

Кристаллография, 2021, T. 66, № 5, стр. 829-835
Определение околоатомной структуры олигомерных белков E. coli без предварительной очистки
Е. Б. Пичкур 1, 2, В. И. Микиртумов 3, О. В. Тихонова 4, Н. И. Деркачева 5, Л. П. Курочкина 6, О. С. Соколова 3, *
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
2 Петербургский институт ядерной физики им. Б.П. Константинова НИЦ “Курчатовский институт”
Гатчина, Россия
3 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
4 Научно-исследовательский институт биомедицинской химии им. В.Н. Ореховича
Москва, Россия
5 Московский государственный медико-стоматологический университет им. А.И. Евдокимова
Москва, Россия
6 Московский государственный университет им. М.В. Ломоносова,
НИИ физико-химической биологии им. А.Н. Белозерского
Москва, Россия
* E-mail: sokolova@mail.bio.msu.ru
Поступила в редакцию 04.05.2020
После доработки 04.05.2020
Принята к публикации 29.05.2020
Аннотация
С помощью криоэлектронной просвечивающей микроскопии изучен грубо очищенный экстракт белков E. coli, на основе 2D-классификации проекций выделены классовые суммы, содержащие проекции двух белков: β-галактозидазы и каталитического дигидролипоилтранссукцинилазного домена 2-оксоглутаратдегидрогеназного комплекса, идентифицированных в экстракте методом тандемной масс-спектрометрии. Решены их структуры с разрешением, близким к атомному. В результате моделирования структуры каталитического домена 2-оксоглутатдегидрогеназного комплекса de novo получена атомная модель, выявляющая различия в положении некоторых аминокислотных остатков активного центра по сравнению с ранее полученными кристаллическими структурами.
ВВЕДЕНИЕ
В последние десятилетия структурные методы биологии претерпели бурное развитие, что выразилось в определении новых атомных структур различных белков. Криоэлектронная просвечивающая микроскопия (крио-ПЭМ) не так давно была признана в качестве одного из основных инструментов при изучении белковых структур с высоким разрешением наряду с рентгеноструктурным анализом и ядерно-магнитной спектроскопией (ЯМР) [1]. Использование этого метода резко возросло в связи с определенными технологическими достижениями. К ним относятся не только новые приборы, прямые детекторы электронов, фазовые пластинки, но и такие программные средства, как Relion [2], cryoSPARC [3], Chimera [4, 5] и Coot [6]. В результате удалось добиться впечатляющих результатов при изучении белков и их комплексов. Ярким примером преимущества крио-ПЭМ может служить полученная за месяц структура S-белка коронавируса 2019-nCoV [7]. Эти данные позволили выявить принципиальные раличия в структуре близкородственных вирусов, вызывающих инфекции SARS и COVID-19.
В настоящее время узким местом при получении данных с высоким разрешением остается биохимическая очистка белков. Этот этап трудно автоматизировать, поскольку он в значительной степени опирается на человеческий опыт. Сохраняется ряд проблем, связанных с трудностями выбора системы экспрессии белка, метода его очистки и концентрации функционального белка. Цель состоит в том, чтобы свести к минимуму количество шагов очистки, минимизировать их влияние на белок и получить достаточное количество гомогенного препарата для дальнейшего структурного анализа.
В представленной работе к решению этой проблемы подошли с технологической стороны. При осаждении рекомбинантного белка из экстракта продуцировавших его клеток E. coli сульфатом аммония получен препарат, содержащий наряду с целевым некоторые клеточные белки. Препарат был изучен в крио-ПЭМ и решены структуры двух идентифицированных масс-спектрометрией белков E. coli: β-галактозидазы и каталитического дигидролипоилтранссукцинилазного домена 2-оксоглутаратдегидрогеназного комплекса (КД-ОДК) с разрешением, близким к атомному. В результате моделирования структуры КД-ОДК de novo построена атомная модель, выявляющая различия в положении некоторых аминокислотных остатков (а.о.) активного центра по сравнению с известными кристаллическими структурами [8, 9].
МЕТОДИКА ИССЛЕДОВАНИЙ
Подготовка белков E. coli. Выделение и осаждение белков E. сoli проводили в соответствии с процедурой, описанной в [10]. Клетки BL21(DE3) E. сoli, продуцирующие рекомбинантный шаперонин бактериофага ОВР, наращивали в большом объеме среды 2xTY при температуре 37°C до оптической плотности A600 = 0.7. Экспрессию белка индуцировали добавлением изопропил-β-D-тиогалактопиранозида до конечной концентрации 1 мМ и растили клеточную культуру в течение 3.5 ч при 25°C. Клетки осаждали центрифугированием, ресуспендировали в 50 мМ трис-HCl-буфере (pH 7.5), подвергали ультразвуковой обработке с помощью дезинтегратора Virsonic 100 (Vertis, США) и центрифугировали (13000 g) для удаления клеточного дебриса. Нуклеиновые кислоты осаждали путем добавления в супернатант 1/10 объема 30% (w/v) раствора сульфата стрептомицина с последующим центрифугированием. Белки осаждали из супернатанта путем добавления насыщенного раствора сульфата аммония до конечной концентрации 30% (w/v). После центрифугирования осадок белка растворяли в 50 мМ Трис-HCl-буфере (рН 7.5), содержавшем 100 мМ NaCl, и промывали 30 объемами буфера (50 мМ Трис-HCl, рН 7.5 с добавлением 10 мМ MgCl2 и 100 мМ KCl) на фильтрах Amicon (Millipore, США). Состав белков анализировали с помощью тандемной масс-спектрометрии.
Масс-спектрометрический анализ. Гидролиз белка в растворе проводили с использованием трипсина (Sequencing Grade Modified, Promega, Madison, WI, USA), как описано в [12]. Анализ полученных при гидролизе пептидов осуществляли с использованием системы высокоэффективной жидкостной хроматографии Ultimate 3000 RSLCnano (Thermo Scientific, США), соединенной с масс-спектрометром Q ExactiveTM HF (Thermo Scientific, США). Пептиды перед аналитическим разделением наносили на обогащающую колонку Accalaim μ-Precolumn (0.5 × 3 мм, 5 мкм размер частиц; Thermo Scientific) при скорости потока 10 мкл/мин в течение 5 мин в изократическом режиме подвижной фазы Б (2% ацетонитрила, 0.1% муравьиной кислоты). Затем пептиды разделяли на колонке Acclaim Pepmap C18 (75 мкм × 150 мм, 2 мкм размер частиц; Thermo Scientific) в градиентном режиме элюирования. Градиент формировали подвижными фазами А (0.1% муравьиной кислоты) и Б (80% ацетонитрила, 0.1% муравьиной кислоты) при скорости потока 0.3 мкл/мин. Колонку промывали 2%-ной подвижной фазой Б в течение 5 мин, после чего концентрацию подвижной фазы Б линейно увеличивали до 35% в течение 40 мин, затем за 5 мин линейно увеличивали концентрацию фазы Б до 99%. После промывки колонки в течение 5 мин при 99% фазы Б концентрацию Б снижали до начальных условий – 2% фазы Б за 5 мин. Общая длительность анализа составила 60 мин.
Масс-спектрометрический анализ проводили в трех технических повторах на гибридном орбитальном масс-спектрометре Q ExactiveTM HF (Thermo Scientific, США) в режиме положительной ионизации с использованием источника NESI (Thermo Scientific, США). Для масс-спектрометрического анализа были установлены следующие параметры настроек: напряжение на эмиттере 2.1 кВ, температура капилляра 240°C. Панорамное сканирование проводили в диапазоне масс от 300 до 1500 m/z, тандемное сканирование фрагментных ионов от нижней границы 100 m/z до верхней границы, определяемой зарядным состоянием прекурсорного иона, но не более 2000 m/z. Для тандемного сканирования учитывали только ионы от z = 2+ до z = 6+ по зарядному состоянию. Максимальное число разрешенных для синхронной изоляции ионов в режиме MS2 было установлено не более 20-ти. Максимальное время накопления для прекурсорных ионов составило не более 50 мс, для фрагментных ионов – не более 110 мс.
Идентификацию белков по масс-спектрам проводили в поисковой системе MASCOT [13] с использованием базы данных аминокислотных последовательностей SwissProt с ограничением видовой принадлежности организма исследуемого образца E. coli. Для оценки FDR была сгенерирована база данных ложных последовательностей белков в виде обратных последовательностей аминокислот протеома E. coli. Параметры поиска установлены следующим образом: расщепляющий фермент – трипсин с возможностью пропуска одного сайта расщепления последовательности, точность определения масс-моноизотопных пиков пептидов ±5 ppm, точность определения масс в спектрах MS/MS ±0.01 Да. В качестве обязательной и возможной модификации было учтено карбамидометилирование цистеина и окисление метионина соответственно. Для валидации сопоставлений спектров и пептидов PSM, идентификации пептидов и идентификации белков использовали величину FDR не более 1.0%. Белки рассматривали в качестве достоверно идентифицированных, если для них было обнаружено по крайней мере два уникальных пептида, прошедших валидацию. Безметковая количественная оценка содержания белков проходила на основе эмпирического показателя emPAI [14].
Криоэлектронная просвечивающая микроскопия. Препарат белков E. coli (3.0 мкл) наносили на сетку с углеродной подложкой, имеющей регулярные отверстия диаметром 1.2 мкм и периодом 1.3 мкм (R1.2/1.3, Quantifoil). Cетки предварительно в течение 30 с обрабатывали тлеющим разрядом в установке для гидрофилизации PELCO easyGlow (Ted Pella, США) при давлении в камере 0.26 мБар и силе тока 25 мА. Сетки с нанесенным белком моментально замораживали в жидком этане в приборе Vitrobot Mark IV (Thermo Fischer Scientific) при 100%-ной влажности и температуре +4.5°С. Стеки микрографий по 38 кадров (4017 стеков) снимали в крио-микроскопе Titan Krios (Thermo Fisher Scientific) с прямым детектором электронов Falcon II в следующих условиях: ускоряющее напряжение – 300 кВ, увеличение – 75 000×, размер пикселя – 0.86 Å, поток электронов ~100 э/Å2. Стеки были предварительно обработаны в программе Warp [11] для оценки параметров функции передачи контраста (CTF) и коррекции дрейфа на отдельных кадрах стека. Изображения белковых молекул были выбраны с помощью нейронной сети BoxNet и вырезаны из микрографий с помощью рамки 256 × 256 пикселей с получением набора из 470 117 частиц. Из них более 250 000 частиц представляли OBP шаперонин [10]. 2D-классификацию проводили в программе CryoSPARC [3]. 2D-классы, соответствующие различным белкам, выбирали вручную с последующим генерированием модели ab-initio для каждого класса в отдельности. Затем выполнили 3D-уточнение структуры каждого класса с использованием предварительной модели без наложения симметрии. На следующем этапе полученные структуры были использованы в качестве ссылок для дальнейшей 3D-классификации исходного набора частиц. Финальные 3D-структуры белка реконструировали с наложенной симметрией: диэдральная D2 для β-галактозидазы, октаэдрическая (O) для КД-ОДК. Окончательные реконструкции включали 99 246 частиц для β-галактозидазы и 13 770 частиц для КД-ОДК. Разрешение реконструкций оценивали в соответствии с критерием FSC = 0.143, оно составило 2.6 Å для β-галактозидазы и 3.2 Å для КД-ОДК.
De novo моделирование структуры белка. Моделирование проводили с помощью программ Coot [6] и Phenix [15]. Разрешение карты КД-ОДК, полученное в крио-ПЭМ, было достаточным для идентификации боковых цепей и однозначного определения последовательности данного белка. Уникальная часть карты КД-ОДК извлечена с помощью алгоритма phenix.mapbox и NCS-операторов, полученных с помощью алгоритма phenix.mapsymmetry после задания симметрии группы точек “O”, атомная модель этой части построена автоматически с помощью алгоритма phenix.maptomodel. Полученную модель проверяли вручную в Coot, большие диапазоны остатков несоответствия и пустые зоны заполняли аланиновыми остатками; при необходимости формировали новые пептидные связи таким образом, чтобы модель состояла из одной цепи. После этого боковые цепи аланинов в модели были заменены на боковые цепи аминокислот, соответствующих последовательности С-концевой части белка КД-ОДК (174–405 а.о.). С помощью программы Coot провели ручную правку всех остатков для максимально точного соответствия карте электростатического потенциала при сохранении правильной геометрии остатков; для всех боковых цепей были выбраны ротамеры с их оптимальной ориентацией. Для отредактированной модели уникальной части структуры были применены операторы NCS, полученные с помощью алгоритма phenix.mapsymmetry.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Цель работы заключалась в том, чтобы показать, что крио-ПЭМ и обработка изображений позволяют получить 3D-структуры белковых молекул без тонкой очистки целевых белков. Для этого получили коктейль белков из клеток E. сoli, гетерологично экспрессирующих вирусный шаперонин (OBP) [10]. Рекомбинантный шаперонин осаждали из клеточного экстракта 30%-ным сульфатом аммония [16], при этом некоторое количество клеточных белков также оказалось в осадке. Идентификацию смеси осажденных белков E. coli проводили с использованием высокоэффективной жидкостной хроматографии с тандемной масс-спектрометрией. Результаты представлены в табл. 1. Относительные количества совместно осажденных белков оценивали с помощью показателя emPAI, рассчитанного в программе MASCOT. Наибольшее соответствие последовательности (39%) получено для β-галактозидазы (P00722), остальные семь белков представлены несколькими пептидами каждый.
Таблица 1.
Список достоверно идентифицированных белков E. coli
| Uniprot ID | Белок | Score | MW, kDa | Sequence Coverage, % | emPAI |
|---|---|---|---|---|---|
| P00722 | Beta-galactosidase (BGAL_ECOLI) | 4240 | 117.35 | 39 | 3.06 |
| P0A9L8 | Pyrroline-5-carboxylate reductase (P5CR_ECOLI) | 395 | 28.36 | 21 | 1.18 |
| P0A9P0 | Dihydrolipoyl dehydrogenase (DLDH_ECOLI) | 484 | 50.94 | 18 | 0.99 |
| P05055 | Polyribonucleotide nucleotidyltransferase (PNP_ECOLI) | 394 | 77.11 | 14 | 0.65 |
| P0A6F5 | 60 kDa chaperonin (CH60_ECOLI) | 396 | 57.46 | 15 | 0.65 |
| P0A9H3 | Inducible lysine decarboxylase (LDCI_ECOLI) | 275 | 81.61 | 7 | 0.54 |
| P0AFG7 | Succinyltransferase component of 2-oxoglutarate dehydrogenase complex (ODO2_ECOLI) | 311 | 43.98 | 6 | 0.34 |
| P0AES4 | DNA gyrase subunit A (GYRA_ECOLI) | 41 | 97.13 | 3 | 0.07 |
Смесь белков после осаждения и отмывки моментально заморозили в Vitrobot MARK IV (Thermo Fisher Scientific) и исследовали в крио-ПЭМ с использованием стандартного протокола (описан выше). Для идентификации различных белков провели два последовательных раунда 2D-классификации в CryoSPARC [3]. Первую классификация использовали для исключения из набора “мусорных” классов, после второй (рис. 1) выделили 40 классов, разделенных визуально на три набора данных, каждый из которых представлял отдельный белок.
Рис. 1.
Крио-ПЭМ-анализ экстракта белков E. coli: 2D-классификация и выделенные классы отдельных белков. Масштабные отрезки – 10 нм.
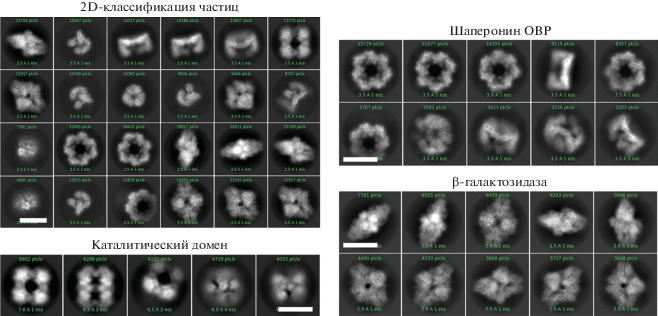
Три олигомерных белка: транзиентно экспрессируемый гептамерный шаперонин фага OBP P. fluorescens [10] и два белка E. сoli – тетрамерная β-галактозидаза и 24-мерный КД-ОДК, имеют частицы сопоставимого размера, 12–15 нм в поперечнике (рис. 1). Кроме этого, наблюдались классы, соответствующие более мелкому асимметричному белку неопределенной природы. Другие белки, пептиды которых были обнаружены при помощи масс-спектрометрии (табл. 1), не смогли идентифицировать на крио-ПЭМ-изображениях. Возможно, эти белки не образуют в используемых условиях стабильные олигомерные комплексы или их концентрации слишком малы для исследования методом крио-ПЭМ.
Наиболее распространенным белком E. coli в исследуемой пробе по данным тандемной масс-спектрометрии (табл. 1) и 2D-классификации (рис. 1) была β-галактозидаза (99 246 частиц). Она кодируется геном lacZ и образует стабильный гомотетрамер с молекулярной массой ∼464 кДа [17]. На крио-ПЭМ-изображениях он имеет хорошо узнаваемую форму и может быть легко отделен при помощи 2D-классификации (рис. 1). Стандартный подход к анализу изображений [18] позволил получить 3D-реконструкцию β-галактозидазы с симметрией D2 и разрешением 2.6 Å (рис. 2а–2в).
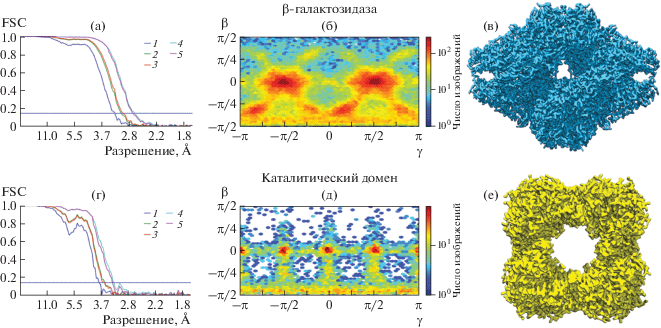
Рис. 2.
Построение трехмерных структур белков E. coli: β-галактозидазы (а–в) и КД-ОДК (г–е). Кривые функций корреляции (FSC) для соответствующих структур (а, г): 1 – без маски, 2 – сферическая маска, 3 – свободная маска, 4 – узкая маска, 5 – после коррекции; распределение проекций частиц относительно вертикальной оси z сферы Эйлера (б, д). β – угол между проекцией и осью z; γ – угол поворота вокруг оси z; изображения выполнены в cryoSPARC [3]. 3D-реконструкция β-галактозидазы (в); 3D-реконструкция КД-ОДК (е).
В состав 2-оксоглутаратдегидрогеназного комплекса E. coli входят три различных фермента: E1 (2-оксоглутаратдекарбоксилаза), E2 (дигидролипоилтранссукцинилаза) и E3 (дигидролипоилдегидрогеназа). Фермент Е2 катализирует перенос сукциниловой группы из S-сукцинил-дигидролипоила на кофермент А (КоА) [19]. Его полноразмерный мономер длиной 405 а.о. имеет структуру, в которой можно выделить три домена: N-концевой липоилсвязывающий домен, Е3-связывающий домен и С-концевой каталитический домен. Молекулярная масса всего белка ∼44 кДа.
В полученном белковом коктейле E. coli с помощью тандемной масс-спектрометрии идентифицировано только два значимых пептида для фермента E2 (один из N-концевого липоилсвязывающего домена и один из С-концевого каталитического домена). Тем не менее на полученных крио-ПЭМ-изображениях С-концевой домен Е2 (КД-ОДК) оказался вторым по численности белком после 2D-классификации (13 770 частиц). 2D-классификация показала характерные частицы с октаэдрической симметрией (рис. 1). Линейные размеры 2D-классов схожи с размерами ранее определенных кристаллических структур КД-ОДК [8, 9]. При сопоставлении полученных данных с результатами [8, 20] оказалось, что в полученной криоэлектронной структуре отсутствует плотность для N-концевых E3-связывающих и липоилсвязывающих доменов, а также соединенных с ними линкеров. Ранее предполагалось, что эти домены могут быть удалены эндогенными протеазами [9]. Структуры изолированных N-концевых доменов были решены с помощью ЯМР [21, 22].
Отметим, что кристаллы этого домена были ранее случайно получены при попытке кристаллизации рекомбинантной амидазы Arabidopsis thaliana, экспрессированной в гетерологичной системе E. coli [9]. Предполагалось, что остатки гистидина на поверхности каталитического домена могут способствовать связыванию его с никельсодержащей смолой [23], однако в данном исследовании мы не использовали Ni-аффинную хроматографию.
Реконструкция в cryoSPARC с октаэдрической симметрией КД-ОДК имеет разрешение 3.2 Å (рис. 2г–2е). Высокое разрешение позволило определить структуру каталитического домена de novo. Модель структуры создавали с помощью программы Coot [6] и трехмерных координат для каждого атома (рис. 3а).
Рис. 3.
Структура каталитического домена 2-оксоглутарат-дегидрогеназного комплекса. Атомная модель КД-ОДК, выполненная de novo; рамкой отмечено положение активного центра, вставки – различия в позициях аргининов R185 и R382 активного центра. Показаны кристаллическая структура из [9] и структура из данного исследования. Карта электростатического потенциала показана на уровне 2ϭ (а). Расстояния между ключевыми аминокислотами активного центра КД-ОДК (б).
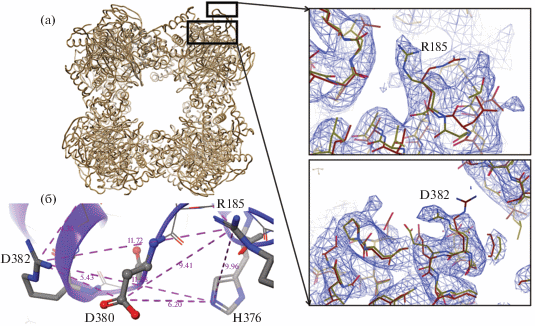
Сравнение крио-ПЭМ-структуры с рентгеновской структурой (PDB ID 6prb) показало, что существуют некоторые различия в ориентациях боковых цепей ключевых аминокислот в активном центре: D382 и D185 (рис. 3а, вставки). Основной, пока не решенный, вопрос относительно структуры КД-ОДК – это причины изменений ориентаций боковых цепей, происходящие в активном участке при связывании субстрата. Ранее было высказано предположение, что остаток H375 инициирует первую ступень катализа путем депротонирования тиоловой группы КоА, которая затем атакует карбонильный атом сукцинильного остатка, связанного с дигидролипамидом. Разрушение промежуточного соединения приводит к образованию сукцинил-КоА и протонированной дигидролипоиловой группы [8]. Также предполагалось, что при связывании КоА в активном центре аспарагин D380 образует солевые мостики с гистидином H376 и с одним из двух остатков аргинина: R185 или R382 [20]. Для этого D380 должен менять свою ориентацию в зависимости от субстрата. Полученная с помощью крио-ПЭМ структура позволила точно локализовать боковые цепи R185 и R382; эти боковые цепи были не упорядочены в раннем рентгеновском исследовании (PDB ID 1e2o) и выявлены позже (PDB ID 6prb). Сравнение модели PDB:6prb с полученной в данной работе (рис. 3а, вставки) показало, что в модели de novo остатки Arg повернуты. Измеренные расстояния между ключевыми а.о. в активном центре составили: D380-H376 = 6.2, D380-R382 = 5.4, D380-R185 = 9.4 Å (рис. 3б). Таким образом, аспарагин D380 располагается примерно посередине между гистидином H376 и аргинином R380 и его взаимодействия с этими остатками, по-видимому, носят ионный характер. С аргинином R185 аспарагин D380 согласно рассмотренной модели не взаимодействует. Для более точного определения структуры активного центра фермента необходимо получить реконструкцию КД-ОДК с субстратом.
ЗАКЛЮЧЕНИЕ
Крио-ПЭМ становится незаменимым инструментом в структурных исследованиях биологических макромолекул. До сих пор для получения структур с высоким разрешением требовались высоко очищенные препараты белков. Однако 3D-классификация гетерогенных частиц, реализованная в программах Relion [2] и cryoSPARC [3] в сочетании с масс-спектрометрией, как показывает практика, позволяет обойтись без специальной очистки [24]. В настоящей работе использовали крио-ПЭМ и тандемную масс-спектрометрию для идентификации белковых молекул в грубо очищенной белковой пробе. С помощью анализа изображений удалось выделить две выборки частиц, соответствующие двум белкам E. coli и получить две 3D-структуры: β-галактозидазы и КД-ОДК. Оба белка находились в нативном состоянии и не имели рекомбинантных тегов, используемых для очистки. Разрешение, близкое к атомному, позволило выполнить de novo-моделирование атомной модели КД-ОДК. Сравнение полученной структуры с опубликованной кристаллической структурой этого же домена [9] показало, что активный сайт в растворе находится в несколько иной конформации, чем в кристалле.
Авторы выражают благодарность В.Н. Новоселецкому за помощь в подготовке рис. 3б.
Эксперименты проведены на оборудовании Уникальной научной установки “3D-EMС” МГУ (при поддержке Министерства науки и высшего образования РФ, уникальный идентификатор RFMEFI61919X0014). Получение крио-ПЭМ-данных с высоким разрешением проведено с использованием оборудования Ресурсного центра зондовой и электронной микроскопии “Нанозонд” НИЦ “Курчатовский институт”. Расчеты проведены с использованием вычислительных ресурсов Федерального центра коллективного пользования “Комплекс моделирования и обработки данных исследовательских установок мега-класса” НИЦ “Курчатовский институт” (http://ckp.nrcki.ru/). Масс-спектрометрические измерения проведены на оборудовании центра коллективного пользования “Протеом человека” ИБМХ.
Исследование выполнено при поддержке Междисциплинарной научно-образовательной школы Московского государственного университета “Молекулярные технологии живых систем и синтетическая биология” в рамках научного проекта государственного задания МГУ № 121032500056-3.
Список литературы
Соколова О.С., Кирпичников М.П., Шайтан К.В. и др. Современные методы изучения структуры и функций ионных каналов. М.: Товарищество научных изданий КМК, 2020. 316 с.
Scheres S.H. // Methods Enzymol. 2016. V. 579. P. 125. https://doi.org/10.1016/bs.mie.2016.04.012
Punjani A., Rubinstein J.L., Fleet D.J. et al. // Nat. Methods. 2017. V. 14. P. 290. https://doi.org/10.1038/nmeth.4169
Yang Z., Lasker K., Schneidman-Duhovny D. et al. // J. Struct. Biol. 2012. V. 179. P. 269. https://doi.org/10.1016/j.jsb.2011.09.006
Pettersen E.F., Goddard T.D., Huang C.C. et al. // J. Comput. Chem. 2004. V. 25. P. 1605. https://doi.org/10.1002/jcc.20084
Emsley P., Lohkamp B., Scott W.G. et al. // Acta Cryst. D. 2010. V. 66. P. 486. https://doi.org/10.1107/S0907444904019158
Wrapp D., Wang N., Corbett K.S. et al. // Science. 2020. V. 367. P. 1260. https://doi.org/10.1126/science.abb2507
Knapp J.E., Carroll D., Lawson J.E. et al. // Protein Sci. 2000. V. 9. P. 37. https://doi.org/10.1110/ps.9.1.37
Andi B., Soares A. S., Shi W. et al. // Acta Cryst. F. 2019. V. 75. P. 616. https://doi.org/10.1107/S2053230X19011488
Stanishneva-Konovalova T.B., Semenyuk P.I., Kurochkina L.P. et al. // J. Struct. Biol. 2020. V. 209. P. 107439. https://doi.org/10.1016/j.jsb.2019.107439
Tegunov D., Cramer P. // Nat. Methods. 2019. V. 16. P. 1146. https://doi.org/10.1038/s41592-019-0580-y
Anselm V., Novikova S., Zgoda V. // Int. J. Mol. Sci. 2017. V. 18. https://doi.org/10.3390/ijms18081763
Perkins D.N., Pappin D.J., Creasy D.M. et al. // Electrophoresis. 1999. V. 20. P. 3551. https://doi.org/10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2
Ishihama Y., Oda, Y., Tabata T. et al. // Mol. Cell. Proteomics. 2005. V. 4. P. 1265. https://doi.org/10.1074/mcp.M500061-MCP200
Afonine P.V., Poon B.K., Read R.J. et al. // Acta Cryst. D. 2018. V. 74. P. 531. https://doi.org/10.1107/S2059798318006551
Wingfield P. // Curr. Protoc. Protein Sci. 2001. Appendix 3. https://doi.org/10.1002/0471140864.psa03fs13
Jacobson R.H., Zhang X.J., DuBose R.F. et al. // Nature. 1994. V. 369. P. 761. https://doi.org/10.1038/369761a0
Zivanov J., Nakane T., Forsberg B.O. et al. // Elife. 2018. P. e42166 https://doi.org/10.7554/eLife.42166
Perham R.N. // Biochemistry. 1991. V. 30. P. 8501. https://doi.org/10.1021/bi00099a001
Knapp J.E., Mitchell D.T., Yazdi M.A. et al. // J. Mol. Biol. 1998. V. 280. P. 655. https://doi.org/10.1110/ps.9.1.37
Ricaud P.M., Howard M.J., Roberts E.L. et al. // J. Mol. Biol. 1996. V. 264. P. 179. https://doi.org/10.1006/jmbi.1996.0632
Robien M.A., Clore G.M., Omichinski J.G. et al. // Biochemistry. 1992. V. 31. P. 3463. https://doi.org/10.1021/bi00128a021
Bolanos-Garcia V.M., Davies O.R. // Biochim. Biophys. Acta. 2006. V. 1760. P. 1304. https://doi.org/10.1016/j.bbagen.2006.03.027
Verbeke E.J., Mallam, A.L., Drew K. et al. // Cell. Rep. 2018. V. 24. P. 259. https://doi.org/10.1016/j.celrep.2018.06.022
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография