

Молекулярная биология, 2019, T. 53, № 1, стр. 28-36
Возможная роль генов, связанных с лизосомными болезнями накопления, в патогенезе болезни Паркинсона
М. М. Руденок a, *, А. Х. Алиева a, М. А. Николаев b, А. А. Колачева c, М. В. Угрюмов c, С. Н. Пчелина b, П. А. Сломинский a, М. И. Шадрина a
a Институт молекулярной генетики Российской академии наук
123182 Москва, Россия
b Санкт-Петербургский национальный исследовательский Академический университет
Российской академии наук
194021 Санкт-Петербург, Россия
c Институт биологии развития им. Н.К. Кольцова Российской академии наук
119334 Москва, Россия
* E-mail: margaritamrudenok@gmail.com
Поступила в редакцию 30.05.2018
После доработки 18.06.2018
Принята к публикации 27.06.2018
Аннотация
Болезнь Паркинсона ‒ одно из самых распространенных нейродегенеративных заболеваний, характеризуется медленной и непрерывной дегенерацией дофаминергических нейронов компактной части черной субстанции мозга. Этиопатогенез болезни Паркинсона, несмотря на его активное изучение, остается не до конца выясненным. В настоящее время показано, что в патогенез болезни Паркинсона вовлечено нарушение процессов лизосомной аутофагии, которое приводит к возникновению ряда лизосомных болезней накопления. Нами проведен отбор генов, связанных с функционированием лизосом, проанализированы изменения экспрессии генов Hspa8, Lamp2, Tfam, Slc18a2 и Vps35 в тканях мозга мышей, которым с целью моделирования самых ранних стадий болезни Паркинсона вводили МФТП (1-метил-4-фенил-1,2,3,6-тетрагидропиридин). Выявлено снижение относительных уровней мРНК генов Hspa8 и Lamp2, что может указывать на связь нарушений лизосомной аутофагии с ранними этапами патогенеза болезни Паркинсона, а снижение интенсивности лизосомной аутофагии может влиять на накопление поврежденных белков и формирование белковых включений при этом заболевании. Полученные результаты позволяют говорить о том, что гены, связанные с функционированием лизосом, могут быть вовлечены в развитие как лизосомных болезней накопления, так и болезни Паркинсона, причем на самых ранних этапах патогенеза.
ВВЕДЕНИЕ
Болезнь Паркинсона (БП), которая входит в число самых распространенных нейродегенеративных заболеваний, характеризуется дегенерацией дофаминергических нейронов (ДА-нейронов) компактной части черной субстанции среднего мозга [1‒5]. Важной особенностью патогенеза БП является хроническое и медленно прогрессирующее течение заболевания. C момента запуска процессов нейродегенерации до появления первых классических двигательных симптомов патологии, по которым в настоящее время ставится диагноз БП, может пройти до 20 лет. Характерные для БП моторные нарушения (ригидность, тремор покоя, постуральная неустойчивость и гипокинезия) проявляются только при гибели примерно 50‒60% ДА-нейронов в черной субстанции и снижении содержания дофамина (ДА) в стриатуме на 70‒80% [6‒8].
Изучение моногенных и спорадических случаев БП позволило выявить большое число различных локусов и генов, ассоциированных с развитием этого заболевания. Исходя из функций выявленных генов, можно сделать вывод, что важную роль в патогенезе БП могут играть нарушения нормального функционирования митохондрий, убиквитинзависимого протеолиза и везикулярного транспорта [9, 10]. Однако несмотря на эти успехи, картина этиопатогенеза БП остается не до конца выясненной и, что не менее важно, молекулярно-генетические механизмы, инициирующие развитие патологического процесса, не установлены.
Известно, что в патогенезе БП может принимать участие лизосомная аутофагия [11] ‒ неспецифический процесс биодеградации крупных белковых комплексов и целых фрагментов клеточных мембран, при этом формируется аутофаголизосома, в которой и происходит деградация частиц. На вовлечение изменений лизосомной аутофагии при БП указывают мутации в генах ATP13A2 и GBA, ассоциированных с БП [12‒14]. Мутации в гене ATP13A2 приводят к развитию атипичной рецессивной формы паркинсонизма с очень ранним началом [15‒17]. Мутации в гене глюкоцереброзидазы (GBA) в гетерозиготном состоянии повышают риск развития БП в 7‒8 раз в различных популяциях, в том числе в России [13, 18, 19]. В гомозиготном состоянии мутации в гене GBA вызывают развитие болезни Гоше (БГ), наиболее распространенного заболевания, относящегося к классу лизосомных болезней накопления (ЛБН) [14, 20, 21].
Под ЛБН подразумевают целый ряд заболеваний, при которых нарушаются функции лизосом и, как следствие, накапливаются белки, не подвергшиеся деградации [22]. Стоит отметить, что при двух ЛБН наблюдается симптоматика, характерная для БП: при БГ ‒ мышечный гипертонус и нарушения мелкой моторики [23], а при болезни Ниманна‒Пика ‒ тремор покоя [24, 25]. Это, по всей видимости, объясняется тем, что в патогенез данных болезней вовлечены те же структуры мозга, что и в патогенез БП, такие как стриатум, кора больших полушарий, таламус и гиппокамп [23, 26‒28].
Необходимо отметить, что роль генов, принимающих участие в функционировании лизосом и при этом вовлеченных в патогенез как ЛБН, так и БП, известна не до конца. В связи с этим нами изучены изменения экспрессии нескольких таких генов. Проанализированы полнотранскриптомные данные, полученные при исследовании тканей мозга мышей, у которых с помощью МФТП индуцировали проявления ранних стадий БП (МФТП-модель) [29]. Определены также изменения относительных уровней мРНК отобранных генов в черной субстанции и стриатуме мозга таких мышей.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Модели болезни Паркинсона. Нами использованы модели досимптомной стадии БП с декапитацией мышей через 6 ч (6ч-ДСС), досимптомной стадии с декапитацией через 24 ч (24ч-ДСС), с поздней досимптомной (ПДСС) и ранней симптомной (РСС) стадиями БП. Модели созданы, как описано ранее [30, 31]. Экспериментальная работа с лабораторными животными проведена в соответствии “The Guide for the Care and Use of Laboratory Animals” и одобрена этическим комитетом Института биологии развития им. Н.К. Кольцова РАН.
Для проведения экспрессионного анализа были сформированы по четыре контрольных и экспериментальных группы мышей-самцов линии C57BL/6 (по 10 животных в каждой группе). Мышей содержали в виварии со свободным доступом к пище и воде с естественной сменой освещения. Для исследования отбирали по 5‒7 мг ткани стриатума и 1‒2 мг черной субстанции каждого животного.
Выделение РНК. Суммарную РНК выделяли из образцов ткани каждого животного с использованием набора RNAeasy Mini Kit (“Qiagen”, Германия) и Quick-RNA MiniPrep Kit™ (“Zymo Research Corp.”, США) согласно рекомендациям производителей. Концентрацию суммарной РНК измеряли с использованием набора Quant-iT RNA BR Assay Kit и флуориметра Qubit 3.0 (“Invitrogen”, США).
Качество РНК оценивали при помощи набора Experion RNA HighSens Analysis Kit и прибора Experion (“Bio-Rad”, США).
Экспрессионный анализ генов-кандидатов. Изменения экспрессии генов анализировали с использованием реакции обратной транскрипции и ПЦР в реальном времени с TaqMan-зондами.
Реакцию обратной транскрипции проводили на амплификаторе T3 Thermocycler (T3 Thermoblock, “Biometra”, Германия) с использованием набора RevertAid™ H Minus Reverse Transcriptase kit (“Thermo Fisher Scientific”, США) согласно рекомендациям производителя. Использовали смеси праймеров Random Hexamer Primer (“Thermo Fisher Scientific”) и Oligo(dT)18 (“Thermo Fisher Scientific”) в соотношении 3 : 2 соответственно.
Праймеры и зонды для ПЦР в реальном времени подбирали с помощью программы Beacon designer 7.0 и нуклеотидных последовательностей генов Tfam, Hspa8, Lamp2, Slc18a2, Vps35, а также генов домашнего хозяйства Bcat2 и Psmd7. Последовательности ген-специфичных праймеров и зондов представлены в табл. 1.
Таблица 1.
Последовательности ген-специфичных праймеров и зондов
| Ген | Нуклеотидная последовательность |
|---|---|
| Tfam NM_009360.4* |
Зонд: 5'-VIC-CCCTGAGCCGAATCATCCTTTGCCTCC-BHQ2-3' Прямой праймер: 5'-AGACCTCGTTCAGCATATAACATTTATG-3' Обратный праймер: 5'-CCAAGCCTCATTTACAAGCTTCA-3' |
| Hspa8 NM_031165.4 |
Зонд: 5'-VIC-TTCACCACCTACTCTGACAACCAGCCTG-BHQ2-3' Прямой праймер: 5'-CCACCAAGCAGACACAGAC-3' Обратный праймер: 5'-AGCAGGTTGTTGTCCTTGG-3' |
| Lamp2 NM_001290485.2 |
Зонд: 5'-VIC-AGGCACCTTCTCCTCAGTGATGTTCAGC-BHQ2-3' Прямой праймер: 5'-ACCTGTCTGCTGGCTACC-3' Обратный праймер: 5'-GGTTGTGGCAGGGTTGATG-3' |
| Slca18a2 NM_172523.3 |
Зонд: 5'-VIC-CTGAAGGACCCATACATTCTCATCGCTGC-BHQ2-3' Прямой праймер: 5'-GTCAGAAGGGGACACCTCTTAC-3' Обратный праймер: 5'-ACATGGTCTCCATCATCCAGATG-3' |
| Vps35 NM_022997.4 |
Зонд: 5'-VIC-TCTTCATCTGTTGGCTCTCCTTCATCAGGT-BHQ2-3' Прямой праймер: 5'-CTTCGAAATTACCTTCTTCAGTGTACTAG-3' Обратный праймер: 5'-CCATGGAATCACTGATATCACCAG-3' |
| Bcat2 NM_001243053.1 | Зонд: 5'-FAM-CGGATACACTCCAACAGCTCCTGCTTGT-BHQ1-3' Прямой праймер: 5'-TCAACATGGACAGGATGCTACG-3' Обратный праймер: 5'-CCAGTCTTTGTCTACTTCAATGAGC-3' |
| Psmd7 NM_010817.2 | Зонд: 5'-FAM-AGTCCTAGGTCCTTTGGCTTCACGTCGA-BHQ1-3' Прямой праймер: 5'-CTGCACAAGAATGATATCGCCATC-3' Обратный праймер: 5'-CTCCACTGAGATGTAGGCTTCG-3' |
При проведении ПЦР в реальном времени в качестве матрицы использовали кДНК, полученные в реакции обратной транскрипции. кДНК перед внесением в реакционную смесь разводили в растворе тРНК E. coli (100 нг/мкл) [32] до концентрации 0.02 нг/мкл. ПЦР в реальном времени проводили на приборах StepOnePlus™ System (“Applied Biosystems”, США). В состав 30 мкл реакционной смеси входило: 5 мкл кДНК (0.02 нг/мкл); 3 мкл ПЦР-буфера (×10) (“Синтол”, Россия); 3 мкл 25 мМ MgCl2; 10 пM праймеров (“Евроген”, Россия); 2.5 пМ зонда (“ДНК-синтез”, Россия); 200 мкM каждого dNTP; 1 ед. акт. Hot-Rescue Taq-ДНК-полимеразы (“Синтол”). При проведении амплификации использовали следующий температурный режим: 50°C, 60 с; 95°C, 600 с; далее 40 циклов 95°C, 20 с и 61°C, 50 с. Каждый образец анализировали трижды для коррекции различий в качестве образцов и эффективности реакции обратной транскрипции.
Биоинформатический анализ. Полнотранскриптомные данные, полученные при исследовании тканей черной субстанции и стриатума МФТП-моделей БП, обрабатывали, используя анализ на сверхпредставленность и ресурс Pathway Studio® 12.0 (“Elsevier”, Нидерланды) [33, 34]. Были отобраны кластеры, связанные с функционированием лизосом, при условии pval < 0.001 и поправке на множественное сравнение FDR < 0.05. Далее были сформированы и проанализированы списки генов, входящих в “лизосомные” кластеры в каждом образце. С помощью ресурса Pathway Studio® 12.0 для экспрессионного анализа отобрали гены, которые могут быть вовлечены в патогенез как БП, так и ЛБН.
Относительные уровни экспрессии рассчитывали с использованием метода сравнения пороговых уровней амплификации ΔΔCt [35]. Статистическую обработку данных проводили с использованием пакета программ “Statistica for Windows 8.0” (StatSoft, Inc. (2007), STATISTICA (version 8.0. www.statsoft.com) и программного обеспечения MS Excel 2013 (Microsoft). Oтносительные уровни экспрессии генов оценивали с помощью непараметрического U-теста Манна‒Уитни. Построение сетей взаимодействий генов выполнено с использованием программы Pathway Studio® 12.0 (“Elsevier”) [33, 34].
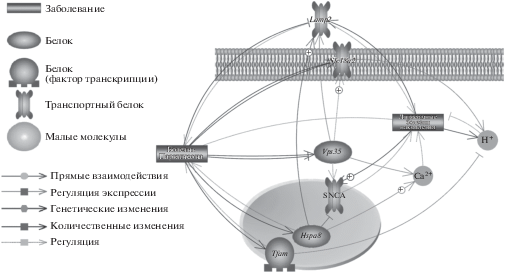
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Нами проведен анализ полнотранскриптомных данных, полученных в результате исследования тканей стриатума и черной субстанции мозга мышей, у которых моделировали ранние стадии БП (МФТП-модели 6ч-ДСС, 24ч-ДСС, ПДСС и РСС БП [29]).
В результате отобрали более 100 генов, которые входили в кластер “лизосома” и были связаны с функционированием лизосом в стриатуме и черной субстанции на всех исследуемых стадиях БП.
Далее отобрали гены, потенциально вовлеченные в патогенез как БП, так и ЛБН (рисунок 1).
Затем был проведен более детальный анализ изменения относительных уровней мРНК генов Tfam, Hspa8, Lamp2, Slc18a2 и Vps35 в тканях стриатума и черной субстанции мозга животных разных групп (моделей 6ч-ДСС, 24ч-ДСС, ПДСС, РСС БП). Результаты экспрессионного анализа генов-кандидатов представлены в табл. 2.
Таблица 2.
Изменение относительных уровней мРНК генов-кандидатов в стриатуме и черной субстанции мозга мышей, у которых смоделированы ранние стадии БП
| Ген | 6ч-ДСС | 24ч-ДСС | ПДСС | РСС | ||||
|---|---|---|---|---|---|---|---|---|
| стриатум | черная субстанция | стриатум | черная субстанция | стриатум | черная субстанция | стриатум | черная субстанция | |
| Tfam | 0.891 0.48‒1.342 |
0.94 0.47–1.43 |
1.16 0.55–1.74 |
1.07 0.54–1.60 |
0.61 0.33–0.92 |
0.78 0.39–1.15 |
0.58 0.28‒0.87 |
0.83 0.43–1.28 |
| Hspa8 | 1.11 0.56‒1.65 |
1.03 0.54–1.55 |
1.63 0.82‒2.41 |
0.98 0.46–1.47 |
0.48 0.24‒0.71 |
0.95 0.49–1.43 |
0.50 0.27‒0.72 |
0.75 0.35–1.13 |
| Lamp2 | 0.69 0.37‒1.04 |
1.18 0.59–1.79 |
0.73 0.39–1.10 |
0.82 0.49–1.33 |
0.64 0.34‒0.96 |
0.92 0.46–1.32 |
0.65 0.36–0.98 |
0.52 0.26–0.74 |
| Slc18a2 | 0.79 0.40–1.16 |
1.52 0.77–2.28 |
0.85 0.43–1.26 |
1.15 0.56–1.73 |
0.88 0.44–1.35 |
0.61 0.33–0.92 |
0.93 0.47–1.32 |
1.05 0.57–1.56 |
| Vps35 | 1.06 0.58‒1.59 |
0.96 0.48–1.42 |
1.09 0.56–1.64 |
0.99 0.50–1.47 |
0.72 0.37–1.08 |
0.86 0.43–1.26 |
0.52 0.26–0.75 |
0.71 0.34–1.02 |
Из представленных данных видно, что экспрессия всех исследуемых генов изменяется статистически значимо на уровне мРНК. Моделирование самых ранних стадий БП выявило повышение относительных уровней мРНК только двух генов: Slc18a2 в черной субстанции мышей (модель 6ч-ДСС) и Hpsa8 в стриатуме (модель 24ч-ДСС). При этом в моделях более развернутых ранних стадий БП выявлено только снижение относительных уровней мРНК всех остальных генов в стриатуме и черной субстанции мозга. Изменения экспрессии генов Hspa8 и Lamp2 были статистически значимыми в трех из восьми образцов, генов Tfam и Slc18a2 – в двух. Значимое изменение уровня мРНК гена Vps35 обнаружено только в стриатуме мышей с ранней симптомной стадией БП (МФТП-моделью РСС).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
На модели БП нами проанализирована экспрессия генов, вовлеченных в функционирование лизосом и ассоциированных с ЛБН, которые могут также участвовать в патогенезе БП.
Эти модели имеют ряд преимуществ. МФТП обладает высокой степенью сродства к транспортеру ДА DAT, благодаря чему он избирательно проникает в ДА-нейроны и вызывает их дегенерацию [36]. Этот токсин проходит через гематоэнцефалический барьер, что позволяет вводить его подкожно, не вызывая повреждений, которыми обычно сопровождается стереотаксическое введение в мозг [37]. МФТП-модели позволяют изучать процессы нейродегенерации на самых ранних этапах патогенеза БП.
Нами выявлено статистически значимое снижение относительных уровней мРНК гена Lamp2 в стриатуме моделей ПДСС и РСС БП и в черной субстанции модели РСС БП (табл. 2). Известно, что белок, кодируемый этим геном, является важным участником шаперонзависимой аутофагии. LAMP2 располагается на мембране лизосомы, он необходим для включения в лизосому белков, которые должны подвергаться аутофагии [38‒40]. В настоящее время белок, кодируемый геном LAMP2, рассматривается в качестве биохимического маркера некоторых ЛБН [41]. Также известно, что LAMP2 может быть вовлечен в патогенез БП. Снижение уровня белка LAMP2 обнаружено в различных тканях пациентов с БП, таких как мозг [42, 43] и лейкоциты периферической крови [44, 45] Выявлена также корреляция падения экспрессии этого белка с накоплением альфа-синуклеина [43].
Нами установлено снижение относительного уровня мРНК Lamp2 в тканях мозга мышей на самых ранних стадиях БП, индуцированных МФТП. Изменения экспрессии гена Lamp2 могут приводить к накоплению белковых агрегатов, в том числе и альфа-синуклеина, при БП. Стоит отметить, что данные о вовлечении LAMP2 на уровне мРНК в патогенез БП на самых ранних этапах получены впервые.
Помимо Lamp2 в наше исследование вошел еще один участник шаперонзависимой аутофагии – Hspa8. Белок теплового шока А8 семейства Hsp70, кодируемый этим геном, является молекулярным шапероном протеасомной деградации белков [46]. HSPA8 обеспечивает доставку белков-мишеней к мембране лизосомы, где они связываются с LAMP2 [47‒49]. Показано, что нарушения в работе белка HSPA8 могут быть связаны с ЛБН [49, 50]. При нейродегенеративных заболеваниях в нервных клетках очень часто происходит накопление недеградированных белков, связанных с нарушением процесса шаперонзависимой аутофагии [51]. Показано повышение экспрессии Hspa8 в стриатуме мышей с 24ч-ДСС БП. Подобные изменения могут быть связаны с участием продукта этого гена в компенсаторных механизмах, в частности, с активацией аутофагии токсических белков, накапливающихся в результате развития окислительного стресса в клетках, в ответ на нейродегенеративные процессы, происходящие на самых ранних этапах патогенеза БП.
Нами выявлено статистически значимое снижение относительных уровней мРНК гена Hspa8 в стриатуме мышей с ПДСС и РСС БП (табл. 2). Стоит отметить, что снижение экспрессии гена Lamp2 в этих образцах согласуется с предположением о функциональной ассоциации белковых продуктов этих генов друг с другом [47‒49].
Обнаруженное нами снижение относительных уровней мРНК генов Hspa8 и Lamp2 в тканях мозга мышей, у которых с помощью токсического воздействия моделировали ранние стадии БП, указывает на вовлечение нарушений процессов лизосомной аутофагии в самые ранние этапы патогенеза БП. Падение экспрессии этих генов в тканях мозга может приводить к снижению интенсивности лизосомной аутофагии, что, в свою очередь, будет влиять на накопление поврежденных белков и формирование белковых включений.
Белок LAMP2, как и HSPA8, взаимодействует с белком VPS35 [52]. Белок VPS35 входит в состав гетеропентамерного комплекса (ретромера) [53], а также участвует в регуляции слияния митохондрий (образование митохондриона) и их фрагментации при помощи белков-регуляторов слияния/деления [54]. Известно, что LAMP2 подвергается убиквитин-протеасомной деградации, при этом VPS35 обеспечивает связывание LAMP2 с убиквитин-Е3-лигазой и дальнейший его протеолиз [52]. Нами выявлено снижение уровня экспрессии гена Vps35 в стриатуме мозга мышей (модель ПДСС БП). Стоит отметить, что мутация p.Asp620Asn в гене VPS35 приводит к развитию семейной формы БП [55]. Кроме того, нокдаун гена Vps35 вызывает дисбаланс лизосомной аутофагии у мышей [56] и у дрозофилы [57]. Также известно, что в составе ретромерного комплекса этот белок взаимодействует с сортилином 1, кодируемым геном SORT1, обеспечивая его ретроградный транспорт к комплексу Гольджи [58]. Показано, что инактивация SORT1 ‒ трансмембранного рецептора некоторых лизосомных белков, приводит к накоплению сфинголипида, маркера ЛБН [59]. VPS35 связывается с рецептором маннозо-6-фосфата CI-MPR, кодируемым геном IGF2R [60]. При недостаточном количестве ретромера в клетке увеличивается поток белка CI-MPR в лизосому, что вызывает ее набухание и потерю функции [61]. Снижение экспрессии гена IGF2R наблюдали также у мышей при генетическом моделировании болезни Помпе, относящейся к ЛБН [62]. Обнаружено накопление и неправильное распределение белка CI-MPR в фибробластах при болезни Помпе [63].
Полученные нами данные указывают на то, что изменение экспрессии гена Vps35 вовлечено в патогенез БП, при этом снижение уровня мРНК этого гена может приводить к снижению интенсивности или даже нарушению лизосомной аутофагии и к последующему накоплению поврежденных белков.
Интересные данные об изменении экспрессии генов Slc18a2 и Tfam в тканях мозга животных получены на МФТП-моделях ранних стадий БП. Известно, что эти гены могут воздействовать на функционирование лизосом через транспорт протонов. Так, нарушение обмена ионов водорода может приводить к развитию ЛБН [64]. Кроме того, дисфункция белка TFAM, одного из активаторов биогенеза митохондрий [65], может быть вовлечена в развитие БП [66, 67]. Нарушение работы TFAM приводит к снижению мембранного потенциала митохондрий, изменению активности всех трех комплексов дыхательной цепи митохондрий в нейронах мышей при генетическом моделировании БГ [68]. К тому же, вследствие делеции Tfam у мышей происходят сбои в функционировании митохондрий, что приводит к наблюдаемому при целом ряде ЛБН образованию избыточного количества сфингомиелина [66]. Нами обнаружено снижение относительной представленности транскрипта Tfam в стриатуме мышей с ПДСС и РСС БП. Скорее всего, такое изменение уровня мРНК Tfam будет приводить к снижению активности биогенеза митохондрий, одного из ключевых нейродегенеративных процессов патогенеза БП на самых ранних его стадиях, а также опосредованно способствует накоплению таких белков, как сфингомиелин, при ЛБН.
Известно, что SLC18A2 это везикулярный переносчик моноаминов, который обеспечивает аккумуляцию моноаминов, в том числе дофамина, в синаптических везикулах [69, 70]. Для функционирования SLC18A2 требуется электрохимический протонный градиент: при доставке моноамина в везикулу в цитоплазму выходят два протона [71]. Показано, что белковые агрегаты, которые могут накапливаться в цитоплазме при некоторых ЛБН, изменяют концентрацию H+ в области лизосом [72]. Нарушение нормального функционирования SLC18A2 влияет на транспорт моноаминов и приводит к их аномальному накоплению в клетках, что способствует окислительному стрессу. Окислительный стресс, в свою очередь, способствует накоплению холестерина, что является причиной некоторых ЛБН [73]. Нами установлено статистически значимое изменение экспрессии гена Slc18a2 в мозге мышей, у которых моделировали БП. Известно, что сверхэкспрессия SLC18A2 увеличивает емкость везикул и их размеров, повышает высвобождение переносчиков, а также улучшает результаты поведенческих тестов в МФТП-моделях БП [74]. Наблюдаемое нами увеличение экспрессии гена Slc18a2 в черной субстанции мышей (модель 6ч-ДСС) указывает на активацию компенсаторных механизмов в ДА-нейронах на самых ранних этапах патогенеза БП. При этом в модели ПДСС БП в черной субстанции снижается относительный уровень мРНК Slc18a2, что указывает на истощение компенсаторных возможностей нервных клеток и усиление процессов нейродегенерации.
Совместный анализ относительных уровней мРНК исследуемых генов позволил выявить повышение относительного уровня экспрессии только двух генов и только в тканях мозга на 6ч-ДСС и 24ч-ДСС БП. Эти гены вовлекаются в патогенез БП на ранних стадиях и могут быть связаны с компенсаторными механизмами. Моделирование более развернутых стадий БП показало снижение представленности транскриптов изучаемых генов. Тенденция к снижению экспрессии генов на более продвинутых стадиях патогенеза БП подтверждает гипотезу, согласно которой на более поздних стадиях БП компенсаторные ресурсы клетки иссякают, и происходит активное развитие нейродегенеративных процессов [29].
Таким образом, полученные нами данные указывают на то, что изменение экспрессии генов, вовлеченных в патогенез ЛБН и функционирование лизосом, может вносить существенный вклад в патогенез БП на самых ранних его этапах.
Авторы данной публикации заявляют об отсутствии конфликта интересов.
Работа выполнена с использованием приборов Центра коллективного пользования ИМГ РАН.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (проект № 17-315-50003 мол_нр) и Российского научного фонда (проект № 16-15-00238). Руденок М.М., Алиева А.Х. и Сломинский П.А. поддержаны Российским научным фондом (проект № 16-15-00238); Николаев М.А. и Шадрина М. И. поддержаны Российским фондом фундаментальных исследований (проект № 17-315-50003 мол_нр).
Список литературы
van de Vijver D.A., Roos R.A., Jansen P.A., Porsius A.J., de Boer A. (2001) Estimation of incidence and prevalence of Parkinson’s disease in the elderly using pharmacy records. Pharmacoepidemiol. Drug Saf. 10(6), 549‒554.
Claveria L.E., Duarte J., Sevillano M.D., Perez-Sempere A., Cabezas C., Rodriguez F., de Pedro-Cuesta J. (2002) Prevalence of Parkinson’s disease in Cantalejo, Spain: a door-to-door survey. Mov. Disord. 17(2), 242‒249.
Morioka S., Sakata K., Yoshida S., Nakai E., Shiba M., Yoshimura N., Hashimoto T. (2002) Incidence of Parkinson disease in Wakayama, Japan. J. Epidemiol. 12(6), 403‒407.
Chan D.K., Cordato D., Karr M., Ong B., Lei H., Liu J., Hung W.T. (2005) Prevalence of Parkinson’s disease in Sydney. Acta Neurol. Scand. 111(1), 7‒11.
Racette B.A., Tabbal S.D., Jennings D., Good L., Perlmutter J.S., Evanoff B. (2005) Prevalence of parkinsonism and relationship to exposure in a large sample of Alabama welders. Neurology. 64(2), 230‒235.
Bernheimer H., Birkmayer W., Hornykiewicz O., Jellinger K., Seitelberger F. (1973) Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 20(4), 415‒455.
Fowler C.J. (2007) Update on the neurology of Parkinson’s disease. Neurourol. Urodyn. 26(1), 103‒109.
Cookson M.R., Hardy J., Lewis P.A. (2008) Genetic neuropathology of Parkinson’s disease. Int. J. Clin. Exp. Pathol. 1(3), 217‒231.
Trinh J., Farrer M. (2013) Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 9(8), 445‒454.
Hunn B.H., Cragg S.J., Bolam J.P., Spillantini M.G., Wade-Martins R. (2015) Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci. 38(3), 178‒188.
Schneider L., Zhang J. (2010) Lysosomal function in macromolecular homeostasis and bioenergetics in Parkinson’s disease. Mol. Neurodegener. 5(14), 1‒13
Park J.S., Koentjoro B., Klein C., Sue C.M. (2018) Single heterozygous ATP13A2 mutations cause cellular dysfunction associated with Parkinson’s disease. Mov. Disord. 33(5), 852‒854.
Sidransky E., Lopez G. (2012) The link between the GBA gene and parkinsonism. Lancet Neurol. 11(11), 986‒998.
Pitcairn C., Wani W.Y., Mazzulli J.R. (2018) Dysregulation of the autophagic-lysosomal pathway in Gaucher and Parkinson’s disease. Neurobiol. Dis. pii S0969-9961(18)30074-3.
Hampshire D.J., Roberts E., Crow Y., Bond J., Mubaidin A., Wriekat A.L., Al-Din A., Woods C.G. (2001) Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J. Med. Genet. 38(10), 680‒682.
Ramirez A., Heimbach A., Gründemann J., Stiller B., Hampshire D., Cid L.P., Goebel I., Mubaidin A.F., Wriekat A.L., Roeper J., Al-Din A., Hillmer A.M., Karsak M., Liss B., Woods C.G., Behrens M.I., Kubisch C. (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38(10), 1184‒1191.
Di Fonzo A., Chien H., Socal M., Giraudo S., Tassorelli C., Iliceto G., Fabbrini G., Marconi R., Fincati E., Abbruzzese G., Marini P., Squitieri F., Horstink M.W., Montagna P., Libera A.D., Stocchi F., Goldwurm S., Ferreira J.J., Meco G., Martignoni E., Lopiano L., Jardim L.B., Oostra B.A., Barbosa E.R., Bonifati V. (2007) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 68(19), 1557‒1562.
Emelyanov A., Boukina T., Yakimovskii A., Usenko T., Drosdova A., Zakharchuk A., Andoskin P., Dubina M., Schwarzman A., Pchelina S. (2012) Glucocerebrosidase gene mutations are associated with Parkinson’s disease in Russia. Mov. Disord. 27(1), 158‒159.
Brockmann, K., Berg D. 2014. The significance of GBA for Parkinson’s disease. J Inherit Metab Dis. 37(4), 643–648.
Dandana A., Ben Khelifa S., Chahed H., Miled A., Ferchichi S. (2016) Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology. 83(1), 13‒23.
Nagral, A. 2014. Gaucher disease. J Clin Exp Hepatol. 4(1), 37‒50.
Parenti G., Andria G., Ballabio A. (2015) Lysosomal storage diseases: from pathophysiology to therapy. Annu. Rev. Med. 66, 471‒486.
Kaye E.M., Ullman M.D., Wilson E.R., Barranger J.A. (1986) Type 2 and type 3 Gaucher disease: A morphological and biochemical study. Ann. Neurol. 120(2), 223‒230.
Henderson L.P., Lin L., Prasad A., Paul C.A., Chang T.Y., Maue R.A. (2000) Embryonic striatal neurons from niemann-pick type C mice exhibit defects in cholesterol metabolism and neurotrophin responsiveness. J. Biol. Chem. 275(26), 20179‒20187.
Josephs K.A., Matsumoto J.Y., Lindor N.M. (2004) Heterozygous Niemann-Pick disease type C presenting with tremor. Neurology. 63(11), 2189‒2190.
Braak H., Ghebremedhin E., Rub U., Bratzke H., Del Tredici K. (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 318(1), 121‒134.
Walterfang M., Patenaude B., Abel L.A., Kluenemann H., Bowman E.A., Fahey M.C., Desmond P., Kelso W., Velakoulis D. (2013) Subcortical volumetric reductions in adult Niemann-Pick disease type C: a cross-sectional study. AJNR Am. J. Neuroradiol. 34(7), 1334‒1340.
Tedeschi G., Bonavita S., Barton N.W., Betolino A., Frank J.A., Patronas N.J., Alger J.R., Schiffmann R. (1998) Proton magnetic resonance spectroscopic imaging in the clinical evaluation of patients with Niemann-Pick type C disease. J. Neurol. Neurosurg. Psychiatry. 65(1), 72‒79.
Alieva A.K., Zyrin V.S., Rudenok M.M., Kolacheva A.A., Shulskaya M.V., Ugryumov M.V., Slominsky P.A., Shadrina M.I. (2018) Whole-transcriptome analysis of mouse models with MPTP-induced Early stages of Parkinson’s disease reveals stage-specific response of transcriptome and a possible role of myelin-linked genes in neurodegeneration. Mol. Neurobiol. 55(9), 7229‒7241.
Колачева А.А., Волина Е.В., Угрюмов М.В. (2014) Дегенерация нигростриатных дофаминергических нейронов на экспериментальной модели ранней клинической стадии болезни Паркинсона. Нейрохимия. 31(3), 225‒235.
Ugrumov M.V., Khaindrava V.G., Kozina E.A., Kucheryanu V.G., Bocharov E.V., Kryzhanovsky G.N., Kudrin V.S., Narkevich V.B., Klodt P.M., Rayevsky K.S., Pronina T.S. (2011) Modeling of presymptomatic and symptomatic stages of parkinsonism in mice. Neuroscience. 181, 175‒188.
Suslov O., Steindler D.A. (2005) PCR inhibition by reverse transcriptase leads to an overestimation of amplification efficiency. Nucl. Acids Res. 33(20), e181.
Hettne K.M., Thompson M., van Haagen H.H., van der Horst E., Kaliyaperumal R., Mina E., Tatum Z., Laros J.F., van Mulligen E.M., Schuemie, M., Aten E., Li T.S., Bruskiewich R., Good B.M., Su A.I., Kors J.A., den Dunnen J., van Ommen G.J., Roos M., t Hoen P.A., Mons B., Schultes, E.A. (2016) The implicitome: A resource for rationalizing gene-disease associations. PLoS One. 11(2), eCollection 2016.
Pathway Studio. Available from: https://mammalcedfx.pathwaystudio.com/login/form.
Livak K.J., Schmittgen T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25(4), 402‒408.
Smeyne R.J., Jackson-Lewis V. (2005) The MPTP model of Parkinson’s disease. Brain Res. Mol. Brain Res. 134(1), 57‒66.
Przedborski S., Jackson-Lewis V., Djaldetti R., Liberatore G., Vila M., Vukosavic S., Almer G. (2000) The parkinsonian toxin MPTP: action and mechanism. Restor. Neurol. Neurosci. 16(2), 135‒142.
Cuervo A.M., Dice J.F. (2000) Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci. 113(24), 4441‒4450.
Bandyopadhyay U., Kaushik S., Varticovski L., Cuervo A.M. (2008) The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 28(18), 5747‒5763.
Hubert V., Peschel A., Langer B., Groger M., Rees A., Kain R. (2016) LAMP-2 is required for incorporating syntaxin-17 into autophagosomes and for their fusion with lysosomes. Biol. Open. 5(10), 1516‒1529.
Hua C.T., Hopwood J.J., Carlsson S.R., Harris R.J., Meikle P.J. (1998) Evaluation of the lysosome-associated membrane protein LAMP-2 as a marker for lysosomal storage disorders. Clin. Chem. 44(10), 2094‒2102.
Alvarez-Erviti L., Rodriguez-Oroz M.C., Cooper J.M., Caballero C., Ferrer I., Obeso J.A., Schapira A.H. (2010) Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 67(12), 1464‒1472.
Murphy K.E. (2015) Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 30(12), 1639‒1647.
Wu G., Wang X., Feng X., Zhang A., Li J., Gu K., Huang J., Pang S., Dong H., Gao H., Yan B1. (2011) Altered expression of autophagic genes in the peripheral leukocytes of patients with sporadic Parkinson’s disease. Brain Res. 1394, 105‒111.
Pang S., Chen D., Zhang A., Qin X., Yan B. (2012) Genetic analysis of the LAMP-2 gene promoter in patients with sporadic Parkinson’s disease. Neurosci. Lett. 526(1), 63‒67.
Stricher F., Macri C., Ruff M., Muller S. (2013) HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy. 9(12), 1937‒1954.
Fujiwara Y., Furuta A., Kikuchi H., Aizawa S., Hatanak Y., Konya C., Uchida K, Yoshimura A., Tamai Y., Wada K., Kabuta T. (2013) Discovery of a novel type of autophagy targeting RNA. Autophagy. 9(3), 403‒409.
Ji M., Lu F., Zhang J., Li H., Cui T., Li Wang X., Tang D., Ji C. (2014) Degradation of AF1Q by chaperone-mediated autophagy. Exp. Cell Res. 327(1), 48‒56.
Catarino S., Pereira P., Girao H. (2017) Molecular control of chaperone-mediated autophagy. Essays Biochem. 61(6), 663‒674.
Ingemann L., Kirkegaard T. (2014) Lysosomal storage diseases and the heat shock response: convergences and therapeutic opportunities. J. Lipid Res. 55(11), 2198‒2210.
Koga H., Cuervo A.M. (2011) Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol. Dis. 43(1), 29‒37.
Yun S.P., Kim H., Ham S., Kwon S.H., Lee G.H., Shin J.H., Lee S.H., Ko H.S., Lee Y. (2017) VPS35 regulates parkin substrate AIMP2 toxicity by facilitating lysosomal clearance of AIMP2. Cell Death Dis. 8(4), eCollection 2017.
Zimprich A., Benet-Pages A., Struhal W., Graf E., Eck S.H., Offman M.N., Haubenberger D., Spielberger S., Schulte E.C., Lichtner P., Rossle S.C., Klopp N., Wolf E., Seppi K., Pirker W., Presslauer S., Mollenhauer B., Katzenschlager R., Foki T., Hotzy C., Reinthaler E., Harutyunyan A., Kralovics R., Peters A., Zimprich F., Brucke T., Poewe W., Auff E., Trenkwalder C., Rost B., Ransmayr G., Winkelmann J., Meitinger T., Strom T.M. (2011) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89(1), 168‒175.
Tang F.L., Liu W., Hu J.X., Erion J.R., Ye J., Mei L., Xiong W.C. (2015) VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 12(10), 1631‒1643.
Struhal W., Presslauer S., Spielberger S., Zimprich A., Auff E., Bruecke T., Poewe W., Ransmayr G., Austrian V.P.S. Investigators Team. (2014) VPS35 Parkinson’s disease phenotype resembles the sporadic disease. J. Neural Transm. (Vienna). 121(7), 755‒759.
Tang F.L., Erion J.R., Tian Y., Liu W., Yin D.M., Ye J., Tang B., Mei L., Xiong W.C. (2015) VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J. Neurosci. 35(29), 10613‒10628.
Miura E., Hasegawa T., Konno M., Suzuki M., Sugeno N., Fujikake N., Geisler S., Tabuchi M., Oshima R., Kikuchi A., Baba T., Wada K., Nagai Y., Takeda A., Aoki M. (2014) VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 71, 1‒13.
Kim E., Lee Y., Lee H.J., Kim J.S., Song B.S., Huh J.W., Lee S.R., Kim S.U., Kim S.H., Hong Y., Shim I., Chang K.T. (2010) Implication of mouse Vps26b-Vps29-Vps35 retromer complex in sortilin trafficking. Biochem. Biophys. Res. Commun. 403(2), 167‒171.
Coutinho M.F., Prata M.J., Alves S. (2012) A shortcut to the lysosome: the mannose-6-phosphate-independent pathway. Mol. Genet. Metab. 107(3), 257‒266.
Damen E., Krieger E., Nielsen J.E., Eygensteyn J., van Leeuwen J.E. (2006) The human Vps29 retromer component is a metallo-phosphoesterase for a cation-independent mannose 6-phosphate receptor substrate peptide. Biochem. J. 398(3), 399‒409.
Arighi C.N., Hartnell L.M., Aguilar R.C., Haft C.R., Bonifacino J.S. (2004) Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J.Cell Biol. 165(1), 123‒133.
Koeberl D.D., Li S., Dai J., Thurberg B.L., Bali D., Kishnani P.S. 2012. Beta2 Agonists enhance the efficacy of simultaneous enzyme replacement therapy in murine Pompe disease. Mol. Genet. Metab. 105(2), 221‒227.
Cardone M., Porto C., Tarallo A., Vicinanza M., Rossi B., Polishchuk E., Donaudy F., Andria G., De Matteis M.A., Parenti G. (2008) Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics. 1(1), 6.
Weinert S., Jabs S., Hohensee S., Chan W.L., Kornak U., Jentsch T.J. (2014) Transport activity and presence of ClC-7/Ostm1 complex account for different cellular functions. EMBO Rep. 15(7), 784‒791.
Gangelhoff T.A., Mungalachetty P.S., Nix J.C., Churchill M.E. (2009) Structural analysis and DNA binding of the HMG domains of the human mitochondrial transcription factor A. Nucl. Acids Res. 37(10), 3153‒3164.
Baixauli F., Acin-Perez R., Villarroya-Beltri C., Mazzeo C., Nunez-Andrade N., Gabande-Rodriguez E., Ledesma M.D., Blazquez A., Martin M.A., Falcon-Perez J.M., Redondo J.M., Enriquez J.A., Mittelbrunn M. (2015) Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 22(3), 485‒498.
Alvarez V., Corao A.I., Sanchez-Ferrero E., De Mena L., Alonso-Montes C., Huerta C., Blazquez M., Ribacoba R., Guisasola L.M., Salvador C., Garcia-Castro M., Coto E. (2008) Mitochondrial transcription factor A (TFAM) gene variation in Parkinson’s disease. Neurosci. Lett. 432(1), 79‒82.
Osellame L.D., Rahim A.A., Hargreaves I.P., Gegg M.E., Richard-Londt A., Brandner S., Waddington S.N., Schapira A.H., Duchen M.R. (2013) Mitochondria and quality control defects in a mouse model of Gaucher disease links to Parkinson’s disease. Cell Metab. 17(6), 941‒953.
Nickell J.R., Culver J.P., Janganati V., Zheng G., Dwoskin L.P., Crooks P.A. (2016) Synthesis and in vitro evaluation of water-soluble 1,4-diphenethylpiperazine analogs as novel inhibitors of the vesicular monoamine transporter-2. Bioorganic Med. Chem. Lett. 26(18), 4441‒4445.
Volz T.J., Hanson G.R., Fleckenstein A.E. (2006) Measurement of kinetically resolved vesicular dopamine uptake and efflux using rotating disk electrode voltammetry. J. Neurosci. Meth. 155(1), 109‒115.
Cliburn R.A., Dunn A.R., Stout K.A., Hoffman C.A., Lohr K.M., Bernstein A.I., Winokur E.J., Burkett J., Schmitz Y., Caudle W.M., Miller G.W. (2017) Immunochemical localization of vesicular monoamine transporter 2 (VMAT2) in mouse brain. J. Chem. Neuroanat. 83‒84, 82‒90.
Poet M., Kornak U., Schweizer M., Zdebik A.A., Scheel O., Hoelter S., Wurst W., Schmitt A., Fuhrmann J.C., Planells-Cases R., Mole S.E., Hubner C.A., Jentsch T.J. (2006) Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc. Natl. Acad. Sci. USA. 103(37), 13854‒13859.
Hu A., Zhao X.T., Tu H., Xiao T., Fu T., Wang Y., Liu Y., Shi X.J., Luo J. (2018) PIP4K2A regulates intracellular cholesterol transport through modulating PI(4,5)P2 homeostasis. J. Lipid Res. 59(3), 507‒514.
Lohr K.M., Bernstein A.I., Stout K.A., Dunn A.R., Lazo C.R., Alter S.P., Wang M., Li Y., Fan X., Hess E.J., Yi H., Vecchio L.M., Goldstein D.S., Guillot T.S., Salahpour A., Miller G.W. (2014) Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc. Natl. Acad. Sci. USA. 111(27), 9977‒9982.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология