

Кристаллография, 2023, T. 68, № 2, стр. 209-222
Структурная сложность молекулярных, цепочечных и слоистых кристаллических структур природных и синтетических сульфидов мышьяка
Д. А. Банару 1, *, С. М. Аксенов 2, **, Н. А. Ямнова 3, А. М. Банару 2, 3
1 Институт геохимии и аналитической химии им. В.И. Вернадского РАН
Москва, Россия
2 Кольский научный центр РАН
Апатиты, Россия
3 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: banaru@geokhi.ru
** E-mail: aks.crys@gmail.com
Поступила в редакцию 31.10.2022
После доработки 08.12.2022
Принята к публикации 08.12.2022
- EDN: BPUACB
- DOI: 10.31857/S0023476123020030
Аннотация
Разработана схема лестничного расчета структурной сложности гетеродесмических кристаллических структур при трактовке кристалла как системы контактирующих молекул, цепочек и слоев. На последней ступени лестничного расчета структурная сложность основного мотива складывается со сложностью контактов вне основного мотива в соответствии с правилом сильной аддитивности. Продемонстрировано использование схемы и получены результаты расчета для кристаллических структур природных и синтетических сульфидов мышьяка. Координация молекул и цепочек, необходимая для расчета сложности контактов вне основного мотива, установлена методом полиэдров Вороного–Дирихле.
ВВЕДЕНИЕ
Молекулярная сеть, создаваемая контактирующими друг с другом молекулами в кристалле, характеризуется информационной сложностью, выражаемой в битах [1] и зависящей как от сложности самой молекулы (Hmol), совпадающей со сложностью кристаллической структуры согласно [2], так и от реберной сложности сети контактов (Hedge):
(1)
${{H}_{{{\text{mol}}}}} = - \mathop \sum \limits_{i = 1}^{{v}{\kern 1pt} {\text{''}}} \frac{{{{{v}}_{i}}}}{{v}}{{\log }_{2}}\frac{{{{{v}}_{i}}}}{{v}}\;\;~({\text{бит/атом}}),$(2)
${{H}_{{{\text{edge}}}}} = - \mathop \sum \limits_{i = 1}^{e''} \frac{{{{e}_{i}}}}{e}{{\log }_{2}}\frac{{{{e}_{i}}}}{e}\;\;({\text{бит/контакт}}),$(3)
${{\Omega }} = \frac{{\sum\limits_{ij} {{{{{\Omega }}}_{{ij}}}} }}{{{{{{\Omega }}}_{{{\Sigma }}}}}} \times 100\% ,$Не следует путать Hmol со сложностью молекулы по группе всех автоморфизмов молекулярного графа [9], так как в общем случае группа автоморфизмов имеет более высокий порядок, чем точечная группа молекулы, и меньшее число занятых орбит. При сложении Hmol и Hedge учитывают добавочное слагаемое, возникающее вследствие объединения двух источников информации согласно правилу сильной аддитивности [10]:
(4)
${{H}_{{{\text{molNet}}}}} = H({v},e) + \frac{{v}}{{{v} + e}}{{H}_{{{\text{mol}}}}} + \frac{e}{{{v} + e}}{{H}_{{{\text{edge}}}}},$(5)
$H({v},e) = - \frac{{v}}{{{v} + e}}{{\log }_{2}}\frac{{v}}{{{v} + e}} - \frac{e}{{{v} + e}}{{\log }_{2}}\frac{e}{{{v} + e}},$Простая аддитивность, при которой шэнноновская сложность целого равна простой сумме значений шэнноновской сложности частей, выполняется лишь при взаимной независимости частей. Но если части целого являются взаимозависимыми (например, содержатся в одной структуре), простая аддитивность заменяется сильной, при которой шэнноновская сложность целого учитывает значения условной шэнноновской сложности частей, выражаемые через условные вероятности [10]. Если все молекулы в кристалле химически одинаковы (гомомолекулярный кристалл) и занимают одну орбиту (моносистемный кристалл), то ${v}$ можно заменить на 2N, где N – число атомов в молекуле, а e – на CNmol [1]. Значения H(${v}$, e) и HmolNet выражаются в бит/степень свободы (с.с.). Как показало статистическое исследование 4152 гомомолекулярных кристаллов с пр. гр. P21/c, в которых четыре молекулы в элементарной ячейке занимают одну общую позицию [11], усредненная доля Hedge в HmolNet составляет 9.5% (SD = 3.6%), а усредненная доля H(${v}$, e) в HmolNet – всего 11.6% (SD = 2.8%), т.е. в среднем почти 80% приходится на Hmol. На критическую сеть приходится в среднем 62.4% Hedge (стандартное отклонение SD = 11.7%). Аналогичные расчеты можно применять не только к молекулярным кристаллам, но и к другим островным гетеродесмическим структурам, таким как полиморфные модификации CaCO3 и его кристаллогидраты [12].
Сложность какого-либо объекта имеет три разновидности [13, 14]:
– сложность описания (сложность по Шэннону, Реньи, Лемпелю–Зиву и т.д.;
– сложность создания (временнáя сложность алгоритма, вычислительная сложность, глубина сложности и др.);
– степень организации (стохастическая сложность, иерархическая сложность, топологический размер “машинного эпсилона” и др.).
Применительно к молекулярной кристаллической структуре сложность второй разновидности отвечает числу опорных/структурообразующих контактов, которые А.И. Китайгородский называл определяющими [15]. Такие контакты могут сохраняться при фазовых переходах. Некоторые указания на опорные контакты можно получить с помощью топологического анализа электронной плотности по Бейдеру (QTAIM) [16], хотя на этот счет имеются серьезные сомнения [17]. Набор опорных контактов является в математическом смысле нечетким [18] и может быть выбран разными способами, так как межмолекулярные взаимодействия обладают свойством кооперативности. Минимально необходимое число симметрически независимых контактов, обеспечивающих трехмерный каркас межмолекулярных взаимодействий при заданных Z ' и Z '' (Z ' – число молекул в симметрически независимой части элементарной ячейки, Z '' – число занятых молекулами орбит [19]), зависит только от пространственной группы кристалла [20] и является инвариантом его структурного класса (СК) по Бельскому–Зоркому [21]. В немолекулярных кристаллах в отличие от молекулярных даже выделение строительного блока (“молекулы”) само по себе является весьма сложной задачей [22–24]. Такие системы здесь рассматривать не будем.
Опорными можно считать любые наборы контактов, формирующие односвязную подсеть критической сети, однако в первую очередь в такой набор следует включать контакты: вносящие наибольший вклад в потенциальную энергию кристалла; наиболее устойчивые к разрыву, что означает маленький индекс ангармоничности и что можно определить по форме потенциальной кривой межатомного взаимодействия [25]. Визуализировать опорные контакты помогает построение энергетического каркаса [26], в котором по результатам расчета энергии межмолекулярного взаимодействия молекулы соединяют ребрами разной толщины, пропорциональной силе взаимодействия. Для образования кристаллической структуры в каждом кристалле достаточно ограниченного набора опорных контактов [27]. Кристаллическую структуру можно описать обобщенным СК, который имеет вид $G_{m}^{n}$, Z = = $k\left( {\bigcup\nolimits_{i = 1}^{Z''} {G_{{0,i}}^{n}} } \right)$ и аналогичен СК по Бельскому–Зоркому [21], где $G_{m}^{n}$ – n-мерная пространственная группа, периодическая в m ≤ n-измерениях, $G_{{0,i}}^{n}$ – n-мерная точечная группа симметрии (стабилизатор) позиции молекул, занимающих i-ю орбиту. В конструкции Z = $k\left( {\bigcup\nolimits_{i = 1}^{Z''} {G_{{0,i}}^{n}} } \right)$ Z означает число структурных единиц в элементарной ячейке, k – целое число, $\bigcup\nolimits_{i = 1}^{Z''} {G_{{0,i}}^{n}} $ – совокупность позиций, занятых структурными единицами. Операции симметрии, входящие во множество $\bigcup\nolimits_{i = 1}^{Z''} {G_{{0,i}}^{n}} $, порождают некоторую группу $\left\langle {\bigcup\nolimits_{i = 1}^{Z''} {G_{{0,i}}^{n}} } \right\rangle $, которая может быть как точечной, так и периодической не более чем в n измерениях. В общем случае, если $\left| {U\left( {\left\langle {\bigcup\nolimits_{i = 1}^{Z''} {G_{{0,i}}^{n}} } \right\rangle } \right)} \right|$ – мощность минимального порождающего подмножества группы $\left\langle {\bigcup\nolimits_{i = 1}^{Z''} {G_{{0,i}}^{n}} } \right\rangle $, |U($G_{m}^{n}$)| – мощность минимального порождающего подмножества исходной группы, точная нижняя граница допустимых значений e'', обозначаемая inf(e''), равна
(7)
$\inf (e{\kern 1pt} '') = \left| {U(G_{m}^{n})} \right| + Z{\kern 1pt} ''\; - 1 - \left| {U\left( {\left\langle {\bigcup\limits_{i = 1}^{Z''} {G_{{0,i}}^{n}} } \right\rangle } \right)} \right|.$В молекулярных кристаллах inf(e'') отвечает числу опорных/структурообразующих контактов.
Целью настоящей работы является расчет комбинаторной сложности молекулярных сетей в кристаллических структурах минералов состава AsxSy.
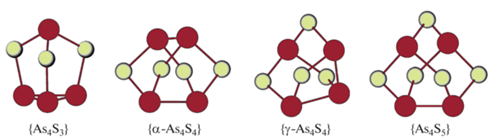
Среди природных сульфидов мышьяка и их полиморфных модификаций большинство относится к молекулярным кристаллам и демонстрирует большое разнообразие не только молекулярных упаковок, но и состава и формы молекул [28] (рис. 1), что повышает ценность данной серии соединений для демонстрации производимых расчетов. На картах электронной плотности молекулярных сульфидов мышьяка видны локальные особенности, отвечающие не валентному взаимодействию Льюисовых пар As⋅⋅⋅S [29], что свидетельствует о направленности опорных контактов и делает построение молекулярной сети в данной серии структур физически обоснованным.
МЕТОДЫ ИССЛЕДОВАНИЯ
Топологический анализ и расчет сложности. Топологические преобразования, а также расчет информационных индексов (1) и (2) выполнены с помощью пакета программ ToposPro [30]. При построении сети молекулярных контактов учитывали все контакты с Ω > 0 согласно (3). Для классификации сетей использовали топологические базы данных RCSR [31] и topcryst [32]. Структурные классы записывали в обозначениях Зоркого [33]. Исходные кристаллоструктурные данные получены из базы данных SpringerMaterials [34] (табл. 1).
Таблица 1.
Кристаллические структуры полиморфов состава AsxSy
| Формула | Название минерала | Образец | R-фактор | Мотив | Литература |
|---|---|---|---|---|---|
| α-As4S4 | реальгар | монокристалл | 0.040 | 0D | [35] |
| β-As4S4 | 0.025 | 0D | [36] | ||
| As4S4(II) | фаза Кутоглу | 0.069 | 0D | [37] | |
| γ-As4S4 | парареальгар | 0.034 | 0D | [38] | |
| As4S5 мон. | узонит | 0.027 | 0D | [39] | |
| As4S5 орт. | 0.099 | 0D | [40] | ||
| As8S9 | алакранит | порошок | 0.045 | 0D | [41] |
| α-As4S3 | α-диморфит | монокристалл | 0.023 | 0D | [42] |
| β-As4S3 | β-диморфит | 0.037 | 0D | [42] | |
| As4S | дуранузит | 0.063 | 2D | [43] | |
| As2S3 | аурипигмент | 0.064 | 2D | [44] | |
| AsS | двойник | 0.084 | 2D | [45] | |
| AsS2 | монокристалл | 1D | [46] |
Немолекулярную кристаллическую структуру помимо минералов состава As4S (дуранузит) и As2S3 (аурипигмент) имеют фазы высокого давления, полученные из аурипигмента при p > 6 ГПа: As2S3 → AsS2 + AsS [46].
Для анализа комбинаторной сложности молекулярных кристаллических структур был использован нижеследующий алгоритм.
1. Рассчитать химическую сложность структуры Hchem, проистекающую из состава соединения $E_{{c1}}^{{(1)}}E_{{c2}}^{{(2)}}{\kern 1pt} \ldots {\kern 1pt} E_{{ck}}^{{(k)}}$, где E(i) – i-й химический элемент, а ci – индекс в простейшей формуле вещества [47]:
(8)
${{H}_{{{\text{chem}}}}} = - \mathop \sum \limits_{i = 1}^k \frac{{{{c}_{i}}}}{c}{{\log }_{2}}\frac{{{{c}_{i}}}}{c}\;\;({\text{бит/атом}}),$2. Рассчитать топологическую сложность молекул ${{H}_{{{\text{mol}},0}}}$ (бит/атом) в наиболее симметричной конформации. Позиции Уайкова точечных групп идеализированных молекул записываются в соответствии с международными обозначениями, табулированными на Кристаллографическом сервере Бильбао [48].
3. В каждой кристаллической структуре (рис. 2а) по ПВД отдельных атомов методом доменов [5] построить матрицу смежности и молекулярный ПВД (рис. 2б). Каждую пару смежных молекулярных ПВД рассматривать как ребро молекулярной сети.
Рис. 2.
Кристаллическая структура алакранита As8S9, проекции вдоль b: а – общий вид, б – ПВД двух симметрически независимых молекул, в – фрагмент сети межмолекулярных контактов, г – фрагмент реберной сети контактов (г).
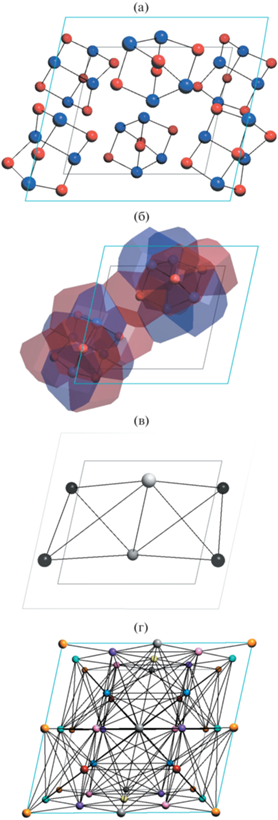
4. Рассчитать согласно [2] сложность структуры Hmol (бит/атом).
3. Каждую молекулу стянуть к своему центру масс с сохранением связности между ними (рис. 2в). В результате получится сеть с e ребрами в приведенной элементарной ячейке, связывающими центры масс и занимающими e'' орбит.
4. Сеть, связывающую центры масс, классифицировать по RCSR.
5. Построить реберную сеть, в которой каждое ребро заменено на вершину, а вершина – на ребро (рис. 2г). Две вершины реберной сети смежны друг с другом тогда и только тогда, когда в исходной сети соответствующие ребра исходят из одной вершины.
6. Рассчитать сложность реберной сети Hedge (бит/контакт).
7. Пункты 5 и 6 повторить для наиболее симметричного вложения сети, полученной после выполнения пункта 4, и рассчитать топологическую сложность сети ${{H}_{{{\text{edge}},0}}}$ (бит/контакт). Под вложением сети в пространство понимается вариант размещения в этом пространстве ее вершин. У кристаллографических сетей наиболее симметричное вложение описывается пространственной группой, изоморфной группе автоморфизмов сети.
8. Рассчитать значения HmolNet (бит/с.с.) и ${{H}_{{{\text{molNet}},{\text{tot}}}}}$ (бит/эл.яч.).
Топологический анализ немолекулярных структур проводили с учетом размерности структурного мотива по аналогичному алгоритму, исключая пункты 3–5 и 7.
Поверхность Хиршфельда и степень ее обогащения контактами. Поверхности Хиршфельда для анализа интегральных характеристик межмолекулярных контактов строили только для молекулярных кристаллов в программе Crystal Explorer [49, 50] при значении весовой функции w(r) = 0.5:
(9)
$w({\mathbf{r}}) = \frac{{\sum\limits_{A \in {\text{mol}}} {{{{{\rho }}}_{A}}({\mathbf{r}})} }}{{\sum\limits_{A \in {\text{cryst}}} {{{{{\rho }}}_{A}}({\mathbf{r}})} }},$Коэффициент упаковки поверхностей Хиршфельда PIH рассчитывали по формуле
(10)
$P{{I}_{{\text{H}}}} = \frac{{\sum\limits_{{\text{u}}.{\text{c}}} {{{V}_{{\text{H}}}}} }}{{{{V}_{{{\text{u}}.{\text{c}}}}}}}~,$Глобулярность G (степень отличия площади поверхности Хиршфельда от площади сферы, ограничивающей такой же объем) рассчитывали по формуле [51]:
где SH – площадь поверхности Хиршфельда.Согласно [52] рассчитывали степень обогащения поверхности Хиршфельда межатомными контактами определенного типа. Пусть межатомным контактам типа X⋅⋅⋅Y (атомы X внутри поверхности, атомы Y – снаружи) отвечает доля поверхности молекулы CXY. Тогда суммарная доля поверхности, отвечающая контактам атомов X, равна
Пусть RXY – доля поверхности, приходящаяся на контакты X⋅⋅⋅Y в предположении, что с химической точки зрения все контакты равновероятны. Тогда RXX = SXSX, RXY = 2SXSY и степень обогащения поверхности контактами X⋅⋅⋅Y и Y⋅⋅⋅X, в том числе при Y = X, рассчитаем по формуле
Если молекулы при взаимной упаковке избегают коротких контактов X⋅⋅⋅Y, то EXY < 1, но если наоборот, такие контакты предпочтительны, EXY > 1.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Сложность молекулярных сульфидов мышьяка. Молекулы AsxSy (рис. 1) имеют различную химическую и топологическую сложность. В общем случае точечная группа молекулы (PG) наивысшего возможного порядка может быть не единственной [53], хотя для молекул AsxSy она единственная (табл. 2). У молекулы α-As4S4, которая является самой стабильной среди молекул состава As4Sy (Ef = –0.58 ккал/моль [54]), топологическая сложность совпадает с химической.
Таблица 2.
Химическая и топологическая сложность молекул AsxSy
| Состав | Hchem, бит/атом | PG | Позиции Уайкова для атомов | ${{H}_{{{\text{mol}},0}}}$, бит/атом |
|---|---|---|---|---|
| α-As4S4 | 1.000 | $\bar {4}2m$ | cb | 1.000 |
| γ-As4S4 | 1.000 | m | b2a4 | 2.500 |
| As4S5 | 0.991 | mm2 | dcba | 1.837 |
| As4S3 | 0.985 | 3m | b2a | 1.449 |
При переходе в кристалл симметрия молекул понижается до m (узонит, α- и β-диморфит), 2 (β-As4S4, алакранит) либо 1. Согласно результатам статистического анализа данных CSDSymmetry [55] для молекул симметрии 3m (диморфит) искажение в кристалле до симметрии m как раз является самым частым (42% молекул). В то же время молекулы симметрии mm2 (узонит) чаще всего теряют все элементы симметрии (61% молекул). Лишь в 17% случаев они сохраняют зеркальную плоскость и лишь в 16% – ось 2, что и происходит с молекулой As4S5 в алакраните. Молекулы симметрии $\bar {4}2m$ чаще всего сохраняют ось 2 (28% случаев), как это происходит в реальгаре, β-As4S4 и алакраните, и реже теряют все элементы симметрии (24% случаев), как это происходит в реальгаре. Наконец, молекулы симметрии m чаще всего понижают свою симметрию до 1 (74% случаев), что наблюдается у парареальгара.
Молекулярные кристаллические структуры состава AsxSy исчерпываются шестью СК (табл. 3), из которых один (P21/c, Z = 4(1)) является “супергигантом” (по Зоркому) и наблюдается более чем у 31% всех гомомолекулярных структур [33] и один – “гигантом” (C2/c, Z = 4(2), 2.38% структур). Структура алакранита является гетеромолекулярной, однако его СК P2/c, Z = 4(22) встречается и у гомомолекулярных кристаллов (0.05% структур).
Таблица 3.
Структурные классы молекулярных сульфидов мышьяка
| СК | Процент от всех гомомоле-кулярных структур* | $\left| {U(G_{3}^{3})} \right|$ | $\left| {U\left( {\left\langle {\bigcup\limits_{i = 1}^{Z''} {G_{{0,i}}^{3}} } \right\rangle } \right)} \right|$ | ${\text{inf}}(e{\kern 1pt} '')$ |
|---|---|---|---|---|
| P21/m, Z = 2(m) | 0.28 | 4 | 1 | 3 |
| P2/c, Z = 4(22) | 0.05 | 4 | 2 | 3 |
| P21/c, Z = 4(1) | 31.26 | 3 | 0 | 3 |
| C2/c, Z = 4(2) | 2.38 | 3 | 1 | 2 |
| Pccn, Z = 8(1) | 0.15 | 3 | 0 | 3 |
| Pnma, Z = 4(m) | 0.69 | 3 | 1 | 2 |
Число опорных контактов у рассмотренных структур ${\text{inf}}(e{\kern 1pt} '')$ равно двум или трем. Примечательно, что в структуре алакранита значение ${\text{inf}}(e{\kern 1pt} '')$ не увеличивается на единицу по сравнению с моносистемным СК P2/c, Z = 4(2), поскольку группы симметрии позиций (2), занятых центрами масс молекул As4S4 и As4S5 (f и e), могут одновременно входить в минимальное порождающее подмножество группы P2/c, т.е. в соответствии с (7) ${\text{inf}}(e{\kern 1pt} '')$ = 4 + 2 –1 – 2 = 3.
Из-за деформации молекул при переходе в кристалл во всех случаях Hmol > ${{H}_{{{\text{mol}},0}}}$ (табл. 4). У алакранита Hmol может быть рассчитана по правилу сильной аддитивности:
(14)
${{H}_{{{\text{mol}},j}}} = - \mathop \sum \limits_{i = 1}^{{{{v}}_{j}}''} \frac{{{{{v}}_{{ij}}}}}{{{{{v}}_{j}}}}{{\log }_{2}}\frac{{{{{v}}_{{ij}}}}}{{{{{v}}_{j}}}}~,$(15)
${{H}_{{{\text{mol}}}}} = \mathop \sum \limits_{j = 1}^{Z''} \frac{{{{{v}}_{j}}}}{{\sum\limits_j {{{{v}}_{j}}} }}{{H}_{{{\text{mol}},j}}} + {{H}_{{Z''}}},$Таблица 4.
Структурные классы и топологические характеристики молекулярной сети сульфидов мышьяка
| Структура | Формула | СК | WS атомов |
Hmol, бит/атом | CNmol | Сеть контактов | WS ребер | Hedge, бит/контакт |
|---|---|---|---|---|---|---|---|---|
| 1 | α-As4S4 | P21/c, Z = 4(1) | e8 | 3.000 | 14 | tcg-x | e5dcba | 3.093 |
| 2 | β-As4S4 | C2/c, Z = 4(2) | f3e2 | 2.250 | 12 | fcu | fdcba | 2.252 |
| 3 | As4S4(II) | P21/c, Z = 4(1) | e8 | 3.000 | 12 | hcp | e4dcba | 2.918 |
| 4 | γ-As4S4 | P21/c, Z = 4(1) | e8 | 3.000 | 14 | bcu-x | e5dcba | 3.093 |
| 5 | As4S5 мон. | P21/m, Z = 2(m) | f3e3 | 2.503 | 14 | bcu-x | e3dcba | 2.807 |
| 6 | As4S5 орт. | Pccn, Z = 8(1) | e9 | 3.170 | 14 | bcu-x | e5dcba | 3.093 |
| 7 | As4S4⋅As4S5 | P2/c, Z = 4(22) | g7f2e | 3.264 | 12, 12 | fcu | g4dcba | 2.918 |
| 8 | α-As4S3 | Pnma, Z = 4(m) | d2c3 | 2.236 | 14 | bcu-x | dc3ba | 2.522 |
| 9 | β-As4S3 | Pnma, Z = 4(m) | d2c3 | 2.236 | 14 | tcg-x | dc3ba | 2.522 |
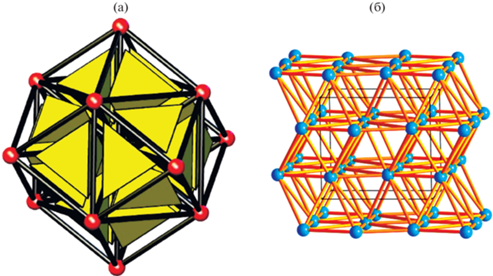
Значения CNmol равны 14 (как чаще всего бывает у молекулярных кристаллов по данным [56]) или 12, но топологический тип молекулярной сети при одинаковых CNmol не всегда одинаков. У парареальгара, As4S5 (обе модификации) и α-диморфита молекулы образуют объемноцентрированную кубическую кладку, в которой число касаний расширено за счет второй координационной сферы (тип bcu-x), в то же время реальгар и β-диморфит относятся к типу tcg-x (рис. 3). Аналогично в β-As4S4 и алакраните упаковка молекул топологически идентична кубической плотнейшей упаковке (fcu), в то время как в фазе Кутоглу – гексагональной плотнейшей упаковке (hcp).
Середины ребер молекулярной сети у рассмотренных структур класса P21/c, Z = 4(1), за исключением фазы Кутоглу, а также у ромбической модификации As4S5 занимают позиции e5dcba, что было характерно для 56.0% выборки 4152 молекулярных структур того же СК [11], в этом случае Hedge = 3.093 бит/контакт, что является наибольшим значением по выборке. Во всех структурах, кроме β-диморфита, сеть контактов является искаженным вариантом идеализированной сети (табл. 5), поэтому в этих случаях Hedge > ${{H}_{{{\text{edge}},0}}}$.
Таблица 5.
Топологическая сложность сетей межмолекулярных контактов
| Тип | Транзи-тивность | WS ребер | ${{H}_{{{\text{edge}},0}}}$, бит/контакт |
|---|---|---|---|
| hcp | 1.2 | hg | 1.000 |
| fcu | 1.1 | d | 0 |
| bcu-x | 1.2 | cb | 0.985 |
| tcg-x | 1.6 | dc3ba | 2.522 |
Примечательно, что в ряду рассмотренных структур Hedge возрастает с увеличением C(inf(e''), e'') – числа сочетаний из e'' по inf(e'') (рис. 4). При этом не все возможные сочетания отвечают inf(e'') наборам опорных контактов, в частности у реальгара – только 62 из 84 сочетаний, у парареальгара – 37 из 84, у As4S5 орт. – 34 из 84, у фазы Кутоглу – 35 из 56, у алакранита – 30 из 56. Помимо этого, в двух сочетаниях сеть контактов алакранита трехмерна, но не односвязна, а представляет собой два взаимопроникающих каркаса алмазного типа dia (рис. 5а), причем каждый каркас образован обеими независимыми молекулами. Такие сочетания тоже не отвечают набору опорных контактов. В структуре узонита наборам опорных контактов отвечают 17 сочетаний из 35, в обеих модификациях диморфита – 8 из 15, а в β-As4S4 – 8 из 10. Таким образом, доля подсетей с e'' = inf(e''), способных соответствовать опорным контактам, для кристаллических структур молекулярных сульфидов мышьяка варьируется в интервале от 40 (для As4S5 орт.) до 80% (для β-As4S4) от C(inf(e''), e''). Эта доля выступает своего рода мерой предопределенности СК при наличии только слабых неспецифических межмолекулярных взаимодействий.
Рис. 4.
Зависимость Hedge (бит/контакт) от числа сочетаний C (inf(e''), e'') для кристаллических структур молекулярных сульфидов мышьяка.
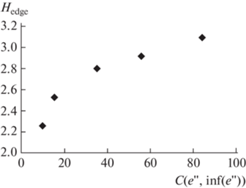
Рис. 5.
Взаимопроникающие алмазные (dia) сети при e'' = 3 (а) и 2,4-координированный каркас межмолекулярных контактов (б) в структуре алакранита, проекции вдоль [100 ] . Центры масс молекул As4S4 и As4S5 показаны разным цветом.
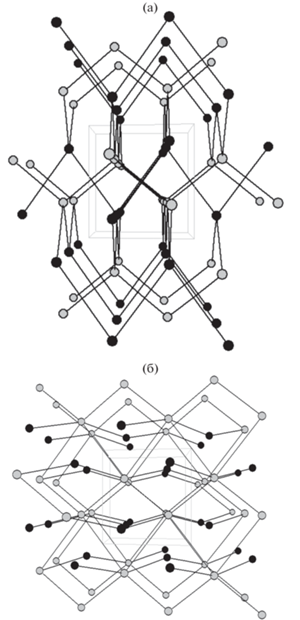
Среди тридцати подсетей, которые могут отвечать опорным контактам в структуре алакранита, десять относятся к типу dia, два – к типу простой шаровой кладки (pcu), восемь – к типу sqc514, четыре – к sqc11 (обе являются 2,2-транзитивными и 4,6-координированными). Остальные восемь, строго говоря, сетями не являются [57], так как содержат двухкоординированные вершины (рис. 5б), однако при удалении последних они приобретают топологический тип pcu.
На рис. 6 представлены вклады Hmol, Hedge и H(${v}$, e) в общую сложность молекулярной сети, рассчитанной по уравнениям (4)–(6). Среди всех структур наименьшие значения HmolNet(β-As4S4) = = 3.236 бит/с.с., HmolNet (β-As4S4) = 90.6 бит/эл.яч., наибольшие значения HmolNet(As4S5 орт.) = = 4.125 бит/с.с., ${{H}_{{{\text{molNet}},{\text{tot}}}}}$ (As4S5 орт.) = = 528.0 бит/эл.яч. Причиной столь низкой сложности структуры β-As4S4 является самый малый среди всех структур (по абсолютной величине) вклад Hedge в HmolNet. Кристаллические структуры неорганических веществ подразделяют на очень простые (<20), простые (20–100), средней сложности (100–500), сложные (500–1000) и очень сложные (>1000) [2]. По этому критерию As4S5 орт. должна относиться к структурам средней сложности, так как ${{H}_{{{\text{mol,tot}}}}}$(As4S5 орт.) = = 72 × 3.17 = 228.2 (бит/эл.яч.), однако с учетом сложности сети контактов эта структура относится к сложным. Точно так же простые (по критерию Кривовичева) структуры β-As4S4, узонита, диморфита (обе модификации) и фазы Кутоглу с учетом сложности контактов оказываются структурами средней сложности.
Рис. 6.
Для кристаллических структур молекулярных сульфидов мышьяка: а – значения Hmol (бит/атом), Hedge (бит/контакт) и Hmix ≡ H(${v}$, e) (бит/степень свободы); б – ${{H}_{{{\text{mol,tot}}}}}$, ${{H}_{{{\text{edge,tot}}}}}$ и ${{H}_{{{\text{mix,tot}}}}}$ ≡ Htot(${v}$, e) (бит/эл.яч.). Нумерация структур соответствует табл. 4.

Структуры α- и β-диморфита имеют одинаковые значения Hmol, Hedge и H(${v}$, e) и, как следствие, HmolNet. Единственное существенное различие между ними заключается в разной топологии сетей (bcu-x и tcg-x соответственно) и разных значениях ${{H}_{{{\text{edge}},0}}}$. Реальгар и парареальгар тоже имеют одинаковые Hmol, Hedge, H(${v}$, e) и HmolNet, но у этих структур различаются значения не только ${{H}_{{{\text{edge}},0}}}$, но и ${{H}_{{{\text{mol}},0}}}$: у парареальгара больше топологическая сложность молекулы (${{H}_{{{\text{mol}},0}}}$), но меньше ее искажение при переходе в кристалл (Hmol – ${{H}_{{{\text{mol}},0}}}$), в то время как у реальгара больше топологическая сложность сети (${{H}_{{{\text{edge}},0}}}$), но меньше ее усложнение (Hedge – ${{H}_{{{\text{edge}},0}}}$). Конфигурационная энтропия кристалла линейно убывает с ростом сложности согласно [58]. Таким образом, у этих модификаций As4S4 наблюдается эффект выравнивания конфигурационной энтропии, что должно способствовать сближению их термодинамической стабильности. Как известно, рыхлая желтая корка, покрывающая природные кристаллы реальгара, состоит именно из парареальгара, [28], а не аурипигмента, как считалось ранее. Реальгар и парареальгар обладают самым большим (по абсолютной величине) вкладом Hedge в HmolNet среди рассмотренных структур.
Структура алакранита, вопреки ожиданиям, не является первой по величине HmolNet, хотя, согласно значению сложности по Кривовичеву, она опережает все остальные структуры благодаря Z '' > 1. По величине Hmol алакранит следует за As4S5 орт., по величине ${{H}_{{{\text{mol,tot}}}}}$ – за As4S5 орт., парареальгаром и реальгаром.
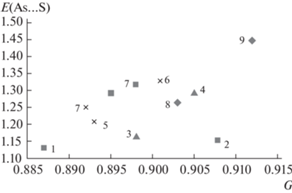
Интегральные характеристики поверхностей Хиршфельда молекул AsxSy, не зависящие напрямую от состава, представлены в табл. 6. Величины PIH, G, а также степени обогащения поверхности Хиршфельда межатомными контактами As⋅⋅⋅As, S⋅⋅⋅S и As⋅⋅⋅S демонстрируют хорошую сходимость к среднему значению по выборке. Во всех структурах EAsAs < 1, ESS < 1 и EAsS > 1, т.е. короткие межатомные контакты As⋅⋅⋅S не случайны, что соответствует ранее полученным результатам топологического анализа электронной плотности [29]. Структура реальгара имеет наименьшие по выборке значения G = 0.887 и EAsS = 1.13, а структура β-диморфита – наибольшие (G = 0.912 и EAsS = 1.45), что может косвенно свидетельствовать о влиянии специфичности контактов As⋅⋅⋅S на форму молекулы. Впрочем, простой корреляции G и EAsS не наблюдается (рис. 7).
Таблица 6.
Интегральные характеристики поверхностей Хиршфельда молекулярных сульфидов мышьяка
| Величина | PIH, % | G, % | EAsAs | ESS | EAsS |
|---|---|---|---|---|---|
| Среднее | 96.9 | 90.0 | 0.67 | 0.79 | 1.26 |
| SD | 0.1 | 0.8 | 0.13 | 0.12 | 0.10 |
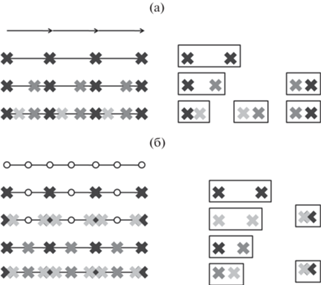
Сложность слоистых сульфидов мышьяка. Структурная сложность дуранузита, аурипигмента и других структур, имеющих слоистое строение, может быть оценена способом, аналогичным (4)–(6), с заменой молекулярной сети на сеть, сформированную контактами соседних слоев. При замене молекул на бесконечные слои система контактов между структурными единицами описывается не федоровской группой $G_{3}^{3}$, а одномерной группой $G_{1}^{1}$, сами слои при этом описываются субпериодическими группами $G_{2}^{3}$. Группа $G_{1}^{1}$ является результатом проекции слоев на направление нормали к ним. Если в группе $G_{3}^{3}$ кристаллической структуры содержится хотя бы одна ρ-операция симметрии, переводящая верхнюю сторону слоя в нижнюю сторону того же или соседнего слоя, т.е. ревертирующая слой, то $G_{1}^{1}$ = p1m. В противном случае $G_{1}^{1}$ = p1. Если имеется ρ-операция, связывающая две стороны одного слоя (λ–ρ-операция), то в группе p1m проекция данного слоя занимает орбиту со стабилизатором m, в противном случае он занимает общую орбиту (рис. 8). В группе $G_{1}^{1}$ возможны разные значения Z и Z ', но число симметрически неэквивалентных слоев в структуре обозначим не Z '', а L'', чтобы подчеркнуть иную размерность группы. При этом
(17)
${{H}_{{{\text{layNet}}}}} = H({v},e) + \frac{{v}}{{{v} + e}}{{H}_{{{\text{lay}}}}} + \frac{e}{{{v} + e}}{{H}_{{{\text{edge}}}}},$(18)
$H({v},e) = - \frac{{v}}{{{v} + e}}{{\log }_{2}}\frac{{v}}{{{v} + e}} - \frac{e}{{{v} + e}}{{\log }_{2}}\frac{e}{{{v} + e}},$(20)
${{H}_{{{\text{lay}},j}}} = - \mathop \sum \limits_{i = 1}^{{{{v}}_{j}}''} \frac{{{{{v}}_{{ij}}}}}{{{{{v}}_{j}}}}{{\log }_{2}}\frac{{{{{v}}_{{ij}}}}}{{{{{v}}_{j}}}},$(21)
${{H}_{{{\text{lay}}}}} = \mathop \sum \limits_{j = 1}^{L''} \frac{{{{{v}}_{j}}}}{{\sum\limits_j {{{{v}}_{j}}} }}{{H}_{{{\text{lay}},j}}} + {{H}_{{L''}}},$Рис. 8.
Слева: а – одномерная группа p1 и схемы ее структурных классов (сверху вниз) p1, Z = 1(1); p1, Z = 2(12); p1, Z = 3(13); б – одномерная группа p1m и схемы ее структурных классов (сверху вниз) p1m, Z = 1(m); p1m, Z = 3(1; m); p1m, Z = 2(m2); p1m, Z = = 4(1; m2). Справа (в прямоугольниках) показаны контакты, порождающие укладку слоев.
У структур дуранузита, аурипигмента и AsS (рис. 9) имеются λ–ρ-операции, вследствие чего контакты между слоями описываются СК p1m, Z = 1(m) (аурипигмент и AsS) и p1m, Z = 2(m2) (дуранузит). Месжслоевые контакты в обоих классах симметрически эквивалентны (e'' = 0), следовательно, Hedge = 0.
Рис. 9.
Слои в структуре: а – дуранузита, вид вдоль X; б – аурипигмента, вид вдоль Z; в – AsS, вид вдоль Y.
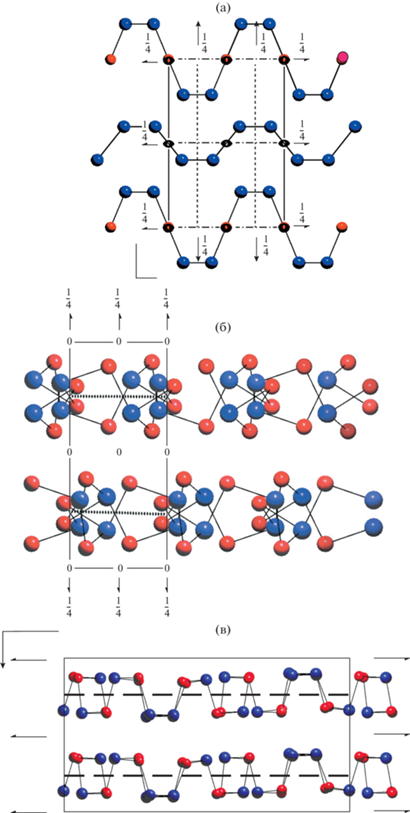
Результаты расчета других вкладов в сложность структуры представлены в табл. 7. Неэквивалентные слои содержатся только в структуре дуранузита. Слой (As)∞, гомеоморфный слоям в структуре черного фосфора, содержит только эквивалентные атомы (As2), ${{H}_{{{\text{lay,1}}}}}$(As4S) = 0. Другой слой имеет состав As2S и содержит два сорта атомов (As1 и S1), ${{H}_{{{\text{lay,2}}}}}$(As4S) = H(2, 1) = 0.918 бит/атом. HL'' = H(2, 3) = 0.971 бит/атом, поэтому согласно (16) Hlay(As4S) = 0 + 3/5 × 0.918 + 0.971 = = 1.522 (бит/атом).
Таблица 7.
Сложность слоистых структур AsxSy
| Структура | Пр. гр. | ${v}$ | e | H(${v}$, e), бит/с.с. | HL'', бит/атом | Hlay, бит/атом | HlayNet, бит/с.с. | ${{H}_{{{\text{layNet,tot}}}}}$, бит/эл.яч. |
|---|---|---|---|---|---|---|---|---|
| As4S | Pmna | 10 | 2 | 0.650 | 0.971 | 1.522 | 1.918 | 23.02 |
| As2S3 | P21/c | 20 | 2 | 0.439 | 0 | 2.322 | 2.550 | 56.10 |
| AsS | Pca21 | 64 | 2 | 0.196 | 0 | 4.000 | 4.075 | 268.95 |
Из-за большого соотношения ${v}$/e относительный вклад Hlay в HlayNet для дуранузита, аурипигмента и AsS составляет 66, 83 и 95% соответственно, в то же время для молекулярных сульфидов мышьяка этот показатель не превышал 50% (среднее 39.7%, SD = 4.5%). Таким образом, сложность слоистых кристаллических структур в отличие от молекулярных определяется главным образом Кривовичевской сложностью.
Сложность цепочечного AsS2. При замене молекул на бесконечные цепочки система контактов между ними снова описывается не федоровской группой $G_{3}^{3}$, а плоской группой $G_{2}^{2}$, сами цепочки при этом описываются субпериодическими группами $G_{1}^{3}$. Группа $G_{2}^{2}$ является результатом проекции цепочек на плоское сечение кристаллической структуры. Перечисление получающихся СК вида $G_{2}^{2}$, Z = $k\left( {\bigcup\nolimits_{j = 1}^{С''} {G_{{0,j}}^{2}} } \right)$, где С'' – число симметрически независимых цепочек, выходит за рамки настоящей работы. Алгоритм расчета структурной сложности (17)–(22) применим и к цепочечным структурам с учетом изменения размерности структурного мотива и при замене L'' → → C'', “lay” → “cha”, “layNet → “chaNet”.
В [46] AsS2 трактовалась как слоистая структура, впрочем, с указанием на то, что слои в этой структуре “…состоят из отдельных слабо связанных зигзагообразных колонок, которые протягиваются вдоль диагоналей в плоскости слоя”. Такая трактовка была удобна для сравнения структур AsS2 и AsS, так как характер строения второй из них – слоистый. Вместе с тем факт цепочечного строения AsS2 остается бесспорным, что подтверждает топологический анализ структуры (рис. 10а). Структура содержит симметрически эквивалентные цепочки AsS2 (HC'' = 0), контакты между которыми относительно группы pg (рис. 10б) распадаются на три класса эквивалентности, так как их середины на плоскости занимают три позиции (a3). Из этого следует, что Hedge = H(2, 2, 2) = = 1.585 бит/контакт, Hcha = 3.585 бит/атом, H(${v}$, e) = H(24, 6) = 0.723 бит/с.с., HchaNet = 24/30 × × 3.585 + 6/30 × 1.585 + 0.723 = 3.908 (бит/с.с.), ${{H}_{{{\text{chaNet,tot}}}}}$ = 30 × 3.908 = 117.24 бит/эл.яч.
ВЫВОДЫ
Для всякой кристаллической структуры разные источники структурной информации аддитивны.
Лестничная сложность кристаллических структур может быть измерена независимо от вида образующих кристалл структурных единиц (молекул, цепочек, слоев) основного структурного мотива в соответствии со схемой: топологическая сложность основного мотива → структурная сложность основного мотива → сложность кристаллической структуры ← структурная сложность контактов за пределами основного мотива ← топологическая сложность контактов за пределами основного мотива.
Данная схема продемонстрирована на примере кристаллических структур состава AsxSy.
Для расчета сложности контактов за пределами основного мотива требуется дополнительная (химическая) информация о взаимодействии структурных единиц кристалла, полученная методами ПВД, поверхностей Хиршфельда и/или анализа электронной плотности.
Предложенная схема может быть полезна для оценки различных структурных вкладов в конфигурационную энтропию кристалла, а также для сравнения сложности полиморфных модификаций одного вещества.
Работа выполнена при частичной поддержке Российского научного фонда (гранты № 20-77-10065 (расчеты информационных индексов) и № 22-13-00122 (топологические расчеты). Концептуализация и теоретический анализ проведены Д.А. Банару в рамках государственного задания ГЕОХИ РАН.
Список литературы
Banaru A.M., Aksenov S.M., Krivovichev S.V. // Symmetry (Basel). 2021. V. 13. P. 1399. https://doi.org/10.3390/sym13081399
Krivovichev S.V. // Angew. Chemie – Int. Ed. 2014. V. 53. P. 654. https://doi.org/10.1002/anie.201304374
Batsanov A.S. // Acta Cryst. E. 2018. V. 74. P. 570. https://doi.org/10.1107/S2056989018005339
Spackman M.A., Jayatilaka D. // CrystEngCommun. 2009. V. 11. P. 19. https://doi.org/10.1039/B818330A
Blatov V.A., Shevchenko A.P., Serenzhkin V.N. // Acta Cryst. A. 1995. V. 51. P. 909. https://doi.org/10.1107/S0108767395006799
Blatov V.A. // Cryst. Rev. 2004. V. 10. P. 249. https://doi.org/10.1080/08893110412331323170
Shevchenko A.P., Blatov V.A. // Struct. Chem. 2021. V. 32. P. 507. https://doi.org/10.1007/s11224-020-01724-4
Banaru A.M., Banaru D.A. // J. Struct. Chem. 2020. V. 61. P. 1485. https://doi.org/10.1134/S0022476620100017
Sabirov D.S., Shepelevich I.S. // Entropy. 2021. V. 23. https://doi.org/10.3390/e23101240
Hornfeck W. // Acta Cryst. A. 2020. V. 76. P. 534. https://doi.org/10.1107/S2053273320006634
Banaru A.M., Aksenov S.M. // Symmetry (Basel). 2022. V. 14. P. 220. https://doi.org/10.3390/sym14020220
Banaru D.A., Banaru A.M., Aksenov S.M. // J. Struct. Chem. 2022. V. 63. https://doi.org/10.26902/JSC_id96300
Lloyd S. // IEEE Control Syst. Mag. 2001. V. 21. P. 7. https://doi.org/10.1109/MCS.2001.939938
Nagaraj N., Balasubramanian K. // Eur. Phys. J. Spec. Top. 2017. V. 226. P. 3251. https://doi.org/10.1140/epjst/e2016-60347-2
Zefirov Y.V., Zorky P.M. // Russ. Chem. Rev. 1995. V. 64. P. 415. https://doi.org/10.1070/rc1995v064n05abeh000157
Bader R.F.W. // Acc. Chem. Res. 1985. V. 18. P. 9. https://doi.org/10.1021/ar00109a003
Jabłoński M. // ChemistryOpen. 2019. V. 8. P. 497. https://doi.org/https://doi.org/10.1002/open.201900109
Banaru A.M. // Moscow Univ. Chem. Bull. 2019. V. 74. P. 101. https://doi.org/10.3103/S0027131419030039
van Eijck B.P., Kroon J. // Acta Cryst. B. 2000. V. 56. P. 535. https://doi.org/10.1107/S0108768100000276
Banaru A.M. // Moscow Univ. Chem. Bull. 2009. V. 64. P. 80. https://doi.org/10.3103/S0027131409020023
Belsky V.K., Zorky P.M. // Acta Cryst. A. 1977. V. 33. P. 1004.
Talis A.L., Everstov A.A., Kraposhin V.S., Simich-Lafitskii N.D. // Met. Sci. Heat Treat. 2021. V. 62. P. 725. https://doi.org/10.1007/s11041-021-00629-1
Talis A.L., Kraposhin V.S., Arestov V. // Met. Sci. Heat Treat. 2022. V. 63. P. 618. https://doi.org/10.1007/s11041-022-00738-5
Talis A.L., Kraposhin V.S., Everstov A.A. // Met. Sci. Heat Treat. 2022. V. 64. P. 338. https://doi.org/10.1007/s11041-022-00811-z
Maleev A.V., Gevorgyan A.A., Potekhin K.A. // J. Struct. Chem. 2018. V. 59. P. 455. https://doi.org/10.1134/S0022476618020294
Mackenzie C.F., Spackman P.R., Jayatilaka D., Spackman M.A. // IUCrJ. 2017. V. 4. P. 575. https://doi.org/10.1107/S205225251700848X
Lord E.A., Banaru A.M. // Moscow Univ. Chem. Bull. 2012. V. 67. P. 50. https://doi.org/10.3103/S0027131412020034
Bonazzi P., Bindi L. // Z. Krist. - Cryst. Mater. 2008. V. 223. P. 132. https://doi.org/doi:10.1524/zkri.2008.0011
Gibbs G.V., Wallace A.F., Downs R.T. et al. // Phys. Chem. Mineral. 2011. V. 38. P. 267. https://doi.org/10.1007/s00269-010-0402-3
Blatov V.A., Shevchenko A.P., Proserpio D.M. // Cryst. Growth Des. 2014. V. 14. P. 3576. https://doi.org/10.1021/cg500498k
O’Keeffe M., Peskov M.A., Ramsden S.J., Yaghi O.M. // Acc. Chem. Res. 2008. V. 41. P. 1782. https://doi.org/10.1021/ar800124u
The Samara Topological Data Center “TopCryst,” available at https://topcryst.com/, n.d.
Zorky P.M. // J. Mol. Struct. 1996. V. 374. P. 9.
Madelung O., Rössler U., Schulz M. 2010 http//www.springermaterials.com
Kyono A. // Am. Mineral. 2009. V. 94. P. 451. https://doi.org/doi:10.2138/am.2009.3075
Lepore G.O., Ballaran T.B., Nestola F. et al. // Mineral. Mag. 2012. V. 76. P. 963. https://doi.org/10.1180/minmag.2012.076.4.12
Kutoglu A. // Z. Anorg. Allg. Chem. 1976. V. 419. P. 176. https://doi.org/https://doi.org/10.1002/zaac.19764190211
Bonazzi P., Menchetti S., Pratesi G. // Am. Mineral. 1995. V. 80. P. 400. https://doi.org/10.2138/am-1995-3-422
Bindi L., Popova V., Bonazzi P. // Can. Mineral. 2003. V. 41. P. 1463. https://doi.org/10.2113/gscanmin.41.6.1463
Bindi L., Bonazzi P. // Am. Mineral. 2007. V. 92. P. 617. https://doi.org/doi:10.2138/am.2007.2332
Pratesi G., Zoppi M. // Am. Mineral. 2015. V. 100. P. 1222. https://doi.org/doi:10.2138/am-2015-5045
Gavezzotti A., Demartin F., Castellano C., Campostrini I. // Phys. Chem. Miner. 2013. V. 40. P. 175. https://doi.org/10.1007/s00269-012-0559-z
Bonazzi P., Lepore G.O., Bindi L. // Eur. J. Mineral. 2016. V. 28. P. 147. https://doi.org/10.1127/ejm/2015/0027-2474
Mullen D.J.E., Nowacki W. // Z. Krist. 1972. B. 136. S. 48. https://doi.org/doi:10.1524/zkri.1972.136.1-2.48
Brazhkin V.V., Bolotina N.B., Dyuzheva T.I. et al. // CrystEngCommun. 2011. V. 13. P. 2599. https://doi.org/10.1039/C0CE00861C
Bolotina N.B., Brazhkin V.V., Dyuzheva T.I. et al. // JETP Lett. 2014. V. 98. P. 539. https://doi.org/10.1134/S0021364013220025
Siidra O.I., Zenko D.S., Krivovichev S. V // Am. Mineral. 2014. V. 99. P. 817.
Aroyo M.I., Perez-Mato J.M., Orobengoa D. et al. // Bulg. Chem. Commun. 2011. V. 43. P. 183.
McKinnon J.J., Mitchell A.S., Spackman M.A. // Chem. – A Eur. J. 1998. V. 4. P. 2136. https://doi.org/10.1002/(SICI)1521-3765(19981102)4:11<2136::AID-CHEM2136>3.0.CO;2-G
Mckinnon J.J., Mark A., Anthony S. // Acta Cryst. B. 2004. V. 60. P. 627. https://doi.org/10.1107/S0108768104020300
Meyer A.Y. // Chem. Soc. Rev. 1986. V. 15. P. 449. https://doi.org/10.1039/CS9861500449
Jelsch C., Ejsmont K., Huder L. // IUCrJ. 2014. V. 1. P. 119. https://doi.org/10.1107/S2052252514003327
O’Keeffe M., Treacy M.M.J. // Symmetry (Basel). 2022. V. 14. P. 822. https://doi.org/10.3390/sym14040822
Shpotyuk O., Hyla M., Shpotyuk Y. et al. // Comput. Mater. Sci. 2021. V. 198. P. 110715. https://doi.org/https://doi.org/10.1016/j.commatsci.2021.110715
Pidcock E., Motherwell W.D.S., Cole J.C. // Acta Cryst. B. 2003. V. 59. P. 634. https://doi.org/10.1107/S0108768103012278
Carugo O., Blatova O.A., Medrish E.O. et al. // Sci. Rep. 2017. V. 7. P. 1. https://doi.org/10.1038/s41598-017-12699-4
Eon J.G. // Acta Cryst. A. 2016. V. 72. P. 376. https://doi.org/10.1107/S2053273316003867
Krivovichev S.V. // Acta Cryst. B. 2016. V. 72. P. 274. https://doi.org/10.1107/s205252061501906x
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография