

Молекулярная биология, 2019, T. 53, № 1, стр. 154-165
Биоинформатический алгоритм обработки данных высокопроизводительного секвенирования для прецизионной идентификации соматических инсерций ретроэлементов
Г. А. Нугманов a, А. Ю. Комков a, М. В. Салютина a, А. А. Минервина a, Ю. Б. Лебедев a, И. З. Мамедов a, *
a Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова
Российской академии наук
117997 Москва, Россия
* E-mail: imamedov78@gmail.com
Поступила в редакцию 01.08.2018
После доработки 12.09.2018
Принята к публикации 12.09.2018
Аннотация
Ретроэлементы считаются одним из важнейших источников генетической вариабельности генома современного человека. Известно, что транспозиционная активность ретроэлементов в герминальных клетках может приводить к появлению новых копий в различных участках генома и в некоторых случаях к врожденным генетическим заболеваниям. В соматических клетках активность ретроэлементов ограничена множеством регуляторных механизмов, хотя ее наличие предполагается в некоторых типах тканей. Предполагается, что соматические инсерции могут вызывать развитие опухолей или быть частью нормальных процессов, связанных с обеспечением функциональной пластичности нейронов. Несмотря на развитие методов высокопроизводительного секвенирования, надежная идентификация соматических инсерций по-прежнему остается непростой задачей, в том числе из-за отсутствия адекватных биоинформатических подходов для анализа полученных последовательностей. В данной работе предлагается биоинформатический алгоритм, позволяющий идентифицировать соматические инсерции ретроэлементов семейства AluYa5 в массивах данных высокопроизводительного секвенирования участков генома, фланкирующих такие инсерции. Особое внимание уделено идентификации различных по природе артефактов, возникающих при подготовке библиотек к высокопроизводительному секвенированию, и определению параметров для их адекватной фильтрации. Высокая специфичность предлагаемого алгоритма показана в экспериментах in silico с использованием искусственного вложенного контроля. Предложенный подход позволяет удалить не менее 80% артефактов, при этом не менее 75% предположительно соматических инсерций сохраняется. Описанные в работе подходы могут быть использованы для исследования инсерционной вариабельности других групп мобильных элементов.
ВВЕДЕНИЕ
Последовательности ретроэлементов (РЭ) составляют более половины человеческого генома [1]. Для данного типа мобильных элементов характерно размножение с использованием механизма копирования–вставки с образованием РНК-интермедиата. РЭ человека в основном представлены тремя классами: 1) LTR (от англ. Long Terminal Repeat) элементы, характеризующиеся длинными концевыми повторами и схожие по структуре c ретровирусами; 2) автономные LINE (от англ. Long Interspersed Nucleic Elements), обладающие двумя открытыми рамками считывания, одна из которых кодирует ревертазу, необходимую для ретротранспозиции, и 3) неавтономные SINE (от англ. Short Interspersed Nucleic Elements), не имеющие открытых рамок считывания и использующие ретротранспозиционную машину LINE. Каждый класс РЭ в свою очередь подразделяется на семейства и подсемейства, каждое из которых обладает определенными структурными особенностями (диагностическими позициями) и появилось в геноме человека в разное время в процессе эволюции приматов. Подавляющее большинство копий (инсерций) РЭ в геноме современного человека неактивно, однако некоторые из них сохраняют способность к ретротранспозиции. Большинство активных копий принадлежит к семействам AluYa5, AluYb8 (класс SINE) [2] и L1Hs (класс LINE) [3]. Активность РЭ в герминальных клетках приводит к появлению de novo инсерций в геноме, которые, как предполагается, происходят с частотой 1 на 200 новорожденных для L1Hs [4] и 1 на 20 новорожденных для Alu [5]. Такие инсерции могут быть нейтральными или приводить к повреждению генов и развитию наследственных заболеваний [6]. Транспозиционная активность РЭ в клетках взрослого организма может стать причиной появления соматических инсерций, несмотря на множество клеточных механизмов, направленных на ее подавление. Предполагается, что активность РЭ связана с опухолеобразованием [7–9], однако, неясно, соматические инсерции становятся причиной развития процесса злокачественной трансформации клетки или это одно из его следствий. Предполагается также, что соматические инсерции возникают в нормальных клетках и участвуют в физиологических процессах, например, в формировании функциональной пластичности нейронов [10].
До появления методов высокопроизводительного секвенирования изучение соматических инсерций РЭ было существенно ограничено: сами элементы часто имеют почти идентичные нуклеотидные последовательности, при этом концентрация соматических копий в исследуемом образце чрезвычайно мала. В связи с этим были разработаны различные методы, позволяющие обогащаться участками ДНК, непосредственно прилегающими к инсерциям РЭ в геноме [11, 12]. Секвенирование таких последовательностей (прилегающих участков, или фланков) позволяет точно определить положение инсерции в геноме, и при этом отпадает необходимость секвенировать весь геном. Развитие технологий секвенирования нового поколения открыло новые возможности в данной области. Многократно возросла чувствительность методов детекции новых ретропозиционных событий, хотя вместе с тем возросла и доля идентифицируемых артефактов, неизбежно возникающих в ходе получения библиотек для секвенирования. Зашумленность данных секвенирования артефактами, имитирующими фланки соматических инсерций РЭ, не позволяет в полной мере оценить реальную частоту соматических инсерций РЭ в клетках человека [13]. Несмотря на существование множества как экспериментальных, так и биоинформатических методов детекции соматических инсерций РЭ, до сих пор не существует ни одного автоматизированного способа выявления и фильтрации артефактов. Глубокий анализ массивов данных, полученных с помощью ранее разработанного метода [14], показал, что такие артефакты возникают на всех ключевых стадиях пробоподготовки, включая лигирование адаптеров и все этапы амплификации полученных библиотек. Часть артефактов имеет очевидную химерную природу и выявляется на стадии картирования на референсный геном, другая же часть артефактов внешне по своей структуре ничем не отличается от фланков соматических инсерций, составляя значительную долю в массивах потенциальных соматических инсерций.
Нами идентифицировано и охарактеризовано подавляющее большинство явных и скрытых артефактов, затрудняющих идентификацию истинных соматических инсерций РЭ. На основе выявленных особенностей артефактов и построенных в ходе данной работы статистических моделей, характеризующих истинные инсерции, разработан ряд биоинформатических фильтров, гарантированно отличающих артефакты от соматических инсерций. Для автоматизации процесса удаления артефактов разработанные фильтры интегрировали в единый алгоритм, позволяющий последовательно и с максимальной эффективностью удалять артефакты всех существующих на сегодня типов.
МЕТОДЫ
Описание массивов данных секвенирования. Для разработки и тестирования алгоритма использованы данные, полученные в рамках продолжения исследования частоты инсерций РЭ в различных областях и тканях мозга человека (SRP155786). Массив сиквенсов, представляющих собой прилегающие геномные участки инсерций Alu ретроэлементов, получен с использованием метода, описанного ранее [14, 15], с некоторыми модификациями. Для получения библиотек, фланкирующих места инсерций AluYa5 в геноме, использовано 4 аликвоты по 150 нг геномной ДНК, полученной из нейрональных и глиальных ядер, выделенных из постмортальных образцов фронтальной коры и зубчатой извилины мозга одного индивида. Фракции нейрональных и глиальных ядер получены с помощью флуоресцентного сортинга препаратов ядер, окрашенных DAPI (“Thermo Fisher Scientific”, США) и антителами против внутриядерного нейронального маркера NeuN (“Abcam”, Великобритания) [16]. Каждый образец геномной ДНК получен из 25000 ядер с помощью ExtractDNA Blood (“Евроген”, Россия).
Основные этапы пробоподготовки включали (рис. 1а): 1) фрагментирование геномной ДНК с помощью эндонуклеазы рестрикции AluI (“Thermo Fisher Scientific”, США); 2) лигирование супрессионных адаптеров, содержащих UMI (Unique Molecular Identifiers) – уникальные молекулярные идентификаторы, представляющие собой случайные 9-нуклеотидные последовательности [17]; 3) селективная ПЦР-амплификация геномных участков, фланкирующих инсерции AluYa5, с использованием специфичного к данному семейству праймера AY107R; 4) вторая, “вложенная” ПЦР, совмещенная с введением адаптеров для секвенирования на платформе Illumina; 5) секвенирование полученной библиотеки на приборе HiSeq с парными прочтениями длиной по 100 н. В результате секвенирования суммарно получено 4 массива в формате FASTQ с 12.1, 9, 8.2 и 11.8 млн. парных прочтений (табл. 1).
Рис. 1.
Получение библиотек для секвенирования. а – Схема получения библиотек последовательностей, фланкирующих инсерции ретроэлементов AluYa5, в геноме человека. AluI – сайты узнавания ретриктазы, AY107, ill-AY19-7, ill-St19, St19 – олигонуклеотидные праймеры, использованные в ПЦР. UMI – уникальные молекулярные идентификаторы, ΔAlu – 5'-фрагмент (начало) Alu, н. – нуклеотиды. б – Структура нуклеотидной последовательности для искусственного вложенного контроля. Ad1 и Ad2 – фрагменты супрессионного адаптера.
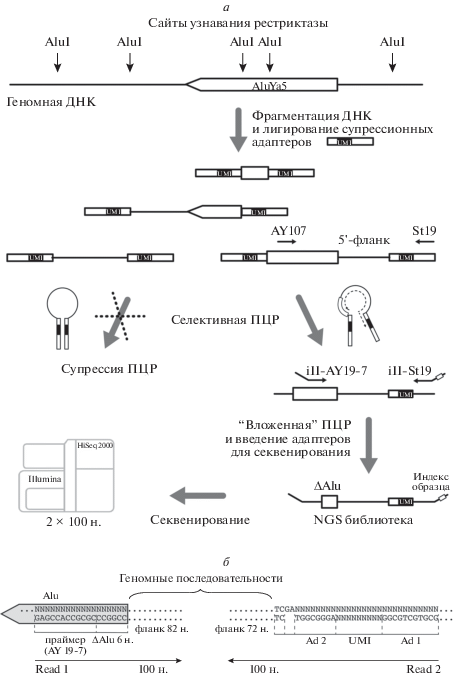
Таблица 1.
Описание использованных данных секвенирования и искусственного вложенного контроля
| Название | Описание | Число ридов | Число реальных ридов после обработки | Число тестовых ридов после обработки | Число предположи-тельно соматических AluYa5 |
|---|---|---|---|---|---|
| Test 1 solo | Искусственный | 1000 | 000000 | 772 | |
| Test 2 solo | Искусственный | 1000 | 000000 | 787 | |
| Test 3 solo | Искусственный | 1000 | 000000 | 782 | |
| Test 4 solo | Искусственный | 1000 | 000000 | 795 | |
| Test 5 solo | Искусственный | 1000 | 000000 | 801 | |
| DGPlus* + test 1 | Реальный + искусственный | 12 148 950 + 1000 | 559 165 | 770 | 197 |
| DGMinus* + test 1 | Реальный + искусственный | 9 014 228 + 1000 | 457 559 | 770 | 137 |
| CorPlus* + test 1 | Реальный + искусственный | 8 208 511 + 1000 | 401 480 (129) | 770 | 129 |
| CorMinus* + test 1 | Реальный + искусственный | 11 775 974 + 1000 | 438 219 (280) | 770 | 280 |
| DGPlus*+ test 2 | Реальный + искусственный | 12 148 950 + 1000 | 559 164 (197) | 787 | 197 |
| DGMinus* + test 3 | Реальный + искусственный | 9 014 228 + 1000 | 457 560 (137) | 779 | 137 |
| CorPlus* + test 4 | Реальный + искусственный | 8 208 511 + 1000 | 401 482 | 793 | 129 |
| CorMinus* + test 5 | Реальный + искусственный | 11 775 974 + 1000 | 438 218 | 799 | 280 |
Read 1 содержит последовательность праймера, использованного во втором раунде амплификации (AY19-7 GAGCCACCGCGC), небольшой фрагмент начала инсерции Alu (ΔAlu 6 н., консенсус GGCCGG) и 82 н. геномного 5'-фланка инсерции (рис. 1б). Read 2 содержит два фиксированных фрагмента адаптера: Ad1 (AGGGCGGT) и Ad2 (GCGTGCTGCGG), – разделенных случайной последовательностью UMI длиной 9 н.
Описание алгоритма анализа данных секвенирования фланков инсерций Alu. Алгоритм реализован на языке Python в виде последовательности операций в программной оболочке Jupyter Notebook с использованием дополнительных консольных программ (bowtie2, bwa). Исходные коды написанных программ доступны в депозитарии Github (https://github.com/labcfg/retropipeline).
Разработанный алгоритм включает следующие стадии (рис. 2):
Рис. 2.
Последовательность этапов анализа данных высокопроизводительного секвенирования библиотек геномных фрагментов, фланкирующих инсерции AluYa5.
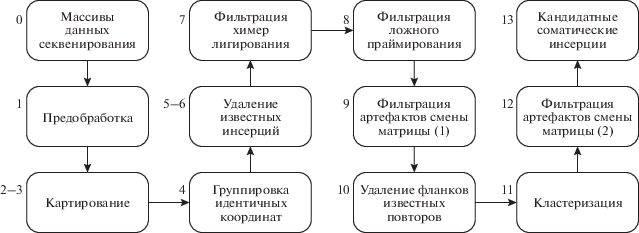
1. В данных секвенирования находятся и удаляются последовательности праймера и адаптеров. Сиквенсы, не содержащие таких последовательностей, удаляются из дальнейшего анализа. Кроме того, проводится поиск UMI и первых 6 н. последовательности Alu, не входящих в состав праймера для амплификации. Найденные последовательности сохраняются в отдельном файле. На этом же этапе удаляются сиквенсы, содержащие сайт рестрикции Alu I (AGCT).
2. Обработанные таким образом данные секвенирования картируются на референсный геном человека (hg38) при помощи программы bowtie2 [18] с параметрами выравнивания для парных прочтений (Read 1 и Read 2) и геномным расстоянием между ними до 1000 н.
3. Последовательности, картировавшиеся некорректно, удаляются, а оставшиеся конвертируются в таблицу координат предполагаемых инсерций. К некорректно картировавшимся последовательностям относятся пары сиквенсовых ридов, картирующиеся дискордантно (на разные хромосомы или на одну и ту же хромосому на расстоянии больше 1000 н. друг от друга), а также пары ридов, взаимное направление картирования которых на хромосоме отличается от ожидаемого.
4. Полностью совпадающие координаты группируются с учетом направления предполагаемой инсерции. Полученный список координат предполагаемых инсерций формирует список А; при этом в качестве дополнительной информации для каждой координаты записываются те последовательности из группы, которые картировались с наилучшим качеством (имеют наименьшее число отличий от соответствующей геномной последовательности). Если таких последовательностей много, то выбирается одна из них случайным образом. В качестве последовательности ΔAlu используется наиболее часто встречаемая последовательность из группы.
5. Координаты фиксированных и полиморфных инсерций Alu, аннотированных в геномной сборке hg38, пересекаются с окном +/–1 нуклеотид с координатами из списка А. Координаты фиксированных и полиморфных инсерций, которые пересеклись с координатами из списка А, записываются в отдельный файл для каждой библиотеки (список Б).
6. Координаты известных инсерций из списка Б используются для удаления известных инсерций Alu, а также артефактов, возникающих в результате смены матрицы в ходе ПЦР. Для каждой координаты из списка Б берется интервал 15 н. в сторону предполагаемой инсерции и 0.9 от длины фланка (до позиции ближайшего рестриктного сайта в геноме) в противоположную сторону. Координаты из списка А, попадающие в такие интервалы, удаляются.
7. Удаляются координаты предполагаемых инсерций, вблизи которых (в интервале 3 н. в сторону инсерции и до ближайшего геномного сайта рестрикции, определяемого Read 2) в референсном геноме находится сайт рестрикции.
8. Удаляются координаты предполагаемых инсерций, вблизи которых (20 н. в сторону инсерции) в референсном геноме обнаруживаются последовательности с расстояниями Хэмминга, меньшими или равными 4, от последовательности праймера GAGCCACCGCGC, использованного для второй ПЦР.
9. Удаляются координаты предполагаемых инсерций, вблизи которых (окно в 8 н. в сторону инсерции) в референсном геноме встречаются последовательности с расстояниями Хэмминга, меньшими или равными 2, от последовательности ΔAlu (записанной на этапе 4).
10. Координаты предполагаемых инсерций пересекаются со списком геномных координат повторяющихся последовательностей. Если фланк предполагаемой инсерции перекрывается с каким-либо геномным повтором, то считается значение К = (1 – D/1000) × overlap (где overlap – длина перекрытия в нуклеотидах, а D – дивергенция повтора). Удаляются все координаты, для которых К > 0.8.
11. До этого этапа все библиотеки обрабатываются параллельно и независимо друг от друга. На этапе кластеризации все координаты предполагаемых инсерций из разных библиотек, принадлежащих одному человеку, сортируются по координате, хромосоме и направлению предполагаемой инсерции в порядке возрастания. Затем с окном в 100 нуклеотидов предполагаемые инсерции группируются в кластеры. На этом этапе определяются предполагаемые соматические инсерции – координата которых присутствует в одной библиотеке и отсутствует в других библиотеках того же индивида. Для каждой соматической инсерции производится подсчет уникальных UMI; при этом UMI, отличающиеся друг от друга на 1 замену, учитываются как один UMI.
12. Предполагаемые соматические инсерции подвергаются дополнительной фильтрации от артефактов смены матрицы. Для этого создаются последовательности нуклеотидов, состоящие из праймера AY19-7, ΔAlu и 11 букв фланка из реальных прочтений. Такие последовательности записываются в файл формата FASTQ с качеством 40 (кодируется символом I) и картируются на референсный геном человека с использованием BWA [19]. Удаляются кластеры, для которых такие последовательности картировались на геном без нуклеотидных замен.
Искусственный вложенный контроль. Создание искусственного массива включает следующие стадии:
1. Случайно выбирают координату на хромосоме.
2. При помощи бинарного поиска по базе сайтов рестрикции, находят ближайший рестриктный сайт к выбранной координате (в сторону начала или конца хромосомы, в зависимости от направления имитируемой инсерции AluYa5).
3. Если расстояние между ближайшим рестриктным сайтом и выбранной координатой находится в пределах от 250 до 750 н., что соответствует распределению длин фрагментов в реальных библиотеках, такую координату сохраняют. В противном случае, операцию (1–3) повторяют до тех пор, пока не будет получено 1000 координат.
4. Из референсного генома выписывают 2 последовательности нуклеотидов: одна, длиной 82 н., прилегает к выбранной координате и направлена в сторону ближайшего геномного рестриктного сайта (выбранного в шаге 2); другая, длиной 72 н., прилегает к ближайшему геномному рестриктному сайту и направлена в обратную сторону относительно первой последовательности (рис. 1б).
5. К обеим последовательностям добавляют последовательности праймеров и адаптеров, идентичные олигонуклеотидам, использованным при получении реальных библиотек. К началу первой последовательности добавляют начало консенсусной последовательности AluYa5 (ΔAlu CCGGCC), а затем праймер AY19-7. К началу второй последовательности добавляют первый фрагмент адаптера (AGGGCGGT), случайную последовательность UMI длиной 9 н. и второй фрагмент адаптера (GCGTGCTGCGG).
Из таких парных фрагментов для каждого искусственного массива создали парные файлы в формате FASTQ с качеством чтения каждого нуклеотида 40. Полученные массивы анализировали с использованием разработанного алгоритма как отдельно, так и в смеси с реальными данными сек-венирования (см. табл. 1).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Создание алгоритма анализа данных и поиска соматических инсерций Alu
Для обработки данных секвенирования фланков инсерций Alu в геноме человека и идентификации соматических инсерций разработан специализированный биоинформатический алгоритм (детальное описание приведено в разделе “Методы” и представлено на рис. 2). На первом этапе полученные последовательности, представляющие собой в основном геномные фланки инсерций AluYa5, картируют на геном человека; удаляют координаты последовательностей, картирующихся неоднозначно, и координаты, соответствующие известным (фиксированным и полиморфным) инсерциям Alu, а также многочисленные артефакты, подробное описание и способы фильтрации которых приведены далее. После этого координаты инсерций в разных анализируемых образцах, полученных от одного и того же индивида (в данном случае 4 образца нейронов и глии из коры головного мозга и зубчатой извилины гиппокампа), сравнивают между собой. Если координата присутствует в одной ткани и отсутствует во всех остальных, такая инсерция считается соматической. Для подсчета числа клеток, несущих данную соматическую инсерцию, используют UMI – уникальные молекулярные идентификаторы.
Фильтрация артефактов
В процессе подготовки библиотек к секвенированию исходную геномную ДНК фрагментируют с использованием рестриктазы, после чего к полученным фрагментам лигируют супрессионные адаптеры, которые берут в большом избытке. Несмотря на избыток адаптеров, в небольшой доле случаев происходит лигирование между фрагментами из разных частей генома, что приводит к образованию химерных последовательностей. В большинстве случаев такие химерные последовательности могут быть удалены в ходе картирования, поскольку картируются дискордантно. Однако, если один из фрагментов ДНК содержит копию Alu и небольшой прилегающий геномный участок (0–20 н.), а второй фрагмент принадлежит геномной области, не содержащей инсерцию, получившаяся химера будет картироваться в геномную область второго фрагмента (рис. 3а). Координата такой псевдоинсерции будет ошибочно принята за координату соматической инсерции при сравнении с другими образцами ДНК того же индивида.
Рис. 3.
Механизмы образования химерных последовательностей и определение параметров их фильтрации. а – Образование химерной последовательности на этапе лигирования. R – участок узнавания рестриктазой (AGCT), серым квадратом обозначен 5'-фрагмент Alu и праймер AY19-7. б – Предполагаемый механизм образования химер во время ПЦР со сменой матрицы. Серым обозначен 5'-фрагмент Alu. в – Результаты анализа совпадения 5'-прилегающих фрагментов известных (фиксированных и полиморфных) инсерций AluYa5; н. – нуклеотиды. г – Анализ встречаемости последовательностей, похожих на 5'-начальный фрагмент Alu (GGCCGG), рядом с координатами предположительно соматических инсерций (черные точки) в сравнении со случайно выбранными геномными координатами (серые окружности).
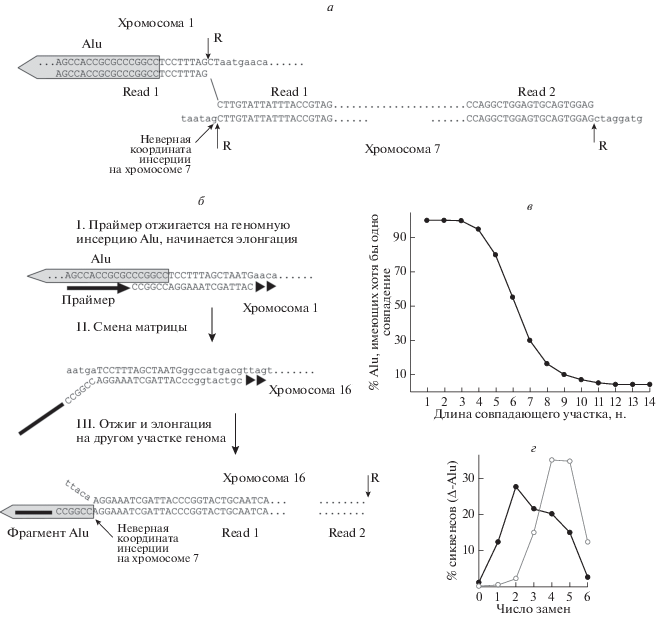
Для удаления таких химерных последовательностей в полученных сиквенсах проводится поиск последовательности рестриктного сайта AGCT. В некоторых случаях из-за ошибок секвенирования и присутствия сайта рестрикции непосредственно в точке предполагаемой инсерции тетрануклеотид AGCT не может быть определен в последовательностях полученных прочтений. Для удаления таких химер проводится поиск сайтов рестрикции также в референсном геноме человека на участке от предполагаемой точки инсерции + 3 н. в сторону инсерции и до точки рестриктного сайта, определяемой Read 2.
Анализ полученных данных секвенирования показал, что в библиотеках присутствуют предположительно соматические инсерции Alu, в которых Read 1 картируется на референсный геном не полностью; при этом небольшая часть Read 1 (как правило, 10–20 н.), прилегающая непосредственно к Alu в полученном сиквенсе, картируется в другое место генома, где присутствует фиксированная или полиморфная инсерция. В отличие от вышеописанных химер лигирования, в данном случае на стыке ΔAlu и фланка не содержится рестриктного сайта или последовательности, на него похожей. Такие химеры могут образоваться в результате смены матрицы в ходе ПЦР. Вероятно, праймер, комплементарный последовательности Alu, отжигается на участок, содержащий фиксированную инсерцию, после чего происходит элонгация. В результате случайного преждевременного обрыва синтеза целевой цепи ДНК образуется очень короткий одноцепочечный ПЦР-продукт, состоящий из фрагмента Alu и 10–30 н. геномной последовательности. Образовавшийся фрагмент отжигается на другой участок ДНК, комплементарный его 3'-концу, и элонгация продолжается уже в другом месте генома. Большинство таких химер может быть удалено при анализе результатов картирования, так как последовательность Read 1 картируется на геном не полностью. Однако в случае полного или почти полного совпадения последовательностей в точке смены матрицы (рис. 3б) этого не удается сделать. Такого рода артефакты можно было бы идентифицировать по совпадению участка полученной последовательности (Read 1), прилегающего к Alu, с любым из участков, прилегающих к фиксированным и полиморфным инсерциям Alu в референсном геноме. Следует заметить, что такой подход имеет определенные ограничения, так как по мере уменьшения длины совпадающего участка возрастает вероятность того, что это совпадение случайно. Для определения минимальной длины совпадения были проанализированы участки, прилегающие к 5'-концу всех известных инсерций AluYa5 в геноме (рис. 3в). Выявлено, что при длине участка менее 11 н. более чем для 5% известных инсерций Alu последовательность прилегающего участка совпадает случайно. Такое значение (≥11 н.) используется в качестве порогового при определении координат, полученных при картировании химерных сиквенсов. Кроме того, при определении вероятности смены матрицы следует учитывать не только последовательность, прилегающую к предполагаемой точке инсерции Alu, но и геномную последовательность непосредственно перед этой точкой, которая может совпадать полностью или частично с последовательностью 5'-начального участка Alu (GGCCGG). В результате анализа полученных данных показано, что последовательности, отличающиеся от GGCCGG 1–2 заменами, встречаются рядом с координатами предположительно соматических инсерций существенно чаще, чем рядом со случайно выбранными геномными координатами (рис. 3г). Проведенный анализ позволяет идентифицировать и исключить из дальнейшего рассмотрения артефакты, образовавшиеся в результате смены матрицы ПЦР. Удаляются координаты предполагаемых инсерций, в непосредственной близости от которых (окно 8 н. в сторону инсерции) находятся гексануклеотиды, отличающиеся от 5'-начального фрагмента Alu 0–2 позициями. Кроме того, для каждой кандидатной соматической инсерции в геноме проводится поиск последовательности, состоящей из праймера AY19-7 (12 н.), начала Alu (6 н.) и прилегающего фланка (11 н.). Координаты, для которых в геноме выявлено полное соответствие с такой последовательностью, исключаются.
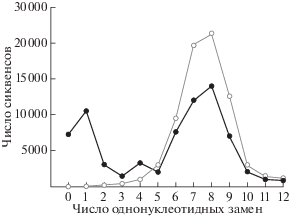
В ходе подготовки библиотек для селективной амплификации используется праймер AY107, отжигающийся на участок Alu c тремя диагностическими позициями, по которым эволюционно молодое семейство AluYa5 отличается от более старых представителей AluY. Ранее показана [11, 14] высокая селективность такого праймера, так как до 90% сиквенсов составляют целевые последовательности – фланки AluYa5. Остальные 10% представлены в основном фланками Alu других семейств, причем каждый небольшим числом сиквенсов. Такие последовательности могут быть легко исключены из дальнейшего анализа путем сравнения полученных координат инсерций с геномными координатами фиксированных Alu других семейств. Однако при анализе полученных координат инсерций обнаружено, что в небольшом числе случаев праймер, используемый на второй стадии амплификации, может отжигаться на геномную последовательность, не содержащую Alu. Для удаления таких артефактов проводят сравнение последовательности праймера AY19-7 с геномной последовательностью, расположенной рядом с точкой предполагаемой инсерции. Для определения порогового значения фильтрации таких артефактов проанализированы точки предполагаемых соматических инсерций из реальных данных (после удаления всех известных Alu) и такое же число точек, случайно взятых из генома. Определено минимальное число нуклеотидных отличий (дистанция Хэмминга) от последовательности праймера на участке генома длиной 20 н., который прилегает к предполагаемой точке инсерции (рис. 4). Как видно из рисунка, реальные координаты предполагаемых инсерций существенно отличаются от случайно взятых координат: число сиквенсов, отличающихся от праймера на 4 нуклеотидных замены и меньше в случайно выбранных геномных участках, существенно ниже, чем для реальных данных секвенирования. Из этого следует, что координаты предположительно соматических инсерций, рядом с которыми в геноме находится последовательность, отличающаяся от праймера на 4 н. и меньше, могут быть артефактами неправильного праймирования.
Создание искусственного вложенного контроля
При фильтрации артефактов пробоподготовки необходимо соблюдать определенный баланс. С одной стороны, биоинформатические фильтры должны эффективно удалять артефакты пробоподготовки, имитирующие настоящие соматические инсерции, с другой стороны, они не должны удалять существенную часть реальных соматических инсерций. Для нахождения такого баланса необходимо тестирование пороговых значений фильтрации с использованием сиквенсов с известными характеристиками (положение в геноме, присутствие или отсутствие в анализируемом образце и т.д.). Для проверки работы биоинформатического алгоритма были созданы пять искусственных массивов данных, имитирующих реальные соматические инсерции Alu. Для этого в геноме человека выбрали 1000 случайных координат. Каждая такая координата имитировала точку инсерции Alu. Далее определяли ближайший геномный сайт узнавания эндонуклеазой рестрикции AluI, использованной для приготовления реальных библиотек. Участок от координаты инсерции до сайта рестрикции, представляющий собой фланк, использовали для создания искусственных сиквенсов в формате FASTQ, содержащих точно такие же элементы, как и реальные данные, полученные в результате секвенирования библиотек 5'-фланков Alu.
Тестирование предлагаемого алгоритма с использованием виртуального вложенного контроля
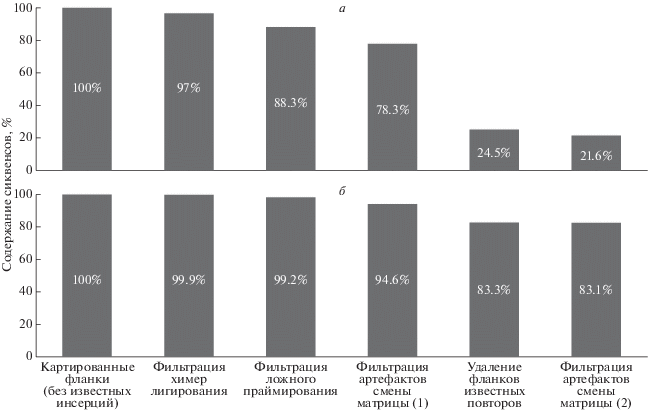
Описанный алгоритм обработки данных секвенирования, фильтрации артефактов и поиска соматических инсерций РЭ протестирован на различных массивах сиквенсовых данных (табл. 1). Искусственные массивы анализировали как по отдельности, так и в смеси с реальными данными секвенирования фланков AluYa5 (табл. 1). На рис. 5 показанo усредненное процентное содержание сиквенсов, удаляемых разработанными фильтрами из искусственных и реальных данных (при анализе в смеси), на каждом этапе фильтрации артефактов. Как видно из рисунка, более 80% сиквенсов искусственного массива остается в данных после фильтрации артефактов, а причиной удаления сиквенсов чаще всего было наличие в них повторов с низкой степенью дивергенции от консенсуса. В то же время из реальных данных, при использовании заданных параметров на всех этапах, суммарно удалено около 75% сиквенсов.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Развитие методов массированного секвенирования вместе с беспрецедентным повышением производительности и чувствительности методов анализа вариабельности геномов привело к обострению проблемы зашумленности данных ложноположительными сигналами. Особенно это актуально в случае поиска и анализа редких аллельных вариантов. Однонуклеотидные ошибки, возникающие в ходе ПЦР [20–22], маскируют точечные мутации. Наряду с процессом лигирования, смена матрицы при ПЦР может приводить к образованию химерных молекул, имитирующих рекомбинационные события [23], включая транслокации [24] и новые аллели HLA [25]. Также описаны механизмы и частоты возникновения химерных структур при полногеномной амплификации [26] и однонуклеотидных ошибок непосредственно при секвенировании [27]. Часть подобных артефактов, прежде всего точечные замены, с высокой степенью надежности может быть удалена при помощи коррекции с использованием уникальных молекулярных идентификаторов [28]. Однако универсальных инструментов для селективной детекции и удаления остальных артефактов (химерных молекул) на сегодняшний день не существует.
Нами проведен всесторонний анализ всех возможных типов артефактов, имитирующих инсерции РЭ и возникающих в процессе получения библиотек для массированного секвенирования. На основе модели случайного распределения сайтов вновь возникающих инсерций РЭ в геноме человека и учета особенностей фланков существующих эволюционно недавних инсерций РЭ, нами выявлены, идентифицированы и формализованы характерные отличия ложных инсерций от истинных. Выявленные отличия легли в основу разработанного в данном исследовании биоинформатического алгоритма удаления артефактов при идентификации соматических инсерций РЭ в геноме человека.
Разработанный алгоритм можно считать первым подобным инструментом, который позволяет в автоматическом режиме удалять подавляющее большинство как “явных”, так и “скрытых” артефактов при минимальных потерях целевых соматических инсерций.
Применение разработанного алгоритма в сочетании с существующими биоинформатическими методами идентификации соматических инсерций РЭ в полных геномах позволит существенно снизить уровень зашумленности данных ложноположительными результатами и повысить тем самым чувствительность этих методов, прежде всего при исследовании дифференциальной ретропозиционной активности в разных типах тканей и клеток, включая опухолевые и нормальные клетки.
Таким образом, применение предлагаемого алгоритма позволит многократно повысить производительность как биоинформатических, так и экспериментальных подходов идентификации соматических инсерций РЭ.
В данном исследовании внимание было сосредоточено на выявлении и фильтрации ложных инсерций Alu, однако принципиальная схема разработанного алгоритма в полной мере может быть использована для создания алгоритмов фильтрации артефактов при идентификации соматических инсерций и других семейств РЭ человека, включая L1 и SVA, а также инсерций мобильных элементов, специфичных для других организмов.
Исследование поддержано грантом РНФ 18-14-00244.
Список литературы
Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., Kann L., Lehoczky J., LeVine R., McEwan P., McKernan K., Meldrim J., Mesirov J.P., Miranda C., Morris W., Naylor J., Raymond C., Rosetti M., Santos R., Sheridan A., Sougnez C., Stange-Thomann Y., Stojanovic N., Subramanian A., Wyman D., Rogers J., Sulston J., Ainscough R., Beck S., Bentley D., Burton J., Clee C., Carter N., Coulson A., Deadman R., Deloukas P., Dunham A., Dunham I., Durbin R., French L., Grafham D., Gregory S., Hubbard T., Humphray S., Hunt A., Jones M., Lloyd C., McMurray A., Matthews L., Mercer S., Milne S., Mullikin J.C., Mungall A., Plumb R., Ross M., Shownkeen R., Sims S., Waterston R.H., Wilson R.K., Hillier L.W., McPherson J.D., Marra M.A., Mardis E.R., Fulton L.A., Chinwalla A.T., Pepin K.H., Gish W.R., Chissoe S.L., Wendl M.C., Delehaunty K.D., Miner T.L., Delehaunty A., Kramer J.B., Cook L.L., Fulton R.S., Johnson D.L., Minx P.J., Clifton S.W., Hawkins T., Branscomb E., Predki P., Richardson P., Wenning S., Slezak T., Doggett N., Cheng J.F., Olsen A., Lucas S., Elkin C., Uberbacher E., Frazier M., Gibbs R.A., Muzny D.M., Scherer S.E., Bouck J.B., Sodergren E.J., Worley K.C., Rives C.M., Gorrell J.H., Metzker M.L., Naylor S.L., Kucherlapati R.S., Nelson D.L., Weinstock G.M., Sakaki Y., Fujiyama A., Hattori M., Yada T., Toyoda A., Itoh T., Kawagoe C., Watanabe H., Totoki Y., Taylor T., Weissenbach J., Heilig R., Saurin W., Artiguenave F., Brottier P., Bruls T., Pelletier E., Robert C., Wincker P., Smith D.R., Doucette-Stamm L., Rubenfield M., Weinstock K., Lee H.M., Dubois J., Rosenthal A., Platzer M., Nyakatura G., Taudien S., Rump A., Yang H., Yu J., Wang J., Huang G., Gu J., Hood L., Rowen L., Madan A., Qin S., Davis R.W., Federspiel N.A., Abola A.P., Proctor M.J., Myers R.M., Schmutz J., Dickson M., Grimwood J., Cox D.R., Olson M.V., Kaul R., Raymond C., Shimizu N., Kawasaki K., Minoshima S., Evans G.A., Athanasiou M., Schultz R., Roe B.A., Chen F., Pan H., Ramser J., Lehrach H., Reinhardt R., McCombie W.R., de la Bastide M., Dedhia N., Blocker H., Hornischer K., Nordsiek G., Agarwala R., Aravind L., Bailey J.A., Bateman A., Batzoglou S., Birney E., Bork P., Brown D.G., Burge C.B., Cerutti L., Chen H.C., Church D., Clamp M., Copley R.R., Doerks T., Eddy S.R., Eichler E.E., Furey T.S., Galagan J., Gilbert J.G., Harmon C., Hayashizaki Y., Haussler D., Hermjakob H., Hokamp K., Jang W., Johnson L.S., Jones T.A., Kasif S., Kaspryzk A., Kennedy S., Kent W.J., Kitts P., Koonin E.V., Korf I., Kulp D., Lancet D., Lowe T.M., McLysaght A., Mikkelsen T., Moran J.V., Mulder N., Pollara V.J., Ponting C.P., Schuler G., Schultz J., Slater G., Smit A.F., Stupka E., Szustakowki J., Thierry-Mieg D., Thierry-Mieg J., Wagner L., Wallis J., Wheeler R., Williams A., Wolf Y.I., Wolfe K.H., Yang S.P., Yeh R.F., Collins F., Guyer M.S., Peterson J., Felsenfeld A., Wetterstrand K.A., Patrinos A., Morgan M.J., de Jong P., Catanese J.J., Osoegawa K., Shizuya H., Choi S., Chen Y.J., Szustakowki J. (2001) Initial sequencing and analysis of the human genome. Nature. 409, 860–921.
Konkel M.K., Walker J.A., Hotard A.B., Ranck M.C., Fontenot C.C., Storer J., Stewart C., Marth G.T., Batzer M.A. (2015) Sequence analysis and characterization of active human alu subfamilies based on the 1000 genomes pilot project. Genome Biol. Evol. 7, 2608–2622.
Brouha B., Schustak J., Badge R.M., Lutz-Prigge S., Farley A.H., Moran J.V., Kazazian H.H., Jr. (2003) Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA. 100, 5280–5285.
Xing J., Zhang Y., Han K., Salem A.H., Sen S.K., Huff C.D., Zhou Q., Kirkness E.F., Levy S., Batzer M.A., Jorde L.B. (2009) Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 19, 1516–1526.
Cordaux R., Hedges D.J., Herke S.W., Batzer M.A. (2006) Estimating the retrotransposition rate of human Alu elements. Gene. 373, 134–137.
Hancks D.C., Kazazian H.H., Jr. (2016) Roles for retrotransposon insertions in human disease. Mob. DNA. 7, 9.
Burns K.H. (2017) Transposable elements in cancer. Nat. Rev. Cancer. 17, 415–424.
Scott E.C., Gardner E.J., Masood A., Chuang N.T., Vertino P.M., Devine S.E. (2016) A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res. 26, 745–755.
Tubio J.M., Li Y., Ju Y.S., Martincorena I., Cooke S.L., Tojo M., Gundem G., Pipinikas C.P., Zamora J., Raine K., Menzies A., Roman-Garcia P., Fullam A., Gerstung M., Shlien A., Tarpey P.S., Papaemmanuil E., Knappskog S., Van Loo P., Ramakrishna M., Davies H.R., Marshall J., Wedge D.C., Teague J.W., Butler A.P., Nik-Zainal S., Alexandrov L., Behjati S., Yates L.R., Bolli N., Mudie L., Hardy C., Martin S., McLaren S., O’Meara S., Anderson E., Maddison M., Gamble S., Foster C., Warren A.Y., Whitaker H., Brewer D., Eeles R., Cooper C., Neal D., Lynch A.G., Visakorpi T., Isaacs W.B., van’t Veer L., Caldas C., Desmedt C., Sotiriou C., Aparicio S., Foekens J.A., Eyfjord J.E., Lakhani S.R., Thomas G., Myklebost O., Span P.N., Borresen-Dale A.L., Richardson A.L., Van de Vijver M., Vincent-Salomon A., Van den Eynden G.G., Flanagan A.M., Futreal P.A., Janes S.M., Bova G.S., Stratton M.R., McDermott U., Campbell P.J. (2014) Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 345, 1251343.
Erwin J.A., Marchetto M.C., Gage F.H. (2014) Mobile DNA elements in the generation of diversity and complexity in the brain. Nat. Rev. Neurosci. 15, 497–506.
Mamedov I.Z., Arzumanyan E.S., Amosova A.L., Lebedev Y.B., Sverdlov E.D. (2005) Whole-genome experimental identification of insertion/deletion polymorphisms of interspersed repeats by a new general approach. Nucleic Acids Res. 33, e16.
Tang Z., Steranka J.P., Ma S., Grivainis M., Rodic N., Huang C.R., Shih I.M., Wang T.L., Boeke J.D., Fenyo D., Burns K.H. (2017) Human transposon insertion profiling: analysis, visualization and identification of somatic LINE-1 insertions in ovarian cancer. Proc. Natl. Acad. Sci. USA. 114, E733–E740.
Evrony G.D., Lee E., Park P.J., Walsh C.A. (2016) Resolving rates of mutation in the brain using single-neuron genomics. eLife. 5, e12966.
Kurnosov A.A., Ustyugova S.V., Nazarov V.I., Minervina A.A., Komkov A.Y., Shugay M., Pogorelyy M.V., Khodosevich K.V., Mamedov I.Z., Lebedev Y.B. (2015) The evidence for increased l1 activity in the site of human adult brain neurogenesis. PLoS One. 10, e0117854.
Курносов А.А., Устюгова С.В., Погорелый М.В., Комков А.Ю., Болотин Д.А., Ходосевич К.В., Мамедов И.З., Лебедев Ю.Б. (2013) Стратегия поиска соматических инсерций ретроэлементов в геноме человека. Биоорган. химия. 39(4), 466–476.
Matevossian A., Akbarian S. (2008) Neuronal nuclei isolation from human postmortem brain tissue. J. Vis. Exp. 20, 914.
Kivioja T., Vaharautio A., Karlsson K., Bonke M., Enge M., Linnarsson S., Taipale J. (2012) Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods. 9, 72–74.
Langmead B., Salzberg S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359.
Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 25, 1754–1760.
Arbeithuber B., Makova K.D., Tiemann-Boege I. (2016) Artifactual mutations resulting from DNA lesions limit detection levels in ultrasensitive sequencing applications. DNA Res. 23, 547–559.
Cline J., Braman J.C., Hogrefe H.H. (1996) PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 24, 3546–3551.
Costello M., Pugh T.J., Fennell T.J., Stewart C., Lichtenstein L., Meldrim J.C., Fostel J.L., Friedrich D.C., Perrin D., Dionne D., Kim S., Gabriel S.B., Lander E.S., Fisher S., Getz G. (2013) Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67.
Won M., Dawid I.B. (2017) PCR artifact in testing for homologous recombination in genomic editing in zebrafish. PLoS One. 12, e0172802.
Parker W.T., Phillis S.R., Yeung D.T., Hughes T.P., Scott H.S., Branford S. (2014) Many BCR-ABL1 compound mutations reported in chronic myeloid leukemia patients may actually be artifacts due to PCR-mediated recombination. Blood. 124, 153–155.
McDevitt S.L., Bredeson J.V., Roy S.W., Lane J.A., Noble J.A. (2016) Hapcad: an open-source tool to detect PCR crossovers in next-generation sequencing generated HLA data. Hum. Immunol. 77, 257–263.
Lasken R.S., Stockwell T.B. (2007) Mechanism of chimera formation during the multiple displacement amplification reaction. BMC Biotechnol. 7, 19.
Schirmer M., Ijaz U.Z., D’Amore R., Hall N., Slo-an W.T., Quince C. (2015) Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 43, e37.
Shugay M., Britanova O.V., Merzlyak E.M., Turchaninova M.A., Mamedov I.Z., Tuganbaev T.R., Bolotin D.A., Staroverov D.B., Putintseva E.V., Plevova K., Linnemann C., Shagin D., Pospisilova S., Lukyanov S., Schumacher T.N., Chudakov D.M. (2014) Towards error-free profiling of immune repertoires. Nat. Methods. 11, 653–655.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология