дифракционная картина фиксировалась детектором Pilatus6M. Для расчета стратегии сбора данных использовали программу Best [19], предложившую следующие параметры: длина волны 0.969 Å, угол вращения 287°, угол качания 0.1°, расстояние кристалл–детектор 302 мм. Набор дифракционных данных обработан с использованием программы iMosflm [20]. Статистика набора данных приведена в табл. 1.
Таблица 1.
Кристаллографические данные и параметры съемки кристалла трансаминазы BioA
| Пр. гр. | P1 |
| a, b, c, Å; α, β, γ, град | 61.23, 61.38, 63.07; 106.11, 93.48, 96.47 |
| Т, K | 100 |
| λ, Å | 0.969 |
| Разрешение, Å | 60.56–1.80 (1.84–1.80)* |
| Число независимых рефлексов | 71599 (4097) |
| Повторяемость | 2.7 (2.6) |
| Полнота набора, % | 88.5 (86.1) |
| I/σ (I) | 6.4 (2.4) |
| Rmeas, % | 12.6 (53.3) |
| CC1/2,% | 98.9 (75.5) |
Решение структуры проведено методом молекулярного замещения при помощи программы MOLREP [21]. В качестве стартовой модели использовали структуру холо-формы синтазы диаминопералгоновой кислоты из Bacillus subtilis (код PDB 3DOD). Кристаллографическое уточнение структуры проведено с использованием программ Refmac5 [22] и Coot [23] до достижения R-факторами следующих значений: Rwork = 17.7%, Rfree = 22.2% (табл. 2).
Таблица 2.
Данные уточнения структуры трансаминазы BioA
| Rfact, % | 17.7 |
| Rfree, % | 22.2 |
| Общий средний B-фактор | 22.0 |
| Средний B-фактор по белку | 21.8 |
| Средний B-фактор по лигандам | 23.3 |
| Средний B-фактор по растворителю | 27.2 |
| Число неводородных атомов | |
| Белок | 6958 |
| Лиганды | 3 |
| Растворитель | 446 |
| Всего | 7407 |
| Среднеквадратичные отклонения | |
| Длины связей, Å | 0.013 |
| Валентные углы, град | 1.842 |
| График Рамачандрана | |
| Наиболее благоприятные, % | 94.5 |
| Допустимые, % | 4.4 |
| Код PDB | 6ZHK |
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Активность BioA и кинетические параметры полной реакции. Удельная активность выделенных препаратов BioA с R-PEA и S-PEA соответственно составила 42 ± 2 и 21 ± 1 мU/мг при 65°C в 100 мМ бикарбонатном буфере, pH 10. Кинетические параметры катализируемой BioA реакции между R-PEA и изобутаналем приведены в табл. 3. Можно сделать вывод, что BioA стабилен и активен в щелочных условиях при 65°C, однако проявляет низкую стереоспецифичность к неканоническим субстратам.
Таблица 3.
Кинетические параметры катализируемой BioA реакции между R-PEA и изобутаналем при 65°C
| Субстрат | kcat, с–1 | Km, мМ | kcat/Km, с–1 мМ–1 |
|---|---|---|---|
| R-PEA | 0.033 ± 0.001 | 0.9 ± 0.1 | 0.037 ± 0.005 |
| Изобутаналь | 0.0162 ± 0.0005 | 0.19 ± 0.03 | 0.08 ± 0.02 |
Анализ структуры BioA. Кристаллы BioA принадлежат пр. гр. P1. В независимой части элементарной ячейки кристалла находятся две субъединицы белка, образующие функциональный димер [1, 8]. В обеих субъединицах в электронной плотности отсутствуют восемь аминокислот с N-конца, а также участок 190–209, что, по-видимому, связано с их относительной подвижностью. Попарное сравнение субъединиц между собой дает RMSD по Сα-атомам, не превышающее 0.14 Å2, свидетельствующее об отсутствии между ними значимых различий.
Общая архитектура субъединицы BioA типична для ТА I типа укладки и состоит из двух доменов – малого N-концевого (остатки 1–58 и 359–466) и большого С-концевого (остатки 59–358) (рис. 1а) [7]. N-концевой домен BioA содержит пять α-спиралей и два β-слоя по три тяжа каждый, а С-концевой – 9 α-спиралей, окружающих β-слой, состоящий из семи тяжей.
Рис. 1.
Структура BioA. Димер BioA (а). N-концевой домен одной из субъединиц димера показан в виде черной ленточной модели, С-концевой – в виде серой ленточной модели. Вторая субъединица димера показана в виде прерывистой ленточной модели. Ионы магния показаны сферами. Субстратный канал BioA (б). Серым показана поверхность димера BioA (центральный срез) и каналы, ведущие от поверхности молекулы в активный центр фермента. Молекула PMP взята из структуры 6IO1 и показана палочковой моделью для одной субъединицы. Активный центр каждой субъединицы обозначен стрелкой.
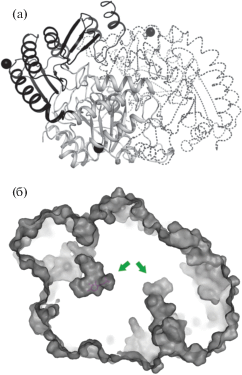
Анализ контактов между субъединицами из независимой части показал, что BioA аналогично многим другим ТА [1, 8, 15] является димером в кристалле. Скрытая при формировании димера поверхность (buried surface area) составляет 5678 Å2 (28.5% поверхности субъединицы). Димер стабилизирован девятью солевыми мостиками и 50 водородными связями. Оценка гидрофобных взаимодействий, сделанная по выигрышу свободной энергии при формировании димерного межмолекулярного контакта, дает величину ‒96.3 ккал/моль (табл. 4).
Таблица 4.
Сравнение димерных межмолекулярных контактов BioA и гомологичных ферментов
Димер BioA является минимальной каталитической единицей фермента, что связано с устройством активного центра ТА, включающего в себя остатки обеих субъединиц, а именно – двух доменов одной субъединицы и С-концевого домена соседней. Активный центр BioA находится глубоко в центре каждой из субъединиц димера и соединен с поверхностью протяженным каналом длиной ∼15 Å (рис. 1б). Полость активного центра сформирована остатками W63, D121, G122, A123, E124, V126, Y155, H156, G157, E234, D268, V270, A271, G336, H337 и T338, а также каталитическим лизином K301. Для данного класса ферментов известно, что помимо каталитических остатков в активном центре находится молекула кофактора – PLP, ковалентно связанная в структуре холо-формы с каталитическим остатком лизина и отсоединяющаяся от него в процессе катализа с образованием свободной PMP-формы, способной покидать активный центр. Анализ активного центра BioA показал, что в обеих субъединицах фермента отсутствует разностная электронная плотность для кофактора, поэтому каталитический остаток лизина K301 находится в свободной форме. Возможно, в случае BioA отсутствие кофактора вызвано условиями кристаллизации.
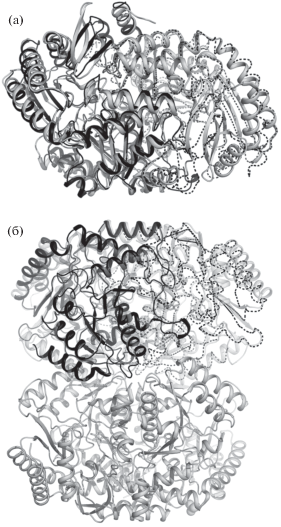
Сравнение субъединицы BioA с гомологичными структурами показало, что наибольшее сходство фермент имеет со структурами синтазы диаминопералгоновой кислоты из Bacillus subtilis (RMSD – 1.08 Å2, код PDB – 3DRD), термостабильной ω-трансаминазы из Thermomicrobium roseum (RMSD – 1.50 Å2, код PDB – 6IO1) и ТА из Pseudomonas fluorescens (RMSD – 1.57 Å2, код PDB – 6S54) (рис. 2а). Несмотря на сходство структур субъединиц, указанные ферменты имеют различное олигомерное состояние в кристалле – димер (3DRD и 6IO1) и тетрамер (6S54), что, впрочем, типично для ТА [1]. Тем не менее все указанные олигомерные состояния включают в себя аналогичный BioA димер (рис. 2б), что позволяет провести их взаимное сравнение. Анализ контактов упомянутых димеров (табл. 4) показывает, что димерное межмолекулярное взаимодействие данного класса ферментов весьма прочно даже в случае белков из мезофильных организмов (например, 6S54). При этом в случае тетрамера 6S54 межмолекулярный контакт, аналогичный димеру BioA, значительно превосходит по прочности остальные, что подчеркивает ключевую роль димеризации для функционирования фермента.
Рис. 2.
Сравнение структур BioA и ближайших гомологов. Наложение димеров BioA (субъединицы показаны в виде сплошной и прерывистой ленточных моделей соответственно) и 3DRD (светло-серый) (а). Наложение димера BioA (окраска аналогична панели А) и тетрамера 6S54 (светло-серый) (б). Димер 6S54, аналогичный димеру BioA, показан полупрозрачно для ясности.
Сравнение активных центров BioA и гомологичных ферментов позволило установить, что связанный кофактор присутствует не во всех сравниваемых структурах. Так, помимо BioA он отсутствует в структурах 3DRD и в одной из субъединиц 6IO1, при этом во второй субъединице обнаружена PMP-форма кофактора. В структуре 6S54 ковалентно связанный PLP находится во всех четырех субъединицах из независимой части. Анализ окружения области связывания кофактора показал, что в случае BioA остатки, формирующие окружение PLP, в основном консервативны, при этом в структуре 3DRD отсутствует петля 143–173 (нумерация по 3DRD), закрывающая активный центр от растворителя. В отсутствие кофактора в BioA вакантное положение его фосфатной группы занято боковой группой остатка H337, имеющей двойное положение, чего не наблюдается в структуре гомологов, где крупная боковая группа гомологичного ароматического остатка отвернута от кофактора. Неконсервативный S240 в BioA (аланин у гомологов) создает полярную область на входе в полость активного центра. Еще одним отличием BioA является наличие вблизи активного центра пары неконсервативных полярных остатков – D158 и Y25, боковые группы которых образуют водородную связь между собой, а также с каталитическим Y155. В положении, аналогичном Y25, у гомологов находится гидрофобный остаток. Таким образом, в случае BioA пара D158–Y25 может дополнительно стабилизировать структуру активного центра, являясь следствием адаптации к повышенной температуре.
Таким образом, исследована структура термостабильной ТА из археи Methanocaldococcus jannaschii DSM 2661, проведено ее сравнение со структурами гомологичных ферментов и выявлены различия в активном центре, которые могут отражать субстратную специфичность BioA.
Работы по экспрессии и выделению белка выполнены при финансовой поддержке Российского научного фонда (грант № 19-14-00164), кристаллизация и рентгеноструктурный эксперимент выполнены при поддержке Российского Федерального космического агентства (проект КЭ (ЦР) “Кристаллизатор”), работы по кристаллографическому уточнению и анализу полученной структуры выполнены при поддержке Министерства науки и высшего образования РФ.
Список литературы
Bezsudnova E.Y., Boyko K.M., Popov V.O. // Biochemistry (Mosc.) 2017. V. 82. P. 1572. https://doi.org/10.1134/S0006297917130028
Fuchs M., Farnberger J.E., Kroutil W. // Eur. J. Org. Chem. 2015. V. 2015. P. 6965. https://doi.org/10.1002/ejoc.201500852
Guo F., Berglund P. // Green Chem. 2017. V. 19. P. 333. https://doi.org/10.1039/C6GC02328B
Slabu I., Galman J.L., Turner N.J. et al. // Acs. Catalysis. 2017. V. 7. P. 8263. https://doi.org/10.1021/acscatal.7b02686
Steffen-Munsberg F., Vickers C., Kohls H. et al. // Biotechnol. Adv. 2015. V. 33. P. 566. https://doi.org/10.1016/j.biotechadv.2014.12.012
Wu S., Snajdrova R., Moore J.C. et al. // Angew. Chem. Int. Ed. Engl. 2020. V. 60. № 1. P. 88. https://doi.org/10.1002/anie.202006648
Grishin N.V., Phillips M.A., Goldsmith E.J. // Protein Sci. 1995. V. 4. P. 1291. https://doi.org/10.1002/pro.5560040705
Bezsudnova E.Y., Popov V.O., Boyko K.M. // Appl. Microbiol. Biotechnol. 2020. V. 104. P. 2343. https://doi.org/10.1007/s00253-020-10369-6
Hult K., Berglund P. // Trends Biotechnol. 2007. V. 25. P. 231. https://doi.org/10.1016/j.tibtech.2007.03.002
Kaulmann U., Smithies K., Smith M.E.B. et al. // Enzyme Microbial Technology. 2007. V. 41. P. 628. https://doi.org/10.1016/j.enzmictec.2007.05.011
Steffen-Munsberg F., Matzel P., Sowa M.A. et al. // Appl. Microbiol. Biotechnol. 2016. V. 100. P. 4511. https://doi.org/10.1007/s00253-015-7275-9
Stoner G.L., Eisenberg M.A. // J. Biol. Chem. 1975. V. 250. P. 4037. https://doi.org/10.1016/S0021-9258(19)41381-1
Cobessi D., Dumas R., Pautre V. et al. // Plant Cell. 2012. V. 24. P.1608. https://doi.org/10.1105/tpc.112.097675
Mann S., Ploux O. // Febs J. 2006. V. 273. P. 4778. https://doi.org/10.1111/j.1742-4658.2006.05479.x
Bezsudnova E.Y., Stekhanova T.N., Popinako A.V. et al. // Appl. Microbiol. Biotechnol. 2018. V. 102. P. 9621. https://doi.org/10.1007/s00253-018-9310-0
Bult C.J., White O., Olsen G.J. et al. // Science. 1996. V. 273. P. 1058. https://doi.org/10.1126/science.273.5278.1058
Boyko K., Gorbacheva M., Rakitina T. et al. // Acta Cryst. F. 2015. V. 71. P. 24. https://doi.org/10.1107/S2053230X14025333
Boyko K.M., Lipkin A.V., Popov V.O. et al. // Crystallography Reports. 2013. V. 58. P. 442. https://doi.org/10.1134/S106377451105004X
Bourenkov G.P., Popov A.N. // Acta Cryst. D. 2006. V. 62. P. 58. https://doi.org/10.1107/S0907444905033998
Battye T.G., Kontogiannis L., Johnson O. et al. // Acta Cryst. D. 2011. V. 67. P. 271. https://doi.org/10.1107/S0907444910048675
Vagin A.A., Isupov M.N. // Acta Cryst. D. 2001. V. 57. P. 1451. https://doi.org/10.1107/s0907444901012409
Vagin A.A., Steiner R.A., Lebedev A.A. et al. // Acta Cryst. D. 2004. V. 60. P. 2184. https://doi.org/10.1107/S0907444904023510
Emsley P., Lohkamp B., Scott W.G. et al. // Acta Cryst. D. 2010. V. 66. P. 486. https://doi.org/10.1107/S0907444910007493
Dey S., Lane J.M., Lee R.E. et al. // Biochemistry. 2010. V. 49. P. 6746. https://doi.org/10.1021/bi902097j
Kwon S., Lee J.H., Kim C.M. et al. // Sci Rep. 2019. V. 9. P. 6958. https://doi.org/10.1038/s41598-019-43490-2
Roura Padrosa D., Alaux R., Smith P. et al. // Front Bioeng Biotechnol. 2019. V. 7. P. 282. https://doi.org/10.3389/fbioe.2019.00282
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография