

Микробиология, 2023, T. 92, № 1, стр. 98-102
Гены, кодирующие НАД+-зависимые формиатдегидрогеназы, в таксономии аэробных метилотрофных бактерий рода Ancylobacter
А. А. Чемодурова a, А. С. Решетников a, Н. В. Агафонова a, Н. В. Доронина a, *
a ФИЦ “Пущинский научный центр биологических исследований РАН” Институт биохимии и физиологии микроорганизмов им. Г.К. Скрябина РАН
142290 Пущино, Московская обл., Россия
* E-mail: doronina@ibpm.pushchino.ru
Поступила в редакцию 29.04.2022
После доработки 29.05.2022
Принята к публикации 30.05.2022
- EDN: NOFTGB
- DOI: 10.31857/S002636562260047X
Аннотация
Проведен сравнительный филогенетический анализ генов НАД+-зависимых формиатдегидрогеназ (НАД+–ФДГ), которые обнаружены во всех доступных геномах метилотрофов родов Ancylobacter, Starkeya и Angulomicrobium, а также у других бактерий семейства Xanthobacteraceae (Xanthobacter, Aquabacter, Azorhizobium). Отмечено, что расположение представителей Xanthobacteraceae на дереве, построенном на основании сравнения аминокислотных последовательностей НАД+–ФДГ, коррелирует с филогенией по гену 16S рРНК. Выявлено, что последовательности белка НАД+–ФДГ родов Ancylobacter, Starkeya и Angulomicrobium имеют уровень идентичности 87.8–98.3%, что свидетельствует о высокой консервативности этого белка в пределах данной группы метилотрофов. Впервые анализ функциональных генов НАД+–ФДГ рекомендован в качестве дополнительного критерия для межвидовой дифференциации метилотрофных бактерий рода Ancylobacter.
НАД+-зависимые формиатдегидрогеназы (НАД+–ФДГ) обнаружены у бактерий, дрожжей, грибов, растений и позвоночных (Alekseeva et al., 2011). У растений, патогенных бактерий и грибов этот фермент является стрессовым белком, у аэробных метилотрофных бактерий (метилотрофов) и дрожжей играет ключевую роль в снабжении клеток энергией (Hatrongjit, Packdibamrung, 2010; Alekseeva et al., 2011). Окисление формиата до CO2, сопряженное восстановлением НАД+ до НАДН, катализируется НАД+–ФДГ и является завершающей стадией цепи реакций прямого С1-окисления у метилотрофов. НАД+–ФДГ относится к надсемейству D-специфических дегидрогеназ 2-оксикислот, имеет гомодимерную структуру, не содержит в активном центре простетических групп и ионов металлов (Shabalin et al., 2010). Данный фермент распространен у метилотрофов с рибулозобисфосфатным путем С1-ассимиляции (Троценко с соавт., 2010). Типичным примером являются представители рода Ancylobacter (семейство Xanthobacteraceae, порядок Hyphomicrobiales), выделяемые из водной среды, донных осадков, активных илов, почвы и растений. В настоящее время род включает 11 валидно описанных видов (https://lpsn.dsmz.de/genus/ancylobacter), очень близких по физиолого-биохимическим, хемотаксономическим свойствам и последовательностям генов 16S рРНК, поэтому актуален поиск новых генетических маркеров, позволяющих осуществить быструю дифференциацию представителей данного рода без привлечения геномного анализа. Известно, что аминокислотные последовательности НАД+–ФДГ довольно консервативны и уровень их сходства у разных организмов составляет ~50%, а у растений эти ферменты имеют до 80% идентичности (Hatrongjit, Packdibamrung, 2010; Alekseeva et al., 2011). Ранее фермент НАД+–ФДГ исследовали только у одного представителя рода Ancylobacter – A. aqua-ticus KNK607M (Nanba et al., 2010), но значение соответствующего функционального гена в таксономии бактерий не рассматривалось.
Цель данной работы – оценка эффективности использования сравнительного анализа последовательностей генов НАД+–ФДГ в таксономии метилотрофных бактерий рода Ancylobacter.
В работе использовали новые метилотрофные изоляты штаммы Ancylobacter sp. VT, Starkeya sp. 3С и 1А, выделенные ранее авторами данной работы, а также ближайший родственник штаммов 3С и 1А – штамм Starkeya sp. HF14-78462, геном которого найден в базе данных NCBI GenBank (https://www.ncbi.nlm.nih.gov/) (CACSAS000000000). Поиск последовательностей генов 16S рРНК, белков НАД+–ФДГ и геномов проводили в базах данных NCBI GenBank и JGI (https://img.jgi.doe.gov/), филогенетический анализ осуществляли с помощью пакетов программ BLAST (https://blast.ncbi.nlm.nih.gov), ClustalW (Thompson et al., 1997) и MEGA5 (метод neighbor-joining) (Tamura et al., 2011), надежность построенных деревьев проверена значением “bootstrap” для 1000 деревьев. Средние значения идентичности нуклеотидов (ANI) и ДНК-ДНК гибридизации in silico (dDDH) рассчитывали с использованием программ Species 1.2.1 (Richter, Rosselló-Móra, 2009) и GGDC 2.1 (https://ggdc.dsmz.de/ggdc.php) (Meier-Kolthoff et al., 2013) соответственно. Амплификацию и секвенирование гена НАД+–ФДГ для штамма 1А проводили с использованием “вырожденных” праймеров, разработанных на консервативные аминокислотные участки НАД+–ФДГ. Анализ доступных аминокислотных последовательностей фермента НАД+–ФДГ из A. aquaticus, Starkeya sp. 3C и ряда других гомологичных ФДГ ферментов из NCBI GenBank позволил выделить две консервативные аминокислотные области, на основе которых составлена пара “вырожденных” праймеров fmd (F2-vir); TGCGTTCTYTACGAYGAYСС и fmd (R-vir); TCTTCCGARCCGCCAGTSGCRTT. Амплифицирован участок гена 1170 п.н., кодирующий фермент НАД+–ФДГ из метилотрофного изолята штамма 1А. Секвенирование и анализ полученного фрагмента выявил 99.9% идентичности аминокислотной последовательности с ферментом НАД+–ФДГ из Starkeya sp. 3C.
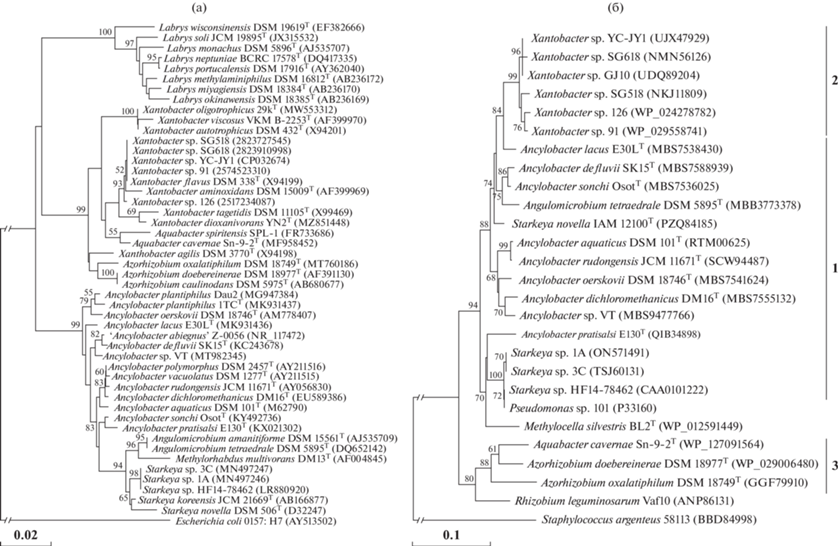
Гены НАД+–ФДГ выявлены нами у некоторых представителей семейства Xanthobacteraceae – родов Xanthobacter, Aquabacter, Azorhizobium, а также во всех доступных геномах метилотрофных бактерий родов Ancylobacter, Starkeya и Angulomicrobium. Установлено, что распределение представителей Xanthobacteraceae на дереве, построенном на основании сравнения аминокислотных последовательностей НАД+–ФДГ, коррелирует с филогенией по гену 16S рРНК (рис. 1). При этом выявлено несколько кластеров на дереве, построенном по результатам сравнительного анализа последовательностей белков НАД+–ФДГ, объединяющих представителей семейства Xanthobacteraceae: (1) группа Ancylobacter, Starkeya и Angulomicrobium; (2) группа Xanthobacter; (3) группа Aquabacter и Azorhizobium.
В группе (1) сходство между видами на основании сравнения нуклеотидных последовательностей генов 16S рРНК составляет 96.2–98.5%, а аминокислотные последовательности белка НАД+–ФДГ проявляют уровень межвидовой идентичности 87.8–98.3% (табл. 1), что свидетельствует о высокой консервативности белка НАД+–ФДГ у представителей родов Ancylobacter, Starkeya и Angulomicrobium. Поиск ближайших родственников Ancylobacter по сходству НАД+–ФДГ выявил также метилотрофа Pseudomonas sp. 101 (91.3–92.8%) (Filippova et al., 2006), НАД+–ФДГ которого является одной из самых изученных ФДГ среди бактерий (Alekseeva et al., 2011). По всей видимости, данный штамм также является представитем Starkeya или Ancylobacter, однако идентификация, проведенная в 1970-х гг. (коллекция кафедры микробиологии Московского государственного университета) (Egorov et al., 1979), устарела и требует пересмотра.
Интересно, что претенденты на новые виды новые изоляты штаммы 3С, 1А, HF14-78462 (~99.9–100% сходства по гену 16S рРНК) и штамм VT также хорошо дифференцируются на видовом уровне по генам НАД+–ФДГ, что коррелирует с их филогенетическим положением по гену 16S рРНК (рис. 1). Анализ геномов штаммов 3С (VMBP00000000) и VT (JAHCQH000000000) подтверждает принадлежность этих микроорганизмов к новым видам, поскольку значение сходства по генам 16S рРНК, уровень ANI и dDDH составило, соответственно: 99.3–99.4, 86.4 и 28.3% между штаммом 3С и S. novella DSM 506T, и 98.3–98.5, 78.0–80.6 и 22.1–24.0% между штаммом VT и ближайшими представителями рода Ancylobacter, что ниже пороговых значений, принятых для разделения видов (ANI = 95%, dDDH = 70%) (Richter et al., 2009; Chun et al., 2018).
Таким образом, показано, что топология филогенетического дерева по белку НАД+–ФДГ коррелирует с таксономическим положением представителей рода Ancylobacter и семейства Xanthobacteraceae, проведенным на основании сравнения последовательностей генов 16S рРНК. Анализ этих достаточно консервативных функциональных генов может быть рекомендован в качестве дополнительного критерия для межвидовой дифференциации прежде всего бактерий рода Ancylobacter.
Предложение использовать функциональные гены ферментов, вовлеченных в метаболизм метилотрофов, не является уникальным. Известно, что у метилотрофов ген mxaF (кодирует большую субъединицу метанолдегидрогеназы) высоко консервативен и используется в качестве функционального гена для их идентификации в различных средах обитания (McDonald, Murrell, 1997). Также в качестве молекулярных проб применяют гены xoxF (Ramachandran, Walsh, 2015; Taubert et al., 2015), но известные последовательности филогенетически разнообразны, делятся на пять групп (XoxF1−5) (Chistoserdova, 2011), а их идентичность в пределах одной группы ~65–70% (Keltjens et al., 2014). Анализ аминокислотных последовательностей белка НАД+–ФДГ представителей Xanthobacteraceae выявил уровень идентичности в пределах семейства 84.0–98.3%, а среди ближайших родственников обнаружены только Methylocella silvestris BL2T (89–90%) и Rhizobium leguminosarum Vaf10 (84.7–86.7%), однако на дереве они держатся обособленно (табл. 1, рис. 1). Кроме того, представители родов Angulomicrobium и Starkeya по исследуемым генам НАД+–ФДГ образуют единый кластер с представителями Ancylobacter, что согласуется с результатами секвенирования генов 16S рРНК и демонстрирует высокое родство представителей этих трех родов. Таким образом, впервые предложено использовать НАД+–ФДГ в качестве функционального маркерного гена для дальнейшего поиска и идентификации метилотрофов рода Ancylobacter, обнаруживаемых в различных местах обитания.
Таблица 1.
Уровни сходства представителей рода Ancylobacter с ближайшими родственниками на основании сравнения нуклеотидных последовательностей гена 16S рРНК и аминокислотных последовательностей белка НАД+–ФДГ, н.д. – нет данных
| Ancylobacter | |||
|---|---|---|---|
| 16S рРНК, % | НАД+–ФДГ, % | ||
| Xanthobacteraceae | Ancylobacter | 96.8–98.5 | 89.0–98.3 |
| Starkeya | 97.0–98.0 | 90.3–96.8 | |
| Angulomicrobium | 96.2–97.7 | 87.8–96.0 | |
| Xanthobacter | 92.1–94.7 | 87.3–94.8 | |
| Azorhizobium | 92.9–94.5 | 84.3–87.5 | |
| Aquabacter | 92.9–94.6 | 84.0–85.8 | |
| Pseudomonas sp. 101 | н.д. | 91.3–92.8 | |
| Methylocella silvestris BL2T | 90.2–91.3 | 89.0–90.0 | |
| Rhizobium leguminosarum Vaf10 | 90.6–91.6 | 84.7–86.7 | |
Рис. 1.
а – филогенетическое положение представителей семейства Xanthobacteraceae на основании сравнительного анализа нуклеотидных последовательностей гена 16S рРНК; б – филогенетическое положение представителей рода Ancylobacter среди ближайших родственников, основанное на сравнении аминокислотных последовательностей белка НАД+–ФДГ.
Список литературы
Троценко Ю.А., Доронина Н.В., Торгонская М.Л. Аэробные метилобактерии. Пущино: ОНТИ ПНЦ РАН, 2010. 325 с.
Trotsenko Y.A., Doronina N.V., Torgonskaya M.L. Aerobic Methylobacteria. Pushchino: ONTI PSC RAS, 2010. 325 р.
Alekseeva A.A., Savin S.S., Tishkov V.I. NAD+-dependent formate dehydrogenase from plants // Acta Naturae. 2011. V. 3. № 4(11). P. 38–54.
Chistoserdova L. Modularity of methylotrophy, revisited // Environ. Microbiol. 2011. V. 13. P. 2603–2622.
Chun J., Oren A., Ventosa A., Christensen H., Arahal D.R., da Costa M.S., Rooney A.P., Yi H., Xu X.-W., De Meyer S., Trujillo M.E. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes // Int. J. Syst. Evol. Microbiol. 2018. V. 68. P. 461–466.
Egorov A.M., Avilova T.V., Dikov M.M., Popov V.O., Rodionov Y.V., Berezin I.V. NAD-dependent formate dehydrogenase from methylotrophic bacterium, strain 1: purification and characterization // Eur. J. Biochem. 1979. V. 99. P. 569–576.
Filippova E.V., Filippova E.V., Polyakov K.M., Tikhonova T.V., Boiko K.M., Tishkov V.I., Popov V.O. Crystal structures of complexes of NAD+-dependent formate dehydrogenase from methylotrophic bacterium Pseudomonas sp. 101 with formate // Crystallography Reports. 2006. V. 51. № 4. P. 627–631.
Hatrongjit R., Packdibamrung K. A novel NADP+-dependent formate dehydrogenase from Burkholderia stabilis 15516: screening, purification and characterization // Enzyme and Microbial Technology. 2010. V. 46. №. 7. P. 557–561.
Keltjens J.T., Pol A., Reimann J., Op den Camp H.J. PQQ-dependent methanol dehydrogenases: rareearth elements make a difference // Appl. Microbiol. Biotechnol. 2014. V. 98. P. 6163–6183.
McDonald I.R., Murrell J.C. The methanol dehydrogenase structural gene mxaF and its use as a functional gene probe for methanotrophs and methylotrophs // Appl. Environ. Microbiol. 1997. V. 63. P. 3218–3224.
Meier-Kolthoff J.P., Auch A.F., Klenk H.-P., Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions // BMC Bioinformatics. 2013. V. 14. P. 1–14.
Nanba H., Takaoka Y., Hasegawa J. Purification and characterization of formate dehydrogenase from Ancylobacter aquaticus strain KNK607M, and cloning of the gene // Bioscience, Biotechnology, and Biochemistry. 2003. V. 67. № 4. P. 720–728.
Ramachandran A., Walsch D.A. Investigation of XoxF methanol dehydrogenases reveals new methylotrophic bacteria in pelagic marine and freshwater ecosystems // FEMS Microbiol. Ecol. 2015. V. 91. P. fiv105.
Richter M., Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition // Proc. Natl. Acad. Sci. USA. 2009. V. 106. P. 19126–19131.
Shabalin I.G., Serov A.E., Skirgello O.E., Timofeev V.I., Samygina V.R., Popov V.O., Tishkov V.I., Kuranova I.P. Recombinant formate dehydrogenase from Arabidopsis thaliana: preparation, crystal growth in microgravity, and preliminary X-ray diffraction study // Crystallography Reports. 2010. V. 55. № 5. P. 806–810.
Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods // Mol. Biol. Evol. 2011. V. 28. P. 2731–2739.
Taubert M., Grob C., Howat A.M., Burns O.J., Dixon J.L., Chen Y., Murrell J.C. XoxF encoding an alternative methanol dehydrogenase is widespread in coastal marine environments // Environ. Microbiol. 2015. V. 17. P. 3937–3948.
Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools // Nucleic Acids Res. 1997. V. 25. P. 4876–4882.
Дополнительные материалы отсутствуют.