

Микроэлектроника, 2021, T. 50, № 2, стр. 146-160
Моделирование кристаллизации, корреляции свойств от состава и размера частиц в двумерных GaSxSe1 – x(0 ≤ х ≤ 1)
С. М. Асадов a, *, С. Н. Мустафаева b, В. Ф. Лукичев c, **
a Институт катализа и неорганической химии Национальной академии наук Азербайджана
AZ-1143 Баку, пр. Г. Джавида, 113, Азербайджан
b Институт физики Национальной академии наук Азербайджана
AZ-1143 Баку, пр. Г. Джавида, 131, Азербайджан
c Физико-технологический институт им. К.А. Валиева Российской академии наук
117218 Москва, Нахимовский просп., 36, корп. 1, Россия
* E-mail: salim7777@gmail.com
** E-mail: lukichev@ftian.ru
Поступила в редакцию 17.07.2020
После доработки 17.07.2020
Принята к публикации 17.07.2020
Аннотация
Представлены результаты теоретико-экспериментальных исследований физико-химических условий синтеза, расчета $T{\kern 1pt} - {\kern 1pt} x$ фазовой диаграммы, термодинамики и кинетики кристаллизации, корреляции свойств от состава и размера частиц полупровдниковых твердых растворов 2D системы GaS–GaSe, где образуется непрерывный ряд твердых растворов GaSxSe1 –x(0 ≤ х ≤ 1). Синтезированы твердые растворы GaSxSe1 –x(0 ≤ х ≤ 1) и с помощью методов физико-химического анализа идентифицированы индивидуальные термодинамические фазы. В рамках разработанной модели растворов, близкий x к идеальным, определены физико-химические и термодинамические закономерности Т–х фазовой диаграммы GaS–GaSe. Термодинамическим способом коррелированы температурно-концентрационные зависимости изменения свободной энергии Гиббса твердых растворов GaSxSe1 – x(0 ≤ х ≤ 1). Исследованы термодинамические и кинетические условия кристаллизации фаз, корреляции свойств от состава и размера частиц в твердых растворах 2D системы GaS–GaSe. Моделированы закономерности кинетики нуклеации и кристаллизации, а также термодинамика образования кластеров в многокомпонентных 2D системах. В рамках теории функционала плотности рассчитаны электронные зонные структуры 2D GaS и GaSe. Из теоретических расчетов определены ширины запрещенных зон для монослоев GaS и GaSe. Анализированы спектры электропоглащения аморфно-пленочных и объемных монокристаллических образцов GaSe.
ВВЕДЕНИЕ
Исследования низкоразмерных, в частности, полупроводниковых материалов носят междисциплинарный характер и охватывают физико-технологию, химию, инженерию и биологию. Синтез, характеристика, сборка и обработка таких материалов и наночастиц значительно продвинулись вперед. В этой области все еще развивающейся темой являются материалы на основе 2D систем. Хотя синтез 2D материалов является одной из развиваемых областей материаловедения и технологии, отсутствуют общие правила синтеза, в т.ч. наночастиц, конструирования микро- и наноструктур и обработки объемных материалов на основе наночастиц [1].
В свете указанного необходимо всесторонне изучить конкретные 2D системы и обеспечить систематическое рассмотрение их для применения в полупроводниковой электронике. Тем не менее, нет общих закономерностей синтеза и корреляции свойств от структуры, состава и важных физико-химических и технологических факторов многокомпонентных 2D систем.
По выше указанной важной и захватывающей, но сложной теме в последние годы интенсивно исследуются системы на основе слоистых халькогенидов III группы, в частности, халькогениды галлия. Сфокусированное на термодинамических и кинетических особенностях получения твердых растворов системы GaS–GaSe, изучение корреляции свойств от состава и размера частиц является задачей настоящей работы.
Поскольку атом имеет размер ~1 Å, кластер с радиусом 1 нм будет состоять из ~25 атомов, и большинство атомов будет находиться на поверхности. Если в таком кластере размеры наночастиц меньше критических размеров, то последние характеризуют многие физические явления или квантовые эффекты в материалах, в частности, длину волны света и длину свободного пробега электрона в полупроводниках, т.е. когда размерность меньше этих критических длин проявляются новые физические и физико-химические свойства. Это делает наночастицы такой интересной и уникальной категорией материала по сравнению с большими молекулами с сотнями или тысячами атомов.
Ниже кратко рассмотрим примеры полупроводниковых систем с участием 2D материалов на основе соединений типа AIIIBVI. Интерес к 2D полупроводниковым материалам на основе соединений типа AIIIBVI, для которых характерны квантовые эффекты, обусловлен потенциалом их применения в наноразмерных устройствах. В 2D кристаллах AIIIBVI в слоях действуют ковалентные связи, а между слоями – слабая Ван-дер-Ваальсовая связь. Благодаря указанному в AIIIBVI проявляются анизотропные свойства. Кристаллы AIIIBVI по оптическим и электрическим свойствам, в частности (GaS [2, 3], GaSe [4, 5], InSe [6, 7], TlS [8, 9]) близки к перспективным материалам нанотехнологии, таким, как графен [10] и топологические изоляторы.
Данные о кристаллической и электронной структуре соединений GaS и GaSe известны [5, 11]. Кристаллы GaS и GaSe принадлежат гексагональной сингонии, характеризуются слоистой структурой и пространственной группой $D_{{3h}}^{1} - P\bar {6}m2$. Они имеют различные политипы. Так, например, GaSe имеет четыре политипа β-, ε-, γ- и δ-GaSe [5, 11]. При комнатной температуре термодинамически более стабильными политипами являются β-GaS и ε-GaSe, соответственно. GaS и GaSe являются широкозонными полупроводниками и при комнатной температуре имеют ширину запрещенной зоны 2.53 и 1.98 эВ соответственно. Они имеют ряд преимуществ перед другими 2D материалами: большая область рабочих температур, возможность создавать на их основе светоизлучающие устройства в видимом спектре, высокие значения критического поля электрического пробоя, радиационная стойкость [12–16]. GaS и GaSe между собой образуют непрерывный ряд твердых растворов GaSxSe1 –x (0 ≤ х ≤ 1) [5, 17–19].
Однако, поли- и монокристаллы GaS и GaSe, а также твердые растворы GaSxSe1 –x, формированные из расплава, как и другие многокомпонентные сплавы [20–25], часто имеют неоднородное распределение плотности дислокаций. Эти нарушения приводят к механическим напряжениям и образованию собственных точечных дефектов в решетке. Как следствие полученный материал имеет невоспроизводимые электрические, оптические, фотоэлектрические, люминесцентные и другие физические характеристики. Кроме того, синтез и изучение свойств GaS и GaSe усложняются также из-за формирования различных политипов.
По сравнению с их объемными аналогами двумерные (2D) кристаллы, в т.ч. GaS и GaSe демонстрируют новые физические свойства и имеют потенциал для использования их в электронных и оптоэлектронных устройствах нового поколения. Тем не менее контролируемый синтез крупных однородных кристаллов и 2D слоев все еще остается сложной технологической задачей. Рассмотрим несколько конкретных примеров.
Двумерные наноструктуры привлекают внимание из-за обнаруженных в них квантовых эффектов. В работе [26] приведены результаты по синтезу 2D материалов на основе GaSe с несколькими нанослоями для высокопроизводительных фотоприемников. Сравнение критических параметров для 2D-наноструктурных фотоприемников показывает, что, например, квантовая эффективность монослоя GaSe в 85 раз больше, чем графена и в 4557 раз – чем оксида графена.
Результаты исследования контролируемого парофазного роста монокристаллических двумерных кристаллов GaSe с высоким фотоответом приведены в работе [27]. Указано, что с использованием осаждения из паровой фазы синтезированы однородные (до 60 мм в поперечном размере) 2D монокристаллы GaSe на подложках SiO2/Si. Процесс контролировался продолжительностью роста, областью роста, температурой роста и потоком газа-носителя аргона. Однако, таким способом трудно получить стехиометрический состав GaSe.
В [28] сообщалось о синтезе многослойных GaS1 –xSex с перестраиваемыми составами во всем диапазоне х = 0–1. Синтез проводили в реакторе из кварцевой трубки методом химического транспорта в паровой фазе. В качестве исходных компонентов использовали порошки Ga2S3 (99.99%, Sigma-Aldrich) и Ga2Se3 (99.99%, Alfa). Для выращивания многослойных нанолистов использовали Si-подложку, на которую при 700°С осаждали слои. Состав контролировался с использованием температуры испарения и соотношения составов двух исходных порошков. Для равномерного роста монослоев на подложках SiOx/Si толщиной 300 нм были использованы двухзонная печь, где зоны I (исходные компоненты) и II (для роста монослоя) нагревали до 950 и 700°С соответственно Затем проводили рост при этих температурах в течение 2–10 мин под давлением 20 Торр в атмосфере аргона. Указано, что полученные многослойные нанопластинки из сплавов системы GaS–GaSe имеют ширину запрещенной зоны ${{E}_{g}}$ = 2.0–2.5 эВ, и величину ${{E}_{g}}$ возможно линейно настраивать в области спектра от красного света до зеленого. Эпитаксиальный рост монослоев приводит к резкому увеличению ${{E}_{g}}$ до 3.3–3.4 эВ, что смещает излучение в УФ область. Расчеты из первых принципов предсказывают, что ${{E}_{g}}$ монослоев GaS и GaSe составляют 3.325 и 3.001 эВ соответственно. При увеличении количества слоев до трех энергии как прямых, так и непрямых переходов ${{E}_{g}}$ значительно уменьшаются и непрямой переход ${{E}_{g}}$ приближается к значению для многослойной пластинки. Отметим, что указанным технологически сложным способом, по-видимому, трудно формировать стехиометрический состав многослойных нанопластинок получить возпроизводимые результаты.
Причины, вызывающие неоднородное распределение структурных дефектов по объему формированных кристаллов в системе GaS–GaSe, определяются несколькими процессами. Основными из них являются кристаллизация, посткристаллизационное охлаждение и термообработка кристалов, в результате чего возникают градиенты концентрации, химического потенциала и температуры. Однако, параметры, влияющие на формирование структуры и физические свойства GaSxSe1 –x, до сих пор не определены с позиций термодинамического моделирования и теории кристаллизации нанофаз.
Из указанного следует, что приложение термодинамической и кинетической теорий кристаллизации к описанию формирования и эволюции конкретных структур и политипов в виде объемных и тонкопленочных образцов на основе системы GaS–GaSe существенно осложняется из-за комплекса нерешенных вопросов. В частности, при создании многокомпонентных материалов с требуемым набором свойств важно не только синтезировать вещество с определенным химическим составом, но также направленно формировать его структуру.
Цель настоящей работы – теоретико-экспериментальное исследование физико-химических условий синтеза, расчет $~T{\kern 1pt} - {\kern 1pt} x$ фазовой диаграммы, термодинамики и кинетики кристаллизации, корреляции свойств от состава фаз и размера частиц твердых растворов 2D GaSxSe1 –x (x = 0–1.0 мольная доля).
2 МЕТОДИЧЕСКАЯ И ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
Материалы. Бинарные соединения GaS и GaSe синтезировали из чистых химических элементов галлия (Ga-5 N), серы (B5) и селена (ос. ч.-17-3). Синтез соединений осуществляли плавлением исходных элементов, взятых в стехиометрических соотношениях, в вакуумированных (10–2 Па) кварцевых ампулах. Ампулы с соответствующими компонентами для синтеза 2D GaS и GaSe помещали в электропечь и температуры поднимали выше температуры плавления этих соединений на 10–15 К (температуры плавления GaS и GaSe – 1288 и 1211 К соответственно). При температурах выше температуры плавления GaS и GaSe ампулы могут разрушаться из-за высокого давления паров халькогенов. Ампулы с расплавами GaS и GaSe выдерживали течение 6–8 ч, а затем их охлаждали до 500 К и выключали печи. Таким образом, ампулы с GaS и GaSe самопроизвольно остывали до комнатной температуры.
Синтезированные полупроводниковые соединения GaS и GaSe p-типа идентифицировали с помощью дифференциального термического анализа (ДТА; дериватограф NETZSCH 404 F1 Pegasus) и порошкового рентгенофазового анализа (РФА; дифрактометр Bruker D8 ADVANCE с CuKα-излучение) [5]. Температурно-фазовые изменения определяли с точностью ±1 К. Погрешность определения параметров ячейки не превышала 0.003 Å.
Затем путем плавления стехиометрических количеств ранее полученных соединений GaS и GaSe в эвакуированных (10–2 Па) кварцевых ампулах синтезировали образцы твердых растворов GaSxSe1 –x (x = 0–1.0) системы GaS–GaSe. Монокристаллы GaSxSe1 –x выращивали методом направленной кристаллизации. Ампулы с образцами нагревали до температуры плавления образцов, а затем проводили кристаллизацию расплавов. Скорость фронта кристаллизации варьировали в интервале 0.5–1.1 мм/ч (метод Бриджмена). Свойства были изучены как на пленочных [12], так и монокристаллических образцах.
Зонная структура и электронные свойств GaS и GaSe со слоистой структурой исследованы в рамках первопринципного метода на основе теории функционала плотности (DFT). Геометрическую оптимизацию и расчет полной энергии проводили с использованием пакета моделирования обменно-корреляционного потенциала версии обобщенного градиентного приближения (GGA) Perdew-Burke-Ernzerhof (PBE) [10]. Анализированы полные и парциальные плотности электронных состояний соединений GaS и GaSe и их монослоев.
Факторы изоморфной взаимозаместимости атомов в кристаллах. При оценке взаимозаместимости изоморфных компонентов GaS и GaSe, рассматривали известные четыре фактора, которые влияют на изоморфную взаимозаместимость атомов в кристаллах твердых растворов: критерий химической индифферентности атомов, фактор размера атомов, фактор сходства природы межатомной связи и фактор изоструктурности кристаллов. В частности, согласно первому условию изоморфные смеси должны образоваться за счет кинетической энергии движения частиц. Такие смеси близки к идеальным растворам, где силы взаимодействия молекулразных компонентов равны силам, с которыми действуют друг на друга молекулы одного вида компонента. Т.е. если частицы GaS образуют изоморфную смесь с частицами GaSe, то для идеального раствора сила взаимодействия частиц GaS и GaSe будет равна силе взаимодействия GaS с GaS и GaSe с GaSe. Это условие исключает образование химических соединений.
Термодинамические свойства. Фазовую диаграмму квазибинарной системы GaS–GaSe с неограниченным диапазоном твердых растворов рассчитывали по термодинамическим данным с использованием энергий Гиббса для соответствующих фаз GaS (1) и GaSe (2). Для термодинамического расчета фазовой диаграммы $T{\kern 1pt} --{\kern 1pt} x$ бинарной системы 1–2 использовали предложенный в [29, 5 ] способ, включающий соотношения между концентрациями исходных компонентов и их термодинамическими функциями плавления $(F_{i}^{{\text{m}}})$. Таким образом, линии ликвидуса (или линии начала первичной кристаллизации) и солидуса (или линии конца первичной кристаллизации) на $T{\kern 1pt} --{\kern 1pt} x$ диаграмме системы 1–2 с непрерывными рядами жидких и твердых растворов описывали формулами (1):
(1)
$\begin{gathered} x_{1}^{l} = \frac{{1 - F_{2}^{{\text{m}}}}}{{F_{1}^{{\text{m}}} - F_{2}^{{\text{m}}}}},\,\,\,\,x_{1}^{s} = F_{1}^{{\text{m}}}x_{1}^{l}, \\ F_{i}^{{\text{m}}} = {\text{exp}}\left[ {\frac{{\Delta H_{i}^{{0,{\text{m}}}}\left( {{{T}^{{ - 1}}}T_{i}^{{ - 1,{\text{m}}}}} \right)}}{R}} \right], \\ \end{gathered} $Концентрационно-температурные зависимости свободной энергии Гиббса образования $\left( {\Delta G_{T}^{0} = f\left( {x,~T} \right)} \right)$ твердых растворов GaSxSe1 –x (0 ≤ $x$ ≤ 1) рассчитывали с использованием модели раствора, включающей немолекулярные соединения [5, 30]. Уравнение для $\Delta G_{T}^{0}$ твердых растворов GaSxSe1 –x из составляющих их бинарных компонентов при p, T = const имеет вид:
(2)
$\begin{gathered} {{\Delta }_{f}}G_{T}^{0}\left( {{\text{Дж}} \cdot {\text{мол}}{{{\text{ь}}}^{{ - 1}}}} \right) = \left( {1 - x} \right){{\Delta }_{f}}H_{{298~}}^{0}\left( {{\text{GaS}}} \right) + x{{\Delta }_{f}}H_{{298}}^{0}\left( {{\text{GaSe}}} \right) - ~~\left( {1 - x} \right)T{{\Delta }_{f}}S_{{298~}}^{0}\left( {{\text{GaS}}} \right) - \\ - \,\,xT{{\Delta }_{f}}S_{{298~}}^{0}\left( {{\text{GaSe}}} \right) - T\Delta C_{{p,298~}}^{0}~\left[ {\ln \left( {\frac{T}{{298}}} \right) + ~\left( {\frac{{298}}{T}} \right) - 1} \right] + ~ \\ + \,\,~8.314~T\left[ {\left( {1 - x} \right)\ln \left( {1 - x} \right) + x\ln \left( x \right)} \right] + ~\,\,{{\Delta }_{f}}G_{T}^{{{\text{0,ex}}}}, \\ \end{gathered} $3. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Физико-химические свойства. Структуры и свойства объемных образцов бинарных соединений GaS (светло-желтого цвета) и GaSe (темно- красного цвета), как известно, в литературе изучены подробно. При синтезе и кристаллизации формируются различные политипы этих полупроводниковых соединений со слоистой структурой. Оба они принадлежат к структурному типу GaS, где каждый атом галлия связан с тремя атомами халькогена и другим атомом галлия.
Данные РФА и ДТА образцов компонентов GaS и GaSe подтверждают, что компоненты неограниченно растворяются друг в друге как в жидком, так и в твердом состоянии. В качестве примера на рис. 1 приведена порошковая рентгенограмма чистого β-GaS, который кристаллизуется в гексагональной сингонии с пространственной группой ${{P{{6}_{3}}} \mathord{\left/ {\vphantom {{P{{6}_{3}}} {mmc}}} \right. \kern-0em} {mmc}}~$. Этот политип кристалла имеет следующие параметры элементарной ячейки: a = 4.002 ± 0.002 Å и с = 15.44 ± 0.005 Å. Для гексагональной структуры β-GaSe параметры решетки составляют: 3.755 ± 0.002 Å и с = 15.475 ± ± 0.005 Å при комнатной температуре. Полученные параметры кристалличесих ячеек GaS и GaSe согласуются с данными [5, 11].

Термодинамические расчеты. Нами изученная и рассчитанная по формуле (1) $T{\kern 1pt} - {\kern 1pt} x$ фазовая диаграмма системы GaS–GaSe показана на рис. 2. По данным ДТА для компонентов GaSe и GaS, получены следующие параметры плавления [5]: ${\Delta }{{H}^{{\text{m}}}}$ (GaSe) = 30300 ± 200 Дж/моль, ${{T}^{{\text{m}}}}$ (GaSe) = = 1211 ± 3 К, ${\Delta }{{H}^{{\text{m}}}}$(GaS) = 34800 ± 200 Дж/моль, ${{T}^{{\text{m}}}}$(GaS) = 1288 ± 3 К. $T{\kern 1pt} --{\kern 1pt} x$ фазовую диаграмму GaS–GaSe, а точнее линии ликвидуса (${{T}^{{\text{l}}}}$) и солидуса (${{T}^{{\text{s}}}}$) рассчитывали с учетом указанных данных процесса плавления компонентов.
Рис. 2.
Расчетная (кривые) и экспериментально (точки) построенная фазовая диаграмма системы GaS–GaSe.
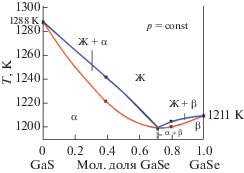
$T{\kern 1pt} - {\kern 1pt} x$ фазовая диаграмма состояния системы GaS–GaSe характеризуется неограниченной взаимной растворимостью исходных компонентов. А точнее компоненты образуют непрерывный ряд твердых растворов и $T{\kern 1pt} - {\kern 1pt} x$ диаграмма имеет минимум на кривой ликвидуса. Минимум соответствует следующим координатам: ~0.7 мольной доли GaSe и 1200 ± 1 К. В указанных координатах составы двух равновесных фаз равны и система GaS–GaSe вырождается в квазиоднокомпонентную, т.е. состав, соответствующий точке минимума на $T{\kern 1pt} {\kern 1pt} - {\kern 1pt} x$ фазовой диаграмме GaS–GaSe, можно рассматривать как отдельный компонент.
$T{\kern 1pt} - {\kern 1pt} x$ фазовая диаграмма GaS–GaSe с точкой минимума относится к третьему типу классификации Росебома. Расчетные температуры солидуса и ликвидуса незначительно отличаются от экспериментальных данных (разница солидуса составляла ~5 K и ликвидуса ~10 K). На $T{\kern 1pt} - {\kern 1pt} x$ фазовой диаграмме в точке минимума расплав соответствующего состава плавится конгруэнтно, т.е. жидкая фаза переходит в твердую фазу того же состава.
Образование непрерывного ряда твердых растворов GaSxSe1 –x в системе GaS–GaSe соответствует эмпирическим критериям: размер атомов: ${{\Delta r} \mathord{\left/ {\vphantom {{\Delta r} r}} \right. \kern-0em} r}$ ≤ 15% (правило Гольдшмидта), одинаковые кристаллические структуры и близкая электроотрицательность: |χA – χB| ≤ 0.6. Кристаллы изоморфной смеси GaSxSe1 – x однородны и однофазны, и частицы изоморфной смеси статистически распределены в элементарных ячейках кристаллов GaS и GaSe. Взаимодействие компонентов GaS и GaSe приводит к образованию фаз переменного состава, и изменение свойств фаз в области однородности непрерывны. РФА GaSxSe1 –x (0 ≤ $x$ ≤ 1) показывает, что эти твердые растворы образуют фазы замещения без изменения симметрии кристаллической структуры.
В точке минимума $~T{\kern 1pt} - {\kern 1pt} x$ диаграммы изменение общей свободной энергии Гиббса $\Delta {{G}_{{{\text{gen}}}}}$ при охлаждении расплава с дальнейшей кристаллизацией происходит за счет двух вкладов. Первая часть $\Delta {{G}_{{{\text{gen}}}}}$ обусловлена понижением энергии системы при кристаллизации. Энергия системы $\Delta {{G}_{{{\text{gen}}}}}$ уменьшается на величину ${{G}_{1}} = V{{G}_{V}}\sim {{n}^{3}}$ ($V$ – объем ядра-зародыша “мономера”, $\Delta {{G}_{V}}$ – удельная разность свободных энергий при переходе расплава в кристаллическое состояние, ${{n}^{3}}$ – число частиц). А вторая часть $\Delta {{G}_{{{\text{gen}}}}}$ наоборот увеличивается на величину, равную работе по образованию поверхности раздела ${{G}_{2}} = s\sigma \sim {{n}^{2}}$($\sigma $ – удельное поверхностное натяжение на границе “кристалл–жидкость”, $s$ – поверхность зародыша). Если предположить, что каждое формированное ядро термодинамической фазы имеет сферическую форму радиуса $r$, тогда можно записать $\Delta {{G}_{{{\text{gen}}}}} = {{r}^{3}}\Delta {{G}_{V}} + 6{{r}^{2}}\sigma $.
Образованные из “мономеров” новые кластеры докритических размеров ($r < r{\text{*}}$) характеризуют зародыши, а кластеры с радиусом больше $r{\text{*}}$ – ядра. Отсюда следует, что зародыши с размером больше критического ($r > r{\text{*}}$) вызывают уменьшение $\Delta {{G}_{{{\text{gen}}}}}$, т.е. такие зародыши будут устойчивыми и способными к росту. Таким образом, в первом приближении процесс возникновения упорядоченной фазы согласуется с критериями термодинамической теории формирования новой фазы.
Растворы системы GaS–GaSe проанализируем термодинамически с учетом свободной энергии Гиббса $G$, которая как и любая экстенсивная (зависящая от массы) термодинамическая функция раствора определяется внешними параметрами ($p,~T$ – давление, температура) и составом раствора: $G = f\left( {p,~T,{{x}_{1}},{{x}_{2}}} \right)$. При условии $p = {\text{const}}$ для образования твердых растворов GaSxSe1 –x по формуле (2) рассчитали зависимости ${{\Delta }_{f}}G_{T}^{0}\left( {x,~T} \right)$. В формуле (2), последние два члена являются свободной энергией смешения образования GaSxSe1 –x из парциальных мольных термодинамических величин компонентов GaS и GaSe.
Согласно теории растворов, функция смешения $\bar {\Phi }_{i}^{{{\text{mix}}}}$ (или относительная парциальная молярная (мольная) величина) характеризует разность между парциальной мольной величиной для компонента в растворе и молярной величиной для чистого компонента, т.е. $\bar {\Phi }_{i}^{{{\text{mix}}}}$ характеризует изменение полной термодинамической функции при смешении 1 моля чистого компонента с бесконечно большим количеством раствора данного состава при постоянных T и p. Отсюда следует, что парциальная молярная функция смешения $\overline {~\Phi } _{i}^{{{\text{mix}}}} = {{\Phi }_{i}} - \Phi _{i}^{0}$ – это функция смешения, характеризующая отдельный компонент раствора, где ${{\Phi }_{i}}$ – полная парциальная молярная (мольная) величина, $\Phi _{i}^{0}$ – молярная (мольная) величина чистого компонента i.
Для описания термодинамических функций состояния, в частности, энергии Гиббса чистых элементов в конденсированном состоянии в CALPHAD-подходе (Calculation of Phase Diagrams – расчет диаграмм состояния), как правило, используют базу данных (БД) SGTE (Scientific Group Thermodata Europe). Данные для расчета энергии Гиббса элементов в различных фазовых состояниях, в том числе метастабильных, для широких температурных интервалов содержатся в БД SGTE. Однако, для аппроксимации $T{\kern 1pt} - {\kern 1pt} x$ фазовой диаграммы GaS–GaSe мы использовали экспериментальные и справочные термодинамические табличные данные. Для расчетов брали следующие усредненные мольные термодинамические функции соединений GaS и GaSe: ${{\Delta }_{f}}H_{{298}}^{0}\left( {{\text{GaS}}} \right)$ = –194 650 ± 14 650 Дж/моль, ${{\Delta }_{f}}S_{{298}}^{0}\left( {{\text{GaS}}} \right)$ = 420 ± 290 Дж/(моль ⋅ K); ${{\Delta }_{f}}H_{{298}}^{0}\left( {{\text{GaSe}}} \right)$ = ‒159 000 Дж/моль и ${{\Delta }_{f}}S_{{298}}^{0}\left( {{\text{GaSe}}} \right) = $ = –11 Дж/(моль ⋅ К).
Концентрационные зависимости теплоемкости $~\Delta C_{{298}}^{0} = f\left( x \right)$ твердых растворов GaSxSe1–x рассчитали с учетом теплоемкостей $C_{{p,298}}^{0}$ бинарных соединений GaS и GaSe; при комнатной температуре $C_{{p,298}}^{0}\left( {{\text{GaS}}} \right)$ = 47 ± 1 Дж/(моль ⋅ К) и $C_{{p,298}}^{0}\left( {{\text{GaSe}}} \right)$ = 48 ± 1 Дж/(моль ⋅ К).
С учетом фазовой диаграммы системы GaS–GaSe для соединений GaS и GaSe рассчитывали парциальные избыточные молярные свободные энергии Гиббса ${{\Delta }_{f}}\bar {G}_{T}^{{0,{\text{ex}}}}$ [5]
(3)
${{\Delta }_{f}}\bar {G}_{T}^{{0,{\text{ex}}}} = {{T}^{s}}\left[ {{{\Delta }_{f}}S_{i}^{m} - R\ln \left( {\frac{{x_{i}^{l}}}{{~~~x_{i}^{s}}}} \right)} \right] - ~{{\Delta }_{f}}H_{i}^{m},$Используя парциальные значения ${{\Delta }_{f}}\bar {G}_{T}^{{0,{\text{ex}}}}$ соединений GaSe и GaS, рассчитали интегральную избыточную молярную свободную энергию ${{\Delta }_{f}}G_{T}^{{{\text{ex}}}}$ твердых растворов GaSxSe1 –x (0 ≤ $x$ ≤ 1). Затем, с учетом молярных энтальпии и энтропии образования соединений GaS и GaSe, ${{\Delta }_{f}}G_{T}^{{{\text{ex}}}}$ и $\Delta C_{{p,298}}^{0}$ с помощью уравнения (2), вычислили свободную энергию смешения ${{\Delta }_{f}}G_{T}^{{{\text{mix}}}}$ при образовании твердых растворов GaSxSe1 –x.
(4)
${{\Delta }_{f}}G_{T}^{{{\text{mix}}}}\left( {{\text{GaS}} - {\text{GaSe}}\,\,{\text{т}}{\text{.р}}{\text{.}}} \right) = \left( {4000 - 3000{{x}^{2}}} \right) \times \left( {1 - x} \right) + RT\left[ {x \cdot \,~{\text{ln}}\left( x \right) + \left( {1 - x} \right){\text{ln}}\left( {1 - x} \right)} \right].$С использованием уравнения (4) в диапазоне температур 300–1200 K провели аппроксимацию температурной зависимости свободной энергии смешения компонентов в системе GaS–GaSe. Вычисленные концентрационно-температурные зависимости, в частности, $~{{\Delta }_{f}}G_{T}^{{{\text{mix}}}} = f\left( {x,~T} \right)$ компонентов согласуются с $T{\kern 1pt} - {\kern 1pt} x~$ фазовой диаграммой GaS–GaSe (рис. 3). Как видно из рисунка при разных температурах чувствительная величина ${{\Delta }_{f}}G_{T}^{{{\text{mix}}}}$ коррелирует концентрационную зависимость свободной энергии “искажения” химических связей в твердых растворах${\text{\;Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}$.
Рис. 3.
Концентрационно-температурная зависимость свободной энергии Гиббса смешения, рассчитанная по формуле (4) для твердых растворов ${\text{Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}$ (x = 0–1 мол. доля) с использованием данных для GaS и GaSe. 1 – 300, 2 – 800, 3 – 1200 K.

Для реакции смешения зависимость энтальпии смешения от температуры вычислили по правилу Кирхгоффа
Образование твердого раствора (т.р.) из смеси соединений GaS и GaSe можем написать в виде реакции $x{\text{GaS}} + \left( {1 - x} \right){\text{GaSe}} \to {\text{Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}$. Для этой реакции смешения (mixing)
(5)
${{\Delta }_{f}}G_{T}^{{{\text{ex}}}} \equiv {{\Delta }_{r}}G_{T}^{{{\text{ex}}}} = {{\Delta }_{f}}G_{T}^{{{\text{mix}}}} = G\left( {{\text{Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}} \right) - \left[ {x\,~{\kern 1pt} \cdot G~\left( {{\text{GaS}}} \right) + \left( {1 - x} \right)~G\left( {{\text{GaSe}}} \right)} \right]~.$Для этой реакции ${{\Delta }_{r}}G_{T}^{{{\text{ex}}}} < ~\,\,0$ и она протекает в направлении от реагентов к продукту (т.р.): $G\left( {{\text{Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}} \right) < G~\left( {{\text{GaS}}} \right) + ~G\left( {{\text{GaSe}}} \right)$, то есть в термодинамическом смысле твердый раствор более устойчив. ${{\Delta }_{f}}G_{{298}}^{{{\text{ex}}}}$ составляла <5% от полной термодинамической величины ${{\Delta }_{f}}G_{{298}}^{0}\left( {{\text{GaS}} - {\text{GaSe}}\,{\text{т}}{\text{.р}}.} \right)$. Поэтому, при синтезе если влияние кинетических факторов незначительно, то будут образовываться твердые растворы, и они не будут распадаться при понижении температуры.
Таким образом, свободная энергия Гиббса смешения близких к идеальным систем, включающих непрерывный ряд твердых растворов, будет представлять собой разность между значениями этой функции для твердого раствора и смеси исходных компонентов. Если взять любую термодинамическую функцию, то для идеальных систем, например А–В, можно написать:
Однако, обнаруживается незначительное отклонение расчетного ликвидуса и солидуса от экспериментальных данных в GaSxSe1 – x(0 ≤ $x$ ≤ 1). Это можно связать с агрегатным состоянием фаз, которые отличаются межмолекулярным взаимодействием молекул. Т.е. это отклонение можно связать с нарушением соотношения между силами притяжения и силами отталкивания между молекулами исходных веществ в разных фазах.
Если учесть, что на расстоянии $r = r_{0}^{{..}}$ (где .. – равновесное расстояние между молекулами) результирующая сила $F = 0$, т.е. силы притяжения и отталкивания уравновешивают друг друга, то в присутствие теплового движения $F \ne ~\,0$. Другими словами при $r < {{r}_{0}}$ преобладают силы отталкивания ($F > \,~0$), a при $r > {{r}_{0}}$ – силы притяжения ($F < ~\,0$). На расстояниях $r$ > 10–9 м межмолекулярные силы взаимодействия отсутствуют ($F$ → 0).
При увеличении расстояния между молекулами на $dr$ элементарная работа $\delta A$ силы $F~$ совершается за счет уменьшения взаимной потенциальной энергии (${{E}_{{{\text{pot}}}}}$) молекул, т.е. $\delta A = Fdr = ~ - d{{E}_{{{\text{pot}}}}}$. Из характерной зависимости ${{E}_{{{\text{pot}}}}} = f~\left( r \right)$ для молекулярных систем следует, что если молекулы находятся друг от друга на расстоянии $r \to \infty $, то ${{E}_{{{\text{pot}}}}} = 0$.
При сближении молекул между ними появляются силы притяжения ($F$< 0) и $\delta A = Fdr > 0$. Тогда, ${{E}_{{{\text{pot}}}}}$ уменьшается, достигая минимума при $r = {{r}_{0}}$. При $r < {{r}_{0}}$ с уменьшением $r$, т.е. когда $r < {{r}_{0}}$ силы отталкивания ($F > ~\,0$) возрастают и совершаемая против них работа будет $\delta A = Fdr < 0$. Также возрастает ${{E}_{{{\text{pot}}}}}$ и становится ${{E}_{{{\text{pot}}}}} > 0$. Таким образом, система из двух взаимодействующих молекул в состоянии $r = {{r}_{0}}$ обладает минимальной потенциальной энергией $(E_{{{\text{pot}}}}^{{{\text{min}}}}$).
Кинетика нуклеации в многокомпонентных системах. В многокомпонентной системе определение условий процесса кластеризации в основном затрудняются с нижеуказанными кинетическими аспектами зарождения: определение вида распределения кластеров, вклада границы раздела и диффузионных потоков на процесс нуклеации и вычисление скорости нуклеации.
В отличие от однокомпонентного случая кинетическое развитие кластера с многокомпонентным составом отличается от состава в начальной фазе. Зарождение и рост этого кластера будет включать в себя как присоединение молекул на границе раздела, так и перенос кластера в исходной фазе в межфазную область. Связь между указанными процессами из-за стохастического характера эволюции кластеров вблизи критического размера услояняжется по сравнению с ростом. При этих условиях межфазные скорости, вероятно будут различными для разных видов атомов из-за дальней диффузии, скорость роста крупных кластеров будет связана с диффузионно-ограниченным ростом. Изменение состава в областях исходной фазы и вблизи каждого кластера будут зависеть от времени. Состав самих кластеров может зависеть от времени и размера. Технологическая история, как известно, также может оказать заметное влияние на последующее поведение нуклеации.
Состав, соответствующий точке минимума на Т–х диаграмме 2D системы GaS–GaSe, можно рассмотреть как квазиоднокомпонентный. Такой термодинамически устойчивый состав GaS0.3Se0.7 проанализируем в рамках теории гомогенной нуклеации с учетом энергетического барьера нуклеации. Примем, что фазовые превращения в области метастабильности инициируются в исходной фазе (GaS0.3Se0.7) зарождением небольших областей новой фазы, которые затем вырастают до макроскопических размеров. Нуклеация характеризуется колебаниями так называемого параметра порядка плотностей, атомных структур или химического состава исходной и новой фазы. Эти флуктуации кластеров из нескольких частиц (или молекул) в конфигурации новой фазы будут локализованы и стохастически распределены как в пространстве, так и во времени. Вероятность возникновения колебаний определяется термодинамическими условиями, в частности, минимальной работой, необходимой для создания колебаний.
Энергетический барьер зародышеобразования возникает из-за потери энергии на создание границы раздела между кластером и исходной фазой. Если фазовый переход является термодинамически выгодным, большие кластеры новой фазы должны иметь меньшую свободную энергию, чем те же частицы, сохраняющие конфигурацию исходной фазы. Однако частицы в области границы раздела между исходной и новой фазами находятся в более высоком энергетическом состоянии, чем в двух макроскопических фазах. Для небольших кластеров новой фазы большинство частиц будут находиться в межфазной области, и поэтому создание таких кластеров потребует работы.
Для критического кластера, имеющего радиус $r{\text{*}}$ и содержащего $n{\text{*}}$ частиц, работа формирования кластера будет максимум $W{\text{*}}$, который составляет барьер для зарождения. По мере того как кластеры выходят за пределы критического размера доля частиц в межфазной области уменьшается, как и работа по формированию кластеров. Поэтому кластеры, большие чем $r{\text{*}}$ (или $n{\text{*}}$), будут расти для завершения фазового превращения. Число кластеров, проходящих этот критический размер за единицу времени, будет скоростью нуклеации.
Рассмотрим термодинамику образования кластера. Из статистических позиций энтропия $S$ определяется числом доступных состояний ($w$) в термодинамической системе. Колебания в небольшом объеме будут сопровождаться изменением энтропии ($\Delta S$) от объемного (или среднего) значения ($\bar {S}$), которое отражает изменение числа доступных состояний от $\bar {S}$, то есть от ${{w}_{0}}$ до $w$: $\Delta S = S - \bar {S} = \,\,~{{{{k}_{{\text{B}}}}\ln w} \mathord{\left/ {\vphantom {{{{k}_{{\text{B}}}}\ln w} {{{w}_{0}}}}} \right. \kern-0em} {{{w}_{0}}}}$, где ${{k}_{{\text{B}}}}$ – постоянная Больцмана. Когда гомогенная среда уравновешивается за время, меньшее, чем требуется для формирования флуктуации, вероятность возникновения флуктуации $P$ равна $P\,\,~ \propto {\text{exp}}\left( {{{\Delta S} \mathord{\left/ {\vphantom {{\Delta S} {{{k}_{{\text{B}}}}}}} \right. \kern-0em} {{{k}_{{\text{B}}}}}}} \right)$, которая выражается через минимальную работу ${{W}^{{{\text{min}}}}}$, требуемую для формирования флуктуации: $~P~ \propto {\text{exp}}\left( {{{ - {{W}^{{{\text{min}}}}}} \mathord{\left/ {\vphantom {{ - {{W}^{{{\text{min}}}}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}} \right)$.
Кинетика кристаллизации. Рассмотрим характерные для получения полупроводниковых материалов твердо-жидкостные реакции. Изучая системы из малых молекул, Оствальд (Ostwald, 1897), по-видимому, впервые в литературе описал правило стадий и условия “созревания” кристаллов. Эта так называемый процесс “созревания по Оствальду”. Согласно правилу стадий кристаллизации процесс превращения нестабильных (или метастабильных) состояний в стабильные состояния происходит поэтапно. При этом первые образовавшиеся продукты обычно не являются термодинамически стабильными продуктами. Они представляют собой промежуточные продукты, имеющие свободные энергии, близкие к начальным состояниям.
Вторым вопросом, связанным с метастабильным состоянием, является вклад межфазной свободной энергии в общую свободную энергию системы. Этот вклад энергии незначителен для кристаллов размером более 1 мкм. В процессе, происходящем в равновесных условиях, т.е. в процессе “созревания по Оствальду”, крупные кластеры растут за счет растворения мелких кластеров. Вначале процесса в системе может образоваться много мелких кристаллов, затем большинство из них постепенно исчезают, за исключением нескольких, которые увеличиваются в результате присоединения других.
Рассмотрим физическую кинетику роста нанокристаллитов в жидкой дисперсионной среде. Для такого случая диффузия является основным кинетическим фактором, влияющим как на зарождение, так и на рост кристаллов. Диффузия описывается коэффициентом диффузии:
Согласно классическим представлениям рост кристаллитов из раствора, представляющего собой гетерогенную дисперсную систему, связан с диффузионными эффектами. Эти эффекты могут вызвать осаждение зерен второй фазы в пересыщенном растворе, содержащем одно или несколько твердых веществ, распределенных в жидкой дисперсионной среде. Такой диффузионный процесс осаждения соответствует “Оствальдовскому созреванию”. Кинетическую модель, отвечающую такому диффузионному механизму, можно представить, как процесс роста некоторых зерен только для того, чтобы потом раствориться. Другие зерна при этом будут увеличиваться в размерах и включат в себя дополнительные зерна, с которыми они будут сталкиваться.
Для указанных процессов кристаллизации в кинетической теории осаждения наночастиц принимается, что радиус частицы меняется в виде степенного закона: ${{\bar {r}}^{n}} - \bar {r}_{0}^{n} \propto t$, где $n$ – коэффициент степенного закона. Его значение зависит от ограничения стадии роста фазы и характеризует зависимость растворимости частицы как функцию от размера частицы.
В условиях разбавленных растворов, где диффузия ионов в растворе является ограничивающим этапом, для степенного закона $n$ = 3. В условиях концентрированных растворов, например, в случае образования зародышей в расплавах, в зависимости от стадии ограничения, участвующей в межфазных реакциях (растворении или переосаждении) коэффициент $n$ = 2–4.
Рост наночастицы из жидкой дисперсионной среды (из раствора суспензии) может протекать не по диффузионному механизму “Оствальдовского созревания”. Если процесс роста представить подобно химической реакции, то частицы, распределенные в жидкой дисперсионной среде, должны будут состоять в контакте друг с другом и тогда появляется возможность их сращивания, т.е. при таком взаимодействии двух частиц с учетом их ориентации должна образоваться третья и осаждаться новая фаза. При такой аналогичной химической реакции и дальнейшем процессе осаждения размер частицы можно определить выражением [31]
(6)
$r_{{{\text{equi}}}}^{3} - \,\,~r_{i}^{3} = ~\frac{{a \cdot t}}{{1 + a \cdot t}} \cdot r_{i}^{3},$Для различных скоростей нуклеации первого зародыша известные [32] выражения приведены в табл. 1.
Таблица 1.
Законы скорости нуклеации и их выражения
| Закон скорости нуклеации | Дифференциальная форма $\left( {J = \frac{{\partial N}}{{\partial T}}} \right)~~$ | Интегральная форма $\left( N \right)$ |
|---|---|---|
| Экспоненциальный | ${{K}_{{{\text{nuc}}}}}{{N}_{0}}{{e}^{{ - {{K}_{{{\text{nuc}}}}} \cdot t}}}$ | ${{N}_{0}}\left( {1 - {{e}^{{ - {{K}_{{{\text{nuc}}}}} \cdot t}}}} \right)$ |
| Постоянная скорость | ${{K}_{{{\text{nuc}}}}}{{N}_{0}}{{K}_{{{\text{nuc}}}}}$ | ${{N}_{0}}t$ |
| Мгновенный | $~\infty $ | $~{{N}_{0}}$ |
В табл. 1 число ядер роста $N$, присутствующих в момент времени $t$; ${{K}_{{{\text{nuc}}}}}$ – константа скорости нуклеации. При силовом влиянии на процесс кристаллизации происходит многоступенчатая кристаллизация и скорость нуклеации зародыша, в частности, при нагреве раствора выражается в виде $J = Z \cdot w \cdot {{t}^{{\left( {w - 1} \right)}}}$ (дифференциальная форма) или $J = Z \cdot {{t}^{w}}$ (интегральная форма), где $w$ – скорость нагрева; $Z$ – сложный параметр, состоящий из ${{{{N}_{0}}{{{({{K}_{{{\text{nuc}}}}} \cdot t)}}^{{~w}}}} \mathord{\left/ {\vphantom {{{{N}_{0}}{{{({{K}_{{{\text{nuc}}}}} \cdot t)}}^{{~w}}}} w}} \right. \kern-0em} w}$. Изотермическую скорость кристаллизации металлов и сплавов оценивают с использованием соотношения Джонсона–Меля–Аврами–Ерофеева–Колмогорова. При этом допускается, что кристаллизация контролируется на границе раздела, а уравнение скорости такой диффузионно-контролируемой кристаллизации выражается уравнением типа Аврами
Предположим, что формированная фаза конечного продукта в объемной форме, например, кристаллы являются внутренне однородными и имеют ту же структуру, состав и свойства. Тогда, свободная энергия Гиббса образования кристаллического зародыша радиуса $r$ равна
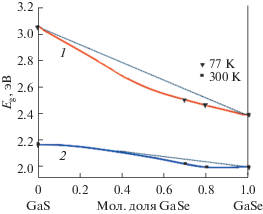
Корреляции свойств с составом, структурой фаз и размером частиц. Экспериментальные результаты зависимости ширины запрещенной зоны${{E}_{g}}$ от состава твердых растворов ${\text{Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}$(x = 0–1) подтверждают изоморфную смесимость между изоструктурными соединениями GaS и GaSe одинакового политипа (ε- и $\beta {\text{ - GaX\;политипы}})$ (рис. 4). С изменением состава непрерывного ряда твердых растворов изовалентные замещения серы (условно ${{{\text{S}}}^{{ - 2}}}$; ионный радиус в кристалле, ${{r}_{{\text{c}}}} = 0.186~\,\,{\text{нм}}$) на селен (условно ${\text{S}}{{{\text{e}}}^{{ - 2}}};\,\,~{{r}_{{\text{c}}}} = 0.198~\,\,{\text{нм}}$) приводит к монотонному изменению ${{E}_{g}}$ для стабильных $\varepsilon $- и $\beta $-политипов GaS и GaSe при разных температурах. При 298 К для GaS и GaSe значение ${{E}_{g}}$ составляет 2.53 и 1.98 эВ соответственно. Концентрация зависимости ширины запрещенной зоны ${{E}_{g}} = f\left( x \right)\,\,{\text{Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}$ (x = 0–1) аппроксимируется уравнением [5]
(7)
$\begin{gathered} {{E}_{g}}\left( x \right) = \left( {1 - x} \right)~\,\, \cdot {{E}_{g}}\left( {{\text{GaSe}}} \right) + \\ + \,\,x \cdot ~{{E}_{g}}\left( {{\text{GaS}}} \right) - bx\left( {1 - x} \right), \\ \end{gathered} $Рис. 4.
Зависимость ширины запрещенной зоны твердых растворов системы GaS–GaSe от состава и температуры. 1 – 77, 2 – 300 K.
Эффективные заряды атомов, оцененные по схеме Бaдера [33], указывают на “перетекание” небольшого (~0.17 e) электронного заряда из [Ga-X] в другой [Ga-X] слой. Такие зарядовые состояния атомов отличаются от состояний, полученных по ионной модели (Ga3+, Х2–). Химические связи в слоях GaX, по-видимому, можно отнести к смешанному ковалентно-ионному типу, в то время как “межслоевые” взаимодействия носят Ван-дер-Ваальсовый характер.
В рамках DFT проведены ab initio расчеты электронно-зонных структур монослоев GaS и GaSe. Для отдельно стоящих слоев GaS и GaSe использовали примитивные супер ячейки. Отбор проб в точке k выполнен с использованием метода DFT [10]. Примитивная ячейка состоит из двух атомов Ga и двух атомов S (или Se) в двух слоях. Чтобы исключить взаимодействие между соседними ячейками вдоль оси c, поддерживали вакуум в 18 Å вдоль направления, нормального к плоскости слоя.
Значения ширины запрещенной зоны для оптимизированных монослойных конфигураций GaS и GaSe, соответственно составляют: 3.8277 и 3.072 эВ для прямого (Γ → Γ) электронного перехода и 0.571 и 0.087 эВ для ${{E}_{g}}$ = ${{E}_{g}}$ (прямой) – ${{E}_{g}}$ (непрямой) с учетом энергии непрямого перехода (рис. 5). Эти значения соответствуют известным данным для GaS и GaSe. Электронные зонные структуры 2D GaS и GaSe и графики энергетических зон монослоов и объемных GaS и GaSe вдоль $k$-точек высокой симметрии аналогичны и отличаются максимумами валентной зоны. Энергия запрещенной зоны – функция числа слоев.
Рис. 5.
Электронные зонные структуры 2D GaS и GaSe из теоретических расчетов. (а) – схема первой зоны Бриллюэна для гексагональной монослойной структуры, где ${{\vec {a}}_{1}}$ и ${{\vec {a}}_{2}}$ векторы. Графики энергетических зон монослоя GaS (б) и GaSe (в) вдоль k-точек высокой симметрии. В рассчитанных зонных структурах уровнь Ферми установлен на ноль энергии.

Полупроводниковые структуры на основе тонкопленочных GaSe, проявляют высокую фоточувствительность [12]. Образец пленки GaSe был получен в ваккуме методом взрывного испарения. Толщина напыленных пленок GaSe составляла порядка одного микрона. В частности, сняты спектры электропоглощения в изготовленном тонком пленочном образце (кривая 1) при приложении к нему переменного (${{F}_{{{\text{ac}}}}}$ = 6 × 104 В/см) и постоянного электрического поля (${{F}_{{{\text{dc}}}}}$ = 106 В/см) (рис. 6). A электропоглощение в монокристаллическом образце GaSe измеряли в электрическом поле ${{F}_{{{\text{dc}}}}}$ = 5 × 104 В см–1, приложенном параллельно оси c кристалла. В обоих образцах в качестве контакта использовали алюминий.
Рис. 6.
Спектры электропоглощения полупроводниковой структуры Al/GaSe/SnO2 с толщиной аморфной пленки GaSe d ≈ 1000 нм (кривая 1) и для монокристалла GaSe (кривая 2), выращенного методом Бриджмена.

Сравнение результатов измерения полупроводниковых образцов моноселенида галлия показывает, что спектры электропоглощения аморфной пленки GaSe (кривая 1) и монокристалла GaSe (кривая 2) сильно различаются. Для экспериментально полученного оптического спектра аморфной пленки GaSe не характерено экситонное электропоглощение (кривая 1), которое наблюдалось в монокристаллическом образце GaSe (кривая 2). Сигнал электропоглощения в пленке GaSe наблюдался в переменном электрическом поле в течение длительного времени после того, как постоянное поле было отключено. Присутствие сигнала электропоглощения, когда к пленочному образцу GaSe приложено только лишь переменное электрическое поле, указывает на существование хаотического внутреннего поля в пленке GaSe.
Существование такого внутреннего поля в пленке GaSe, по-видимому, обусловлено наличием в аморфной структуре таких микроскопических неоднородностей, как границы кристаллитов, создающих потенциальные барьеры для дрейфа и рекомбинации носителей заряда. Кроме того, в приконтактной области пленки GaSe скапливается инжектированный из контакта заряд, захваченный глубокими ловушками в запрещенной зоне. Появление дополнительного отрицательного пика при увеличении электрического поля, вероятно, связано с изменением поглощения в этой области.
В макроскопическом полупроводниковом кристалле в частнице GaSe энергетические уровни образуют полосы. Валентная зона заполнена, а зона проводимости при 0 К пустая. Полосы разделены энергетической щелью. Когда электрон получает воздействие, например, за счет тепловых возбуждений, создается электронно-дырочная пара. Электрон в зоне проводимости и дырка в валентной зоне связываются, когда они приближаются друг к другу на конечном расстоянии.
Эта связанная пара, т.е. экситон, делокализована по всему кристаллу. Радиус Бора экситона задается как: ${{a}_{{\text{B}}}} = {{{{h}^{2}}\varepsilon } \mathord{\left/ {\vphantom {{{{h}^{2}}\varepsilon } {4{{\pi }^{2}}{{e}^{2}}}}} \right. \kern-0em} {4{{\pi }^{2}}{{e}^{2}}}}\left[ {{1 \mathord{\left/ {\vphantom {1 {{{m}_{e}}}}} \right. \kern-0em} {{{m}_{e}}}} + {1 \mathord{\left/ {\vphantom {1 {{{m}_{h}}}}} \right. \kern-0em} {{{m}_{h}}}}} \right]$, где ε – диэлектрическая проницаемость материала, ${{m}_{e}}$ и ${{m}_{h}}$ – эффективные массы электрона и дырки соответственно и $e$ – элементарный заряд.
Квантовые размерные эффекты проявляются при длине нанокристалла радиусом экситона ${{a}_{{\text{B}}}}$. Это ~25 Å для тонкой пленки GaSe. Размеры элементарной ячейки полупроводниковых материалов меньше, чем характерная длина волны де Бройля ($\lambda $). В таких материалах $\lambda ~ = {{~h} \mathord{\left/ {\vphantom {{~h} {mv}}} \right. \kern-0em} {mv}}$ находится в диапазоне нанометров, и сильные эффекты ограничения проявляются только тогда, когда размер частицы приближается к этому значению. В этом измерении большинство материалов имеют структуры, подобные структурам их объемных аналогов.
Электронная структура материалов тесно связана с природой материала. В трехмерном объекте большого размера электронная структура не ограничена размером материала. Длина волны электронов намного меньше, чем длина материала. Когда электронное движение ограничено в одном измерении и оно свободно в двух других измерениях, это приводит к созданию “квантовых ям” или “квантовых пленок”. При этом электроны чувствуют потенциальную яму, поскольку они будут захвачены “квантовой пленкой”. В таких пленках плотность состояний показывает ступенчатое поведение. В случае одномерной системы, то есть когда электроны могут свободно перемещаться только в одном направлении, получается ситуация, когда плотность состояний показывает форму линии Лоренца. Такая ситуация наблюдается в углеродных нанотрубках. Если электроны ограничены какой-либо точкой, получается система нулевой размерности, в которой электроны не могут свободно двигаться. Здесь получаются состояния, которые имеют молекулярный характер.
В объемных материалах плотность состояний изменяется плавно, тогда как в замкнутых квантовых системах наблюдаются разрывы. Это приведет к шагам в двумерном ограничении, особенностям в одномерном, удержанию и дискретным линиям в ограничении нулевого измерения.
Одними из свойств, которые резко изменяются в зависимости от размера, являются оптические свойства, включая как поглощение, так и излучение света, что видно из рис. 6. Пленки GaSe аналогично нанокристаллам имеют дискретные орбитали. Как известно блочная модель предсказывает для частиц энергии с уровнями ${{{{n}^{2}}{{h}^{2}}} \mathord{\left/ {\vphantom {{{{n}^{2}}{{h}^{2}}} {8{{m}_{e}}{{a}^{2}}}}} \right. \kern-0em} {8{{m}_{e}}{{a}^{2}}}}\left( {n = 1,2, \ldots } \right)$. Тогда энергия первого уровня частицы будет смещена от положения объемного значения для этих частиц на ${{{{h}^{2}}} \mathord{\left/ {\vphantom {{{{h}^{2}}} {8{{m}_{e}}{{a}^{2}}}}} \right. \kern-0em} {8{{m}_{e}}{{a}^{2}}}}$, где $a$ – диаметр частицы. Энергетическая щель увеличивается с уменьшением размера ($a$) частицы. Вследствие этого тонкие пленки GaSe могут чувствовать свет в широком диапазоне, и поэтому цветовой диапазон можно менять в зависимости от размера частицы.
Ширина запрещенной зоны увеличивается с уменьшением размера частицы, что объясняется указанным выше размерным эффектом. Ширина запрещенной зоны для пленки GaSe при комнатной температуре составляет 2.2 эВ, в то время как для монокристалла GaSe эта величина составляла 1.98 эВ, а для всех составов твердых растворов GaSxSe1 –x измеренная ширина запрещенной зоны по мере возрастания х увеличивалась до 2.5 эВ.
ЗАКЛЮЧЕНИЕ
В системе GaS–GaSe в условиях проведенного эксперимента, с учетом литературных данных, обнаружено образование непрерывных фазовых областей, в которых в равновесии с жидкой фазой сосуществует конденсированная фаза. Результаты расчетов показывают, что кривые равновесия $T{\kern 1pt} - {\kern 1pt} x$ фазовой диаграммы квазибинарной системы GaS–GaSe в жидкой и твердой фазах достаточно хорошо аппроксимируются термодинамическими параметрами плавления (${\Delta }{{H}^{{\text{m}}}}$(GaS) = = 34 800 ± 200 Дж/моль, ${{T}^{{\text{m}}}}$(GaS) = 1288 ± 1 К; ${\Delta }{{H}^{{\text{m}}}}$ (GaSe) = 30300 ± 200 Дж/моль). Вычисленные температуры солидуса и ликвидуса в пределах погрешности отличаются на ≤3–5% от экспериментальных. Такой результат объясняется плавным усилением межмолекулярного взаимодействия между бинарными соединениями GaS и GaSe, а также ионами халькогенов, возникающего благодаря аналогичной электронной структуре вблизи фазовых переходов (жидкость ↔ твердый) в непрерывных твердых растворах GaSxSe1 –x(0 ≤ $x$ ≤ 1). Это значит, что энергия межмолекулярного взаимодействия между исходными компонентами в жидкой фазе по сравнению с ковалентными силами больше, чем в твердой фазе, где действует также дальний порядок между частицами, т.е. в жидко-кристаллических фазах уже в неупорядоченной фазе присутствует относительно сильный ангармонизм колебаний решетки, который значительно усиливается при переходе в жидкое состояние. Так как межмолекулярные взаимодействия имеют электрическую природу и складываются из сил притяжения (ориентационных, индукционных и дисперсионных) и сил отталкивания, то в жидком состоянии последние усиливаются и ликвидус относительно солидусу больше отличается от экспериментально найденных значений. Это приводит к появлению дополнительных минимумов потенциальной энергии, что облегчает ковалентно-ионный транспорт частиц в жидкой фазе. Таким образом, при наличии экспериментальных кривых ДТА представленная нами модель позволяет теоретически предсказать $T{\kern 1pt} - {\kern 1pt} x$ фазовую диаграмму системы, включающей исходные соединения с аналогичной структурой, находящейся в неупорядоченной фазе, возможность непрерывного перехода в упорядоченное состояние. При моделировании $T{\kern 1pt} - {\kern 1pt} x$ фазовой диаграммы GaS–GaSe учитывали наши экспериментальные и термодинамические данные исходных компонентов GaS и GaSe. Аппроксимированная концентрационно-температурная зависимость свободной энергии смешивания Гиббса твердых растворов GaSxSe1 –x (0 ≤ $x$ ≤ 1) согласуется с $T - x$ фазовой диаграммой системы GaS–GaSe.
Из анализа термодинамических и кинетических условий роста и кристаллизации фаз 2D системы GaS–GaSe следует, что структурнохимический механизм образования твердых растворов GaSxSe1 –x(0 ≤ $x$ ≤ 1) не меняется в зависимости от состава при температурах синтеза. Это объясняется устойчивостью фаз при изовалентном замещении, которое не меняет механизм формирования фаз в системе.
Увеличение концентрации одного из компонентов приводит к образованию фаз со слоистой структурой типа GaS, в которых часть ионов халькогена замещена на ионы другого халькогена, ширина запрещенной зоны (${{E}_{g}}$) которых меняются монотонно в области гомогенности.
В рамках теории функционала плотности рассчитанные (GGA приближение) значения ширины запрещенных зон ${{E}_{g}}$ образцов составили, в частности, для монослоев GaS и GaSe 3.8277 и 3.072 эВ соответственно. Эти значения ${{E}_{g}}$ сильно отличаются от экспериментальных данных для объемных образцов GaS (2.53 эВ) и GaSe (1.98 эВ) при 298 К. Несоответствие между значениями $E_{g}^{{{\text{calc}}}}$ и $E_{g}^{{{\text{exp}}}}$, кроме размера частиц можно связать также с проблемой адекватности использованного в расчете локального обменно-корреляционного потенциала.
Сравнение спектров электропоглощения аморфно-пленочных (АП) и объемных образцов GaSe показывает, что исследованный АП образец близок по своим свойствам к нанокристаллическим. Экситонное электропоглощение, которое наблюдалось в монокристаллическом образце GaSe, не характерено для тонкой пленки GaSe. Сигнал электропоглощения в пленке GaSe наблюдался в переменном электрическом поле в течение длительного времени после того, как постоянное поле было отключено.
Список литературы
Лукичев В.Ф., Амиров И.И. Исследования и разработки в области микро- и наносистемной техники // История науки и техники. 2018. № 8. С. 92–99.
Asadov S.M., Mustafaeva S.N., Lukichev V.F. Charge transport in layer gallium monosulfide in direct and alternate electric fields // Russian Microelectronics. 2019. V. 48. № 6. P. 422–427. https://doi.org/10.1134/S1063739719
Mustafaeva S.N., Asadov M.M. Currents of isothermal relaxation in GaS〈Yb〉 single crystals // Solid State Communications. 1983. V. 45. № 6. P. 491–494. https://doi.org/10.1016/0038-1098(83)90159-X
Asadov S.M., Mustafaeva S.N., Lukichev V.F., Guseinov D.T. Effect of the Composition on the Dielectric Properties and Charge Transfer in 2D GaS1 –xSex Materials // Russian Microelectronics. 2019. V. 48. № 4. P. 203–207.https://doi.org/10.1134/S1063739719040024
Asadov S.M., Mustafaeva S.N., Mammadov A.N. Thermodynamic assessment of phase diagram and concentration–temperature dependences of properties of solid solutions of the GaS-GaSe system // J. Thermal Analysis and Calorimetry. 2018. V. 133. №2. P. 1135–1141.
Mustafaeva S.N., Asadov M.M., Ismailov A.A. Charge transfer along localized states in InSe and InSe〈Sn〉 single crystals // Low Temp. Phys. 2010. V. 36. № 4. P. 310–312. https://doi.org/10.1063/1.3388822
Mustafaeva S.N., Asadov M.M., Ismailov A.A. Effect of γ irradiation on the parameters of localized states in p-InSe and n-InSe⟨Sn⟩ single crystals // Low Temp. Phys. 2010. V. 36. № 7. P. 642–644. https://doi.org/10.1063/1.3479690
Mustafaeva S.N., Asadov M.M., Ismailov A.A. Charge transfer over localized states in a TlS single crystal // Phys. Solid State. 2008. V. 50. № 11. P. 2040–2043. https://doi.org/10.1134/S1063783408110073
Mustafaeva S.N., Asadov M.M., Ismailov A.A. Dielectric and baric characteristics of TlS single crystal // Physica B: Physics of Condensed Matter. 2014. V. 453. P. 158–160.https://doi.org/10.1016/j.physb.2014.03.095
Асадов М.М., Гусейнова С.С., Лукичев В.Ф. Ab initio-моделирование электронной и энергетической структуры, а также открытие ширины запрещенной зоны легированного 4p-элементами монослоя графена // Микроэлектроника. 2020. Т. 49. № 5. С. 334–343. https://doi.org/10.31857/S0544126920050038 [Asadov M.M., Guseinova S.S., Lukichev V.F. Ab Initio Modeling of the Electronic and Energy Structure and Opening the Band Gap of a 4p-Element-Doped Graphene Monolayer // Russian Microelectronics. 2020. Vol. 49. No. 5. P. 314–323.https://doi.org/10.1134/S1063739720050030]
Медведева З.С. Халькогениды элементов IIIб подгруппы периодической системы. М.: Наука. 1968. 216 с.
Mustafaeva S.N., Asadov M.M. High field kinetics of photocurrent in GaSe amorphous films // Materials Chemistry and Physics. 1986. V. 15. P. 185–189.https://doi.org/10.1016/0254-0584(86)90123-9
Jung C.S., Shojaei F., Park K., Oh J.Y., Im H.S., Jang D.M., Park J., Kang H.S. Red-to-ultraviolet emission tuning of two-dimensional gallium sulfide/selenide // ACS Nano. 2015. V. 9. No 10. P. 9585–9593.https://doi.org/10.1021/acsnano.5b04876
Hu P.A., Wen Z.Z., Wang L.F., Tan P.H., Xiao K. Synthesis of few-layer GaSe nanosheets for high performance photodetectors // ACS Nano. 2012. V. 6. № 7. P. 5988–5994. https://doi.org/10.1021/nn300889c
Li X.F., Lin M.W., Puretzky A.A., Idrobo J.C., Ma C., Chi M.F., Yoon M., Rouleau C.M., Kravchenko I.I., Geohegan D.B., Xiao K. Controlled vapor phase growth of single crystalline, two-dimensional GaSe crystals with high photoresponse // Sci. Rep. 2015. V. 4. 5497. P. 1–9. https://doi.org/10.1038/srep05497
Tan L., Liu Q., Ding Y., Lin X., Hu W., Cai M.-Q., Zhou H. Effective shape-controlled synthesis of gallium selenide nanosheets by vapor phase deposition // Nano Research. 2020. CN 11-5974/O4. https://doi.org/10.1007/s12274-020-2653-8
Ho C.H., Wang S.T., Huang Y.S., Tiong K.K. Structural and luminescent property of gallium chalcogenides GaSe1−xSx layer compounds // J. Mater. Sci.: Mater. Electron. 2009. V. 20. P. S207–S210. https://doi.org/10.1007/s10854-007-9539-3
Bereznaya S., Korotchenko Z., Redkin R., Sarkisov S., Tolbanov O., Trukhin V., Gorlenko N., Sarkisov Y., Atuchin V. Broadband and narrowband terahertz generation and detection in GaSe1 −x Sx crystals // J. Opt. 2017. V. 19. № 11. P. 115503. https://doi.org/10.1088/2040-8986/aa8e5a
Kolesnikov N.N., Borisenko E.B., Borisenko D.N., Tereshchenko A.N., Timonina A.V. Synthesis and growth of GaSe1 –xSx (x = 0–1) crystals from melt. Phase Composition and properties // Inorganic Materials: Applied Research. 2018. V. 9. № 1. P. 66–69. https://doi.org/10.1134/S2075113318010173
Mullin J.W. Crystallization. 4th ed. Oxford; Boston. Butterworth-Heinemann. 2001. 612 p. ISBN: 9780750648332
Herlach D.M. (Ed.). Phase Transformations in Multicomponent Melts. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. 2008. 450 p. ISBN: 9783527319947
Aaronson H.I., Enomoto M., Lee J.K. Mechanisms of Diffusional Phase Transformations in Metals and Alloys. BocaRaton. 1st Ed. CRC Press. Taylor & Francis Group. 2010. 685 p. ISBN-23: 978-1420062991
Кукушкин С.А., Слезов В.В. Дисперсные системы на поверхности твердых тел (эволюционный подход): механизмы образования тонких пленок. СПб.: Наука. 1996. 304 с. ISBN: 5-02.-024856-8
Chernov A.A. Modern crystallography III. Springer-verlag Berlin, Heidelberg, New York, Tokyo. 1984. 517 p. ISBN 3-540-11516-1. https://doi.org/10.1007/978-3-642-81835-6
Nucleation in Condensed Matter: Applications in Materials and Biology. Kelton K.F. and Greer A.L. (Eds.). Pergamon Materials Series 15. Amsterdam. Elsevier. Pergamon. 2010. 726 p. ISBN: 9780080421476
Hu P.A., Wen Z.Z., Wang L.F., Tan P.H., Xiao K. Synthesis of few-layer GaSe nanosheets for high performance photodetectors // ACS Nano. 2012. V. 6. № 7. P. 5988–5994. https://doi.org/10.1021/nn300889c
Li X.F., Lin M.W., Puretzky A.A., Idrobo J.C., Ma C., Chi M.F., Yoon M., Rouleau C.M., Kravchenko I.I., Geohegan D.B., Xiao K. Controlled Vapor Phase Growth of Single Crystalline, Two-Dimensional GaSe Crystals with High Photoresponse // Sci. Rep. 2015. V. 4. 5497. P. 1–9. https://doi.org/10.1038/srep05497
Jung C.S., Shojaei F., Park K., Oh J.Y., Im H.S., Jang D.M., Kang H.S. Red-to-Ultraviolet Emission Tuning of Two-Dimensional Gallium Sulfide/Selenide // ACS Nano. 2015. V. 9. № 10. P. 9585–9593. https://doi.org/10.1021/acsnano.5b04876
Ахмедли К.М., Асадов М.М. Простой способ расчета диаграмм фазовых равновесий в двухкомпонентных системах с непрерывными рядами жидких и твердых идеальных растворов. // РАН. Неорганические материалы. 1996. Т. 32. № 2. С. 152–153. [Ahmedly K.M., Asadov M.M. Procedure for Calculating Phase Equilibrium in Simple Binary Systems of Ideal Liquid and Solid Solution // Inorganic Materials. 1996. V. 32. № 2. P. 133–134].
Asadov M.M., Mustafaeva S.N., Mamedov A.N., Tagiyev D.B. Effect of Composition on the Properties of (TlInSe2)1 –x(TlGaTe2)x Solid Solutions // Inorganic Materials. 2015. V. 51. № 12. P. 1232–1236.https://doi.org/10.1134/S0020168515110011
Ribeiro C., Lee E.J.H., Longo E., Leite E.R. A kinetic model to describe nanocrystal growth by the oriented attachment mechanism // Chem. Phys. Chem. 2005. V. 6. P. 690–696. https://doi.org/10.1002/cphc.200400505
Illeková E., Šesták J. Crystallization of Metallic Micro, Nano-, and Non-Crystalline Alloys. Thermal Analysis of Micro, Nano- and Non-Crystalline Materials. Springer ScienceCBusiness Media Dordrecht. 2013. P. 257–289. https://doi.org/10.1007/978-90-481-3150-1_13
Bader R.F.W. Atoms in Molecules. A Quantum Theory. A Clarendon Press Publication. UK. 1994. 458 p. ISBN 13: 9780198551683
Дополнительные материалы отсутствуют.
Инструменты
Микроэлектроника