

Молекулярная биология, 2019, T. 53, № 1, стр. 166-176
Сравнительный анализ профиля белков хромосомы 18 в биоптатах тестикулярной ткани человека с помощью алгоритмов Mascot и IdentiPy
А. В. Лисица a, b, Н. А. Петушкова a, *, Л. И. Левицкий c, d, В. Г. Згода a, О. В. Ларина a, Ю. С. Кисриева a, В. Е. Франкевич e, С. И. Гамидов e
a Научно-исследовательский институт биомедицинской химии им. В.Н. Ореховича
119121 Москва, Россия
b East China University of Technology
330013 Nanchang, China
c Институт энергетических проблем химической физики им. В.Л. Тальрозе Российской академии наук
119334 Москва, Россия
d Московский физико-технический институт (государственный университет)
141700 Московская обл, Долгопрудный, Россия
e Национальный медицинский исследовательский центр акушерства, гинекологии и перинатологии
им. академика В.И. Кулакова, Министерства здравоохранения Российской Федерации
117997 Москва, Россия
* E-mail: cyp450@mail.ru
Поступила в редакцию 29.05.2018
После доработки 03.07.2018
Принята к публикации 13.08.2018
Аннотация
Методом тандемной масс-спектрометрии с электроспрейной ионизацией исследовали протеомный профиль биоптатов ткани яичек человека. Проведено сравнение результатов идентификации белков с помощью коммерческой поисковой машины Mascot, свободно-распространяемого пакета SearchGUI и их аналога IdentiProt, основанного на алгоритме IdentiPy. Алгоритм и серверная оболочка доступны в виде открытого исходного кода по ссылке http://hg.theorchromo.ru/identipy. Уникальным преимуществом IdentiPy является автоматическая оптимизация параметров при поиске белков по масс-спектрометрическим данным. Эта особенность алгоритма позволила идентифицировать на треть большее количество белков, чем достигали другие поисковые протеомные машины. Впервые работа IdentiPy интегрирована с программой выравнивания спектров (Progenesis LC-MS) и оценены изменения протеомного профиля в результате выравнивания по сравнению со стандартным конвертером масс-спектров ProteoWizard. Для хромосомы 18 человека найдено в общей сложности 16 белков, в том числе, был выявлен основной белок миелина, не характерный для текстикулярной ткани.
ВВЕДЕНИЕ
Типичный протеомный анализ включает в себя десятки образцов и тысячи пептидов. Результаты жидкостной хроматографии, сопряженной с электроспрейной масс-спектрометрией (LC-ESI-MS/MS), представляют собой пакет исходных данных (raw-файлы), которые содержат информацию о массе пептидов и их фрагментов, а также время удержания пептида на хроматографической колонке [1]. Схема анализа и оценка наборов хромато-масс-спектров должна быть автоматизирована и объективна, так как включает в себя несколько этапов, в том числе поиск ”пептидного” сигнала (“признак” или “feature”), которому присуще характерное изотопное распределение, значение m/z моноизотопного пика, времени его удержания на колонке и интенсивности. Кроме того, для сравнения одного и того же “признака” в разных образцах и/или технических повторах одного и того же образца необходим дополнительный этап обработки, который состоит в выравнивании хроматограмм этих образцов/повторов для учета различий во времени удержания пептида на колонке. При этом, поскольку “feature” определяют по массе и времени удержания после выравнивания, то для идентификации пептида может быть достаточен один хороший MS/MS-спектр [2]. Каждый “признак” обладает определенной интенсивностью масс-спектрометрического сигнала, что и используют для полуколичественного анализа содержания пептида/белка в образце.
Программное обеспечение Progenesis LC-MS© (http://www.nonlinear.com/) позволяет быстро, объективно и достоверно определять “признаки” во всех повторах для всех анализируемых образцов, после чего экспортировать результаты для дальнейшей обработки (идентификации аминокислотной последовательности пептидов, которые определены как “признаки”) в ходе протеомного анализа.
Важнейший компонент анализа наборов данных LC-ESI-MS/MS – поисковая система, алгоритм, с помощью которого происходит идентификация пептидной последовательности молекулярного иона [3]. В настоящее время для идентификации пептидов/белков получили распространение такие поисковые системы, как Mascot [4], Sequest [5], X!Tandem [6, 7], Andromeda [8], MS-GF+ [9] и некоторые другие. Все они рассчитаны на стандартную процедуру эксперимента: гидролиз белковой фракции с помощью сайт-специфичного фермента, разделение на хроматографической колонке, масс-спектрометрический анализ с селективным измерением спектров фрагментации ионов пептидов [10, 11]. Все они действуют по схожей схеме [12], которая включает сопоставление экспериментального MS/MS-спектра с теоретическим спектром фрагментации для каждого пептида из базы данных на основе заданных параметров: заряда пептида, возможных химических и посттрансляционных модификаций, фермента, используемого для гидролиза, количества пропущенных внутренних мест расщепления протеолитического фермента, точность определения ионов пептидов. Получаемый список идентифицированных пептидов можно ранжировать в зависимости от значения индекса совпадения между теоретическим и экспериментальным MS/MS-спектрами.
Большинство протеомных поисковых систем хорошо работают на типичных наборах данных, причем результаты имеют существенное совпадение между алгоритмами [13]. Наиболее высокая степень соответствия выявлена, главным образом, для пептидных последовательностей, идентифицированных с высокой степенью достоверности. Тем не менее, степень расхождения в результатах различных поисковых систем довольно высока [3], так как некоторые поисковые машины используют уникальные методы для рассмотрения пептидов или модификаций, которые не учитывают другие системы.
Недавно разработана новая протеомная поисковая машина (ППМ) IdentiProt, основанная на алгоритме IdentiPy, для обработки данных протеомного анализа методами LC-ESI-MS/MS и идентификации пептидов/белков. Показано, что IdentiPy с функцией автонастройки показывает более высокую чувствительность по сравнению с такими популярными поисковыми системами, как X!Tandem, MS-GF+ [14].
Цель данного исследования – анализ протеомного профиля биоптатов тестикулярной ткани человека с использованием ППМ IdentiProt. Проведено сравнение результатов идентификации белков хромосомы 18 человека, полученных с помощью ППМ IdentiProt и популярной поисковой машины Mascot.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использованы следующие реактивы: додецилсульфат натрия, бычий сывороточный альбумин (БСА) (“Merck”, Германия); трипсин из поджелудочной железы свиньи модифицированный лиофилизированный (“Promega”, США); трифторуксусная кислота (ТФУ, “Fluka”, Германия); ацетонитрил, дитиотреитол (ДТТ) деионизованная вода (“Acros”, США); метанол (“Pierce”, США), а также реактивы отечественного производства квалификации “х. ч.”.
Объектом исследования служили биоптаты тестикулярной ткани пациентов с необструктивной азооспермией, находившихся на лечении в отделении андрологии и урологии ФГБУ “НМИЦ АГП им. В.И. Кулакова” в 2016–2017 гг. Образцы отбирали под местным наркозом методом микрохирургической тестикулярной экстракции. Включение пациентов в клиническое исследование проводили после получения информированного согласия и протоколировали по стандартам Этического комитета Российской Федерации.
Солюбилизаты тестикулярной ткани готовили по методу, описанному ранее в работе [15] с небольшими модификациями. 40 мг ткани подвергали замораживанию-оттаиванию в жидком азоте (5 циклов), а затем солюбилизировали в 6-ти объемах буфера, содержащего 7 М мочевину, 2 М тиомочевину, 65 мМ дитиотреитол и 1%-ный ингибитор протеаз Е64. Образцы инкубировали при +4°С в течение 1 ч при перемешивании. Затем проводили озвучивание при +4°С по программе к ультразвуковой установке BANDELIN sonoplus HD 2070 (Германия). После озвучивания пробы центрифугировали при 15 000 об./мин (Hettich micro 12-24, Германия) при комнатной температуре в течение 15 мин до получения прозрачного супернатанта. В случае необходимости процедуру центрифугирования повторяли 2–3 раза.
Содержание белка в солюбилизатах тестикулярной ткани определяли по методу Бредфорда [16] с использованием БСА в качестве стандарта.
Восстановление, алкилирование, триптический гидролиз проводили, как описано ранее [17]. Смесь пептидов анализировали с использованием хроматографической системы Ultimate 3000 nano-flow HPLC (“Dionex”, США), интегрированной с масс-спектрометром Orbitrap Q Exactive (“Thermo Scientific”, США) с источником электростатической ионизации Nanospray Flex NG ion source (“Thermo Scientific”) [18].
Масс-спектры солюбилизатов тестикулярной ткани в формате “.raw” анализировали с помощью коммерческого программного обеспечения Progenesis LC-MS (“Nonlinear Dynamics Ltd.”) либо конвертировали исходные raw-файлы в соответствующие MGF-файлы с помощью программы ProteoWizard MSConvert (http://proteowizard.sourceforge.net/).
Идентификацию пептидов и белков осуществляли по программам Mascot (www.matrixscience.com) и IdentiProt (ООО КуБ, Россия) со следующими параметрами поиска: база данных “Swiss_Prot” (SP, версия 2012_11, “.fasta”-формат) для вида Homo sapiens; расщепляющий фермент – трипсин; точность совпадения теоретической и экспериментальной массы пептида – ±10 м.д., точность совпадения теоретической и экспериментальной массы фрагментарных ионов – ±0.05 Да; значение зарядового состояния ионов пептида – “2+, 3+, 4+”; количество возможных пропущенных участков расщепления трипсином – 1; фиксированная модификация – пиридилэтилирование цистеина, вариабельная модификация – окисленный метионин. Поиск проводили по базе данных инвертированных и случайных последовательностей аминокислот (decoy), процент ложноположительных результатов (false discovery rate, FDR) ≤1% [19].
Выходные данные Mascot в формате XML или IdentiProt в формате pep.XML реимпортировали в программу Progenesis LC-MS для полуколичественной оценки содержания пептидов/белков.
Выходные данные Mascot в формате HTML или IdentiProt в формате TSV конвертировали в таблицы Excel для удобства анализа списков идентифицированных белков.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Анализ протеома тестикулярной ткани человека с помощью ППМ IdentiProt и Mascot
В настоящее время основная цель характеристики протеома человека – идентификация так называемых “необнаруженных (“missing”) белков”, для которых нет надежных масс-спектрометрических доказательств их существования [20]. В частности, одним из подходов для выявления “необнаруженных белков” предложено использовать необычные органы или типы клеток [21]. На основании данных РНК-секвенирования недавно показано, что тестикулярную ткань относят к наиболее перспективным объектам для поиска “необнаруженных белков” [22]. Кроме того, один из подходов к решению этой проблемы – усовершенствование методов биоинформатической обработки протеомных данных. За последние 15 лет разработаны практически все ныне существующие ППМ, в том числе поисковая система с открытым исходным кодом IdentiPy, реализованная на языке программирования Python [14] с рядом уникальных функций (например, автоматический поиск настроек в сочетании с алгоритмом постобработки данных МР score [23]).
В настоящей работе проведен анализ протеома тестикулярной ткани человека с помощью ППМ IdentiProt, основанной на платформе IdentiPy Server [14]. Результаты идентификации пептидов/белков сравнивали с результатами, полученными при помощи популярной поисковой машины Mascot.
Всего проанализировано 10 raw-файлов одного биоптата, который разделен на две части, для каждой проведена процедура триптического гидролиза. Снятие масс-спектров проводили в разное время, по 2–3 технических повтора. В итоге для сопоставления белкового профиля тестикулярной ткани получено 52760 MS/MS спектров. С помощью программного обеспечения Progenesis LC-MS получен файл в MGF-формате, который параллельно загружали в ППМ IdentiProt и Mascot для поиска пептидов и белков. Входные параметры поиска (относительная точность измерения масс ионов-предшественников – 10 м.д. (рис. 1а), ошибку определения масс ионов ионов-продуктов – 0.05 Да, рис. 1б) выбирали на основании предоставляемых интерфейсом ППМ IdentiProt возможностей алгоритма МР-score (“MP-score descriptors” [23]) для оценки достоверности идентификаций.
Рис. 1.
Распределение двух “дескрипторов” MP score в предварительной обработке хромато-масс-спектров с помощью ППМ IdentiProt: а ‒ ошибка массы родительского иона; б ‒ медианная ошибка массы ионов-фрагментов. 1 – Распределение для идентификаций, прошедших порог достоверности = 1% (частота ложноположительных идентификаций, FDR = 1%); 2 – для идентификаций из “истинной” (target) базы данных, 3 – для “ложных” (decoy) идентификаций.
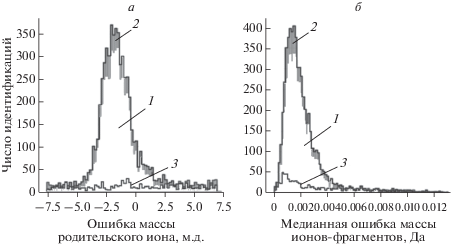
В результате с помощью IdentiProt идентифицировано 1443 пептида, что соответствовало 611 белкам. Поисковая система Mascot выявила 484 белка и 1226 пептидов. 441 белок обнаружен как с помощью IdentiProt (72% от общего числа идентификаций), так и поисковой системой Mascot (91%). По двум и более пептидам идентифицировано около 38 и 47% белков в случае использования IdentiProt и Mascot соответственно. При этом только три белка (0.5%) не имели пептидов, используемых программой Progenesis LC-MS для их количественной оценки в комбинации с IdentiProt, в то время как доля таких белков при анализе Progenesis LC-MS/Mascot составила 3%.
В целом следует отметить, что принципиальных отличий качества идентификации белков между IdentiProt и Mascot не обнаружено. Увеличение количества идентифицированных белков с использованием ППМ IdentiProt происходит главным образом за счет идентификаций по одному пептиду.
Идентификация белков хромосомы 18 человека
В результате анализа протеома солюбилизата тестикулярной ткани зарегистрированы белки, кодируемые генами хромосомы 18 человека (табл. 1). Интерес к белкам этой хромосомы обусловлен тем, что измерение протеома, кодируемого генами хромосомы 18, представляет российскую часть международного проекта “Протеом человека” [24]. Кроме того, для ряда идентифицированных белков этой хромосомы согласно сведениям протеомной базы UniProtKB (http://www.uniprot.org/) есть аннотации о взаимосвязи генов хромосомы 18 и продуктов их экспрессии с возникновением и развитием заболеваний. Эти данные крайне важны в плане перспективы развития методов постгеномного профилирования, и, как следствие, использования результатов для предсказания рисков развития и диагностики заболеваний.
Таблица 1.
Список идентифицированных белков хромосомы 18 в образце тестикулярной ткани человека в зависимости от поисковой машины
| № п/п |
Идентификатор | Название белка | Количество пептидов | Среднее нормализованное количество (average normalized abundance) |
Заболевание*** | ||
|---|---|---|---|---|---|---|---|
| Mascot* | IdentiProt* | Mascot* | IdentiProt* | ||||
| 1 | ATPA_HUMAN | АТФ-синтаза митохондриальная, субъединица альфа | 5(3)** | 4 | 6.11E + 06 ± 8.43E + 05 | 9.18E + 06 ± 8.40E + 05 | Синдром Лея, синдром MELAS |
| 2 | LAMA1_HUMAN | альфа-Ламинин-1 | 5 | 6 | 2.54E + 06 ± 2.25E + 05 | 3.25E + 06 ± 4.78E + 05 | Синдром Поретти-Больтшаузера |
| 3 | TBB6_HUMAN | бета-Тубулин-6 | 8(0)** | 1 | — | 5.99E + 06 ± 3.78E + 05 | |
| 4 | THIM_HUMAN | 3-кетоацил-CoA тиолаза митохондриальная | 3 | 3 | 1.20E + 06 ± 1.05E + 05 | 1.20E + 06 ± 1.05E + 05 | |
| 5 | LMAN1_HUMAN | Белок ERGIC-53 | 1 | 1 | 3.18E + 05 ± 1.06E + 05 | 3.18E + 05 ± 1.06E + 05 | Комбинированныйдефицитфакторов свертывания крови V иVIII |
| 6 | E41L3_HUMAN | Полосы 4.1-подобный белок 3 | 2 | 1 | 6.51E + 05 ± 1.93E + 05 | 1.97E + 05 ± 1.38E + 05 | |
| 7 | TTHY_HUMAN | Транстиретин | 1 | 1 | 3.06E + 05 ± 4.93E + 03 | 3.06E + 05 ± 4.93E + 03 | Транстиретиновый амилоидоз |
| 8 | CNDP2_HUMAN | Неспецифическая цитозольная дипептидаза | 1 | 1 | 5.54E + 05 ± 4.56E + 04 | 5.54E + 05 ± 4.56E + 04 | |
| 9 | MAOM_HUMAN | NAD-зависимая малатдегидрогеназа митохондриальная | 1 | 1 | 1.03E + 06 ± 5.74E + 04 | 1.03E + 06 ± 5.74E + 04 | |
| 10 | MBP_HUMAN | Основной белок миелина | 1 | 1 | 2.59E + 05 ± 4.41E + 05 | 2.59E + 05 ± 4.41E + 05 | Острый рассеянный энцефаломиелит |
| 11 | TPPC8_HUMAN | Субъединица 8 сложного комплекса белка-транспортера TRAPP | Не опр. | 1 | Не опр. | 9.83E + 05 ± 1.49E + 05 | |
| 12 | HDHD2_HUMAN | Домен 2 гидролазы-подобной дегалогеназы галокислоты | Не опр. | 1 | Не опр. | 3.09E + 05 ± 2.27E + 04 | |
Как следует из табл. 1, с помощью Progenesis LC-MS/Mascot идентифицировано 10 белков, в то время как использование ППМ IdentiProt привело к увеличению количества выявленных белков хромосомы 18. Помимо 10 белков, обнаруженных с помощью Mascot, использование ППМ IdentiProt позволило зарегистрировать присутствие в солюбилизате таких белков, как субъединица 8 сложного комплекса белка-транспортера TRAPP (TPPC8_HUMAN) и домен 2 гидролазa-подобной дегалогеназы галокислоты (HDHD2_HUMAN). Три белка (ATPA, LAMA1 и THIM) идентифицированы по двум и более пептидам, как в случае поиска с помощью Mascot, так и при использовании IdentiProt. Большинство белков, представленных в табл. 1, идентифицированы по одному пептиду. Тем не менее, с большой долей вероятности эти идентификации можно считать достоверными, так как во всех случаях выявлены протеотипические для этих белков пептиды [25]. Кроме того, определено содержание большинства из них (Average Normalized Abundances). По данным табл. 1 значения Average Normalized Abundances большинства белков, определенных Progenesis LC-MS/Mascot, сопоставимы или совпадают со значениями содержания белков, найденных Progenesis LC-MS/IdentiProt. Только для одного белка, а именно бета-тубулина-6 (TBB6_HUMAN), при использовании Mascot не обнаружено высокоспецифичных пептидов (т.е. пептидов, автоматически выбираемых программой Progenesis LC-MS для идентификации и полуколичественной оценки белков). В случае IdentiProt идентификация и количественная оценка бета-тубулина-6 произведена с помощью пептида MASTFIGNSTAIQELFKR, который также выявлен поисковой системой Mascot для этого белка, но при этом его количество не установлено.
Можно предположить, что заложенный в ППМ IdentiProt алгоритм позволил соотнести найденный пептид с большим количеством пиков в спектре фрагментации, тем самым увеличив достоверность идентификации, что позволило присвоить этому пептиду статус высококачественного, т.е. используемого для количественной оценки.
Среди белков, идентифицированных по одному пептиду, нами обнаружен основной белок миелина (MBP_HUMAN), специфичный маркер олигодендроцитов и шванновских клеток [26], что могло бы указывать на ошибку, исходя из гистологического представления о ткани яичка. Однако следует учитывать, что с внедрением масс-спектрометрического анализа высокого разрешения и оптимизации алгоритмов поиска пептидов/белков, возможно обнаружение специфичных белков в следовых концентрациях и в других тканях. Нами проведен доскональный анализ протеомных данных, хранящихся в международном общедоступном хранилище PRIDE (the PRoteomics IDEntifications database) Европейского института биоинформатики (http://www.ebi.ac.uk/pride), который показал, что этот белок обнаружен и в таких тканях человека, как сердце и толстая кишка [27, 28]. Кроме того, авторы работы [29] также идентифицировали MBP_HUMAN в тестикулярной ткани методом панорамной протеомики с использованием гибридного масс-спектрометра Q Exactive HF после разделения белков с помощью одномерного электрофореза в трициновом геле.
Чтобы подтвердить результаты идентификации белков хромосомы 18 в солюбилизате тестикулярной ткани, мы применили подход, предложенный в работе [29], где перед масс-спектрометрическим анализом белки разделяли методом одномерного электрофореза в геле. Масс-спектрометрический анализ в срезах геля позволил определить приведенные в табл. 1 белки по двум и более пептидам. Например, неспецифическая цитозольная дипептидаза (CNDP2_HUMAN) и белок ERGIC-53 (LMAN1_HUMAN) идентифицировали по 6 и 13 высокоспецифичным пептидам соответственно. Таким образом, для первоначальной оценки потенциала ППМ IdentiProt по сравнению с другими поисковыми системами, в частности с Mascot, можно учитывать также и результаты, основанные на выявлении одного протеотипического пептида на белок (при условии, что идентификации прошли фильтр по критерию FDR < 1%).
Среди идентифицированных белков, кодируемых генами хромосомы 18, обнаружен белок, специфичный для тестикулярной ткани (E41L3_HUMAN) (табл. 1). Кроме того, нами обнаружен альфа-ламинин-1 (LAMA1), мутация в гене которого приводит к развитию синдрома Поретти–Больтшаузера – аутосомно-рецессивного синдрома, характеризующегося дисплазией мозжечка с кистами и аномальной формой четвертого желудочка [30]. Кроме того, идентифицирован транстиретин (TTHY_HUMAN), мутации гена которого представляют непосредственную причину первичного наследственного амилоидоза [31].
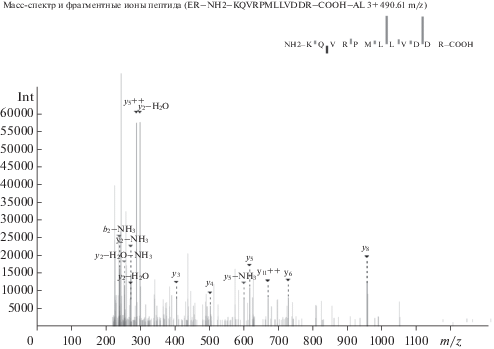
Чтобы дополнительно проверить эффективность ППМ IdentiProt для идентификации белков хромосомы 18, проводили анализ масс-спектров (MGF-файл, полученный при обработке масс-спектров в Progenesis LC-MS) с помощью платформы SearchGUI (v. 3.3.1) [32] с использованием поисковых алгоритмов X!Tandem и MS-GF+. Входящий в состав платформы интегратор PeptideShaker применен для получения текстового файла в формате для загрузки в Progenesis LC-MS. В результате сгенерировано два отчета, по одному для X!Tandem и MS-GF+, соответственно. Табл. 2 отражает результаты идентификации белковых продуктов хромосомы 18 с использованием Progenesis LC-MS/X!Tandem и Progenesis LC-MS/MS-GF+. В обоих случаях выявлено по девять белков, что сопоставимо с количеством белков, идентифицированных с помощью Mascot, но на три белка меньше, чем установлено ППМ IdentiProt. Как и в случае ППМ IdentiProt и Mascot большая часть белков хромосомы 18 идентифицирована по одному протеотипическому пептиду. Восемь белков найдены как поисковой системой X!Tandem, так и алгоритмом MS-GF+. Один белок –E41L3_HUMAN – обнаружен всеми поисковыми алгоритмами, кроме MS-GF+. Однако MS-GF+, как и ППМ IdentiProt, обеспечивал обнаружение пептида KQVRPMLLVDDR (рис. 2), специфичного для одного из доменов дегалогеназы HDHD2_HUMAN, в то время ни X!Tandem ни Mascot не позволили его выявить. В отличие от IdentiProt, оба поисковых алгоритма (X!Tandem и MS-GF+), не выявили пептиды, необходимые для идентификации и количественного анализа бета-тубулина-6 (TBB6_HUMAN).
Таблица 2.
Список белков хромосомы 18 в образце тестикулярной ткани человека, идентифицированных с помощью платформы SearchGUI
| № п/п |
Идентификатор | Название белка | Количество пептидов | Среднее нормализованное количество (average normalized abundance) |
||
|---|---|---|---|---|---|---|
| X!Tandem* | MS-GF+* | X!Tandem* | MS-GF+* | |||
| 1 | ATPA_HUMAN | АТP-синтаза митохондриальная, субъединица альфа | 7 | 7 | 1.34E + 07 ± 1.42E + 06 | 1.36E + 07 ± 1.48E + 06 |
| 2 | LAMA1_HUMAN | альфа-Ламинин-1 | 7 | 8 | 2.88E + 06 ± 3.55E + 05 | 3.44E + 6 ± 4.37E + 05 |
| 3 | THIM_HUMAN | 3-кетоацил-CoA-тиолаза митохондриальная | 5 | 5 | 1.94E + 06 ± 2.25E + 05 | 2.16E + 06 ± 6.03E + 04 |
| 4 | TTHY_HUMAN | Транстиретин | 3 | 2 | 1.51E + 06 ± 1.55E + 05 | 9.80E + 05 ± 8.79E + 04 |
| 5 | LMAN1_HUMAN | Белок ERGIC-53 | 1 | 1 | 3.18E + 05 ± 1.06E + 05 | 3.18E + 05 ± 1.06E + 05 |
| 6 | MAOM_HUMAN | NAD-зависимая малатдегидрогеназа митохондриальная | 1 | 1 | 1.03E + 06 ± 5.74E + 04 | 1.03E + 06 ± 5.74E + 04 |
| 7 | MBP_HUMAN | Основной белок миелина | 1 | 1 | 2.59E + 05 ± 4.56E + 04 | 2.59E + 05 ± 4.41E + 04 |
| 8 | CNDP2_HUMAN | Неспецифическая цитозольная дипептидаза | 1 | 1 | 5.54E + 05 ± 4.56E + 04 | 5.54E + 05 ± 4.56E + 04 |
| 9 | E41L3_HUMAN | Белок, подобный белку полосы 4.1 | 1 | Не опр. | 1.97E + 05 ± 1.38E + 05 | Не опр. |
| 10 | HDHD2_HUMAN | Домен 2 гидролаза-подобной дегалогеназы галокислоты | Не опр. | 1 | Не опр. | 3.09E + 05 ± 2.27E + 04 |
Рис. 2.
Масс-спектр и фрагментные ионы пептида KQVRPMLLVDDR, специфичного для домена дегалогеназы галокислоты (HDHD2_HUMAN).
Таким образом, сравнение показало, что в целом ППМ IdentiProt идентифицирует в солюбилизате тестикулярной ткани человека несколько большее число белков хромосомы 18, в то время как результаты идентификации, полученные с помощью поисковых алгоритмов Mascot, X!Tandem и MS-GF+ сопоставимы.
Известно, что программное обеспечение Progenesis – подходящий инструмент для определения разницы в содержании (average normalized abundance) белка в разных образцах. Однако ему не всегда удается автоматически выравнивать технические повторы в наборе данных, что может снижать число идентифицированных белков [33].
Чтобы исключить стадию выравнивания хромато-масс-спектров между собой, мы использовали ProteoWizard [34] для преобразования исходных raw-файлов в списки пиков, так как поисковые системы принимают результаты конвертации в качестве входных данных [35]. При этом ProteoWizard создает файлы, совместимые с большинством поисковых систем. По результатам анализа конвертированных 10 MGF-файлов получены отчеты для IdentiProt и Mascot по 10 для каждой ППМ. Идентификация пептидов/белков с помощью Mascot не привела к изменению списка белков хромосомы 18 по сравнению с поиском в Mascot в комбинации с анализом raw-файлов в Progenesis LC-MS. В то же время использование IdentiProt способствовало обнаружению дополнительно четырех белков, кодируемых хромосомой 18 (табл. 3). При этом один белок, идентифицированный комбинацией Progenesis LC-MS/IdentiProt (субъединица 8 сложного комплекса белка-транспортера TRAPP, табл. 1), не найден. Анализ клеточной локализации дополнительно идентифицированных белков показал, что кадхерин-2 (CADH2_HUMAN) и С-концевая убиквитин-гидролаза 14 (UBP14_HUMAN) – это мембранные белки, в то время как биспиральный домен-содержащий белок 68 (CCD68_HUMAN) и кальций-связывающий белок, регулирующий фосфорилирование тирозина (CABYR_HUMAN) локализованы в цитоплазме. Два белка, а именно CCD68 и CABYR, специфичны для тестикулярной ткани. Таким образом, суммарно ППМ IdentiProt позволила выявить в солюбилизате тестикулярной ткани человека 16 белков хромосомы 18, т.е. на шесть белков больше, чем поисковый алгоритм Mascot.
Таблица 3.
Список белков хромосомы 18, идентифицированных в образце тестикулярной ткани человека с помощью комбинации ProteoWizard/ IdentiProt
| № п/п |
Идентификатор | Название белка | Последовательность пептида | Эксперимен-тальное значение отношения массы к заряду для пре-курсорного иона пептида (m/z exp) | Количество возможных пропущенных участков расщепления трипсином | Время удержания на колонке (RT exp) |
|---|---|---|---|---|---|---|
| 1 | CADH2_HUMAN | Кадхерин-2 | MFVLTVAAENQVPLAK | 865.979 | 0 | 49.02 |
| 2 | UBP14_HUMAN | С-концевая убиквитин-гидролаза 14 | RVEIMEEESEQ | 689.805 | 1 | 29.20 |
| 3 | CCD68_HUMAN | Биспиральный домен-содержащий белок 68 | AVSTSELKTEGVS PYLMLIR | 737.389 | 1 | 48.91 |
| 4 | CABYR_HUMAN | Кальций-связывающий белок, регулирующий фосфорилирование тирозина | VLEVQVVNQTSVH VDLGSQPK | 569.813 | 0 | 24.58 |
ЗАКЛЮЧЕНИЕ
В работе путем обработки экспериментальных данных, полученных на масс-спектрометре Orbitrap Q Exactive, совмещенном с нанопотоковой хроматографической системой, проведено сравнение результатов идентификации белков тестикулярной ткани человека. Сравнение показало, что ППМ IdentiProt позволяет получить дополнительные идентификации белков. Проведено сопоставление с коммерческим программным продуктом Mascot( компания Matrix Science) с закрытым кодом, а также со свободно распространяемым программным обеспечением, входящим в пакет SearchGUI. На примере белков хромосомы 18 человека проведен детальный разбор случаев, в которых данные различных поисковых машин подтверждают или дополняют друг друга.
Показано, что для получения максимально полного протеомного профиля необходимо объединение результатов, полученных несколькими способами анализа (например, сочетаниями Progenesis LC-MS/IdentiProt или ProteoWizard/IdentiProt) исходных масс-спектрометрических данных.
Работа выполнена в рамках Программы фундаментальных научных исследований государственных академий наук на 2013−2020 гг. В работе использовались данные, полученные ООО “Постгентех” (г. Москва).
Список литературы
Matthiesen R. (2013) LC-MS spectra processing. Methods Mol. Biol. 1007, 47‒63.
Lemeer S., Hahne H., Pachl F., Kuster B. (2012) Software tools for MS-based quantitative proteomics: a brief overview. Methods Mol. Biol. 893, 489‒499.
Shteynberg D., Nesvizhskii A.I., Moritz R.L., Deutsch E.W. (2013) Combining results of multiple search engines in proteomics. Mol. Cell Proteomics. 12(9), 2383‒2393.
Perkins D.N., Pappin D.J., Creasy D.M., Cottrell J.S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 20(18), 3551–3567.
Eng J.K., McCormack A.L., Yates J.R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5(11), 976–989.
Craig R., Beavis R.C. (2004) TANDEM: Matching proteins with tandem mass spectra. Bioinformatics. 20(9), 1466–1467.
Craig R., Beavis R.C. (2003) A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 17(20), 2310–2316.
Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805.
Kim S., Pevzner P.A. (2014) MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277.
Nesvizhskii A.I., Vitek O., Aebersold R. (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nature Methods. 4, 787–797.
Bogdanov B.C., Smith R.D. (2004) Proteomics by FTICR mass spectrometry: top down and bottom up. Mass Spectrometry Reviews. 24(2), 168–200.
Balgley B.M., Laudeman T., Yang L., Song T., Lee C.S. (2007) Comparative evaluation of tandem MS search algorithms using a target-decoy search strategy. Mol. Cell Proteomics. 6(9), 1599–1608.
Kapp E.A., Schütz F., Connolly L.M., Chakel J.A., Meza J.E., Miller C.A., Fenyo D., Eng J.K., Adkins J.N., Omenn G.S., Simpson R.J. (2005) An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 5(13), 3475–3490.
Levitsky L.I., Ivanov M.V., Lobas A.A., Bubis J.A., Tarasova I.A., Solovyeva E.M., Pridatchenko M.L., Gorshkov M.V. (2018) IdentiPy: an extensible search engine for protein identification in shotgun proteomics. J. Proteome Res. (in press), doi 10.1021/acs.jproteome.7b00640
Yu S.M., Cai X., Sun L., Zuo Z.C., Mipam T.D., Cao S.Z., Shen L., Ren Z., Chen X., Yang F., Deng J., Ma X., Wang Y. (2016) Comparative iTRAQ proteomics revealed proteins associated with spermatogenic arrest of cattleyak. J. Proteomics. 142, 102–113.
Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254.
Petushkova N.A., Zgoda V.G., Pyatnitskiy M.A., Larina O.V., Teryaeva N.B., Potapov A.A., Lisitsa A.V. (2017) Post-translational modifications of FDA-approved plasma biomarkers in glioblastoma samples. PLoS One. 12 (5): e0177427.
Кисриева Ю.С., Петушкова Н.А., Саменкова Н.Ф., Кузнецова Г.П., Ларина О.В., Завьялова М.Г., Теряева Н.Б., Беляев А.Ю., Карузина И.И. (2016) Сравнительный анализ протеома плазмы крови больных на ранней стадии хронической церебральной ишемии. Биомедицинская химия. 62, 599–602.
Elias J., Gygi S. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 4(3), 207–214.
Horvatovich P., Lundberg E.K., Chen Y.J., Sung T.Y., He F., Nice E.C., Goode R.J., Yu S., Ranganathan S., Baker M.S., Domont G.B., Velasquez E., Li D., Liu S., Wang Q., He Q.Y., Menon R., Guan Y., Corrales F.J., Segura V., Casal J.I., Pascual-Montano A., Albar J.P., Fuentes M., Gonzalez-Gonzalez M., Diez P., Ibarrola N., Degano R.M., Mohammed Y., Borchers C.H., Urbani A., Soggiu A., Yamamoto T., Salekdeh G.H., Archakov A., Ponomarenko E., Lisitsa A., Lichti C.F., Mostovenko E., Kroes R.A., Rezeli M., Végvári Á., Fehniger T.E., Bischoff R., Vizcaíno J.A., Deutsch E.W., Lane L., Nilsson C.L., Marko-Varga G., Omenn G.S., Jeong S.K., Lim J.S., Paik Y.K., Hancock W.S. (2015) Quest for missing proteins: update 2015 on chromosome-centric human proteome project. J. Proteome Res. 14, 3415–3431.
Lane L., Bairoch A., Beavis R.C., Deutsch E.W., Gaudet P., Lundberg E., Omenn G.S. (2014) Metrics for the Human Proteome Project 2013–2014 and strategies for finding missing proteins. J. Proteome Res. 13(1), 15–20.
Son C.G., Bilke S., Davis S., Greer B.T., Wei J.S., Whiteford C.C., Chen Q.R., Cenacchi N., Khan J. (2005) Database of mRNA gene expression profiles of multiple human organs. Genome Res. 15(3), 443–450.
Ivanov M.V., Levitsky L.I., Lobas A.A., Panic T., Laskay Ü.A., Mitulovic G., Schmid R., Pridatchenko M.L., Tsybin Y.O., Gorshkov M.V. (2014) Empirical Multidimensional Space for Scoring Peptide Spectrum Matches in Shotgun Proteomics. J. Proteome Res. 13(4), 1911–1920.
Пономаренко Е.А., Згода В.Г., Копылов А.Т., Поверенная Е.В., Ильгисонис Е.В., Лисица А.В., Арчаков А.И. (2015) Россия в международном проекте “протеом человека”: первые итоги и перспективы. Биомедицинская химия. 61(2), 169–175.
Craig R., Cortens J.P., Beavis R.C. (2005) The use of proteotypic peptide libraries for protein identification. Rapid Commun. Mass Spectrom. 19, 1844–1850.
Müller C., Bauer N.M., Schäfer I., White R. (2013) Making myelin basic protein -from mRNA transport to localized translation. Front Cell Neurosci. 7, 169.
Barallobre-Barreiro J., Didangelos A., Schoendube F.A., Drozdov I., Yin X., Fernández-Caggiano M., Willeit P., Puntmann V.O., Aldama-López G., Shah A.M., Doménech N., Mayr M. (2012) Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation. 125(6), 789–802.
Mikula M., Rubel T., Karczmarski J., Goryca K., Dadlez M., Ostrowski J. (2011) Integrating proteomic and transcriptomic high-throughput surveys for search of new biomarkers of colon tumors. Funct. Integr. Genomics. 11(2), 215–224.
Wei W., Luo W., Wu F., Peng X., Zhang Y., Zhang M., Zhao Y., Su N., Qi Y., Chen L., Zhang Y., Wen B., He F., Xu P. (2016) Deep coverage proteomics identifies more low-abundance missing proteins in human testis tissue with Q-exactive HF mass spectrometer. J. Proteome Res. 15(11), 3988–3997.
Micalizzi A., Poretti A., Romani M., Ginevrino M., Mazza T., Aiello C., Zanni G., Baumgartner B., Borgatti R., Brockmann K., Camacho A., Cantalupo G., Haeusler M., Hikel C., Klein A., Mandrile G., Mercuri E., Rating D., Romaniello R., Santorelli F.M., Schimmel M., Spaccini L., Teber S., von Moers A., Wente S., Ziegler A., Zonta A., Bertini E., Boltshauser E., Valente E.M. (2016) Clinical, neuroradiological and molecular characterization of cerebellar dysplasia with cysts (Poretti-Boltshauser syndrome). Eur. J. Hum. Genet. 24(9), 1262–1267.
Ueda M., Misumi Y., Mizuguchi M., Nakamura M., Yamashita T., Sekijima Y., Ota K., Shinriki S., Jono H., Ikeda S., Suhr O.B., Ando Y. (2009) SELDI-TOF mass spectrometry evaluation of variant transthyretins for diagnosis and pathogenesis of familial amyloidotic polyneuropathy. Clin. Chem. 55(6), 1223–1227.
Vaudel M., Barsnes H., Berven F.S., Sickmann A., Martens L. (2011) SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 11(5), 996–999.
Välikangas T., Suomi T., Elo L.L. (2017) A comprehensive evaluation of popular proteomics software workflows for label-free proteome quantification and imputation. Brief Bioinform. bbx054, 1–12.
Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 24, 2534–2536.
Verheggen K., Martens L., Berven F.S., Barsnes H., Vaudel M. (2016) Database search engines: paradigms, challenges and solutions. Adv. Exp. Med. Biol. 919, 147–156.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология