

Молекулярная биология, 2019, T. 53, № 1, стр. 132-141
Функция скоринга РНК для прогнозирования третичной структуры РНК на основе многослойных нейронных сетей
Y. Z. Wang a, J. Li a, S. Zhang a, B. Huang a, G. Yao a, J. Zhang a, *
a School of Physics, Collaborative Innovation Center of Advanced Microstructures, and National Laboratory
of Solid State Microstructures, Nanjing University
210093 Nanjing, China
* E-mail: jzhang@nju.edu.cn
Поступила в редакцию 10.01.2018
После доработки 26.04.2018
Принята к публикации 17.05.2018
Аннотация
Чтобы предсказать ab initio третичные структуры РНК, необходима хорошая функция скоринга. В этом исследовании мы изучили возможности применения подхода на основе машинного обучения в качестве функции скоринга. По сравнению с традиционными функциями скоринга данный подход более гибкий в использовании различных функций и, кроме того, он свободен от сложной проблемы выбора эталонного состояния. Построены и обучены две многослойные нейронные сети. В качестве входных данных использованы данные о структуре кандидатной РНК, а на выходе получали оценку подобия, которая выражала сходство кандидата с нативной структурой. Первая сеть работала на крупнозернистом уровне структуры РНК, а вторая – на полноатомном уровне. Мы также создали базу данных РНК и разделили ее на обучающий, валидационный и тестовый наборы, содержащие 322, 70 и 70 РНК соответственно. Путем высокотемпературного молекулярно-динамического моделирования для каждой РНК создано 300 искусственных структур. Сети “натренированы” на специальном обучающем наборе и оптимизированы с помощью стратегии ранней остановки, основанной на исключении из проверочного набора. Затем мы проверили производительность сетей на тестовом наборе. Установлено, что результаты существенно лучше, чем при использовании недавно установленного полноатомного потенциала.
ВВЕДЕНИЕ
РНК – макромолекулы, выполняющие важнейшие и разнообразные биологические функции. Чтобы полностью оценить их функции, часто необходимо знание трехмерных (3D) структур. Хотя экспериментальные данные, полученные методами рентгеновской кристаллографии, ЯМР-спектроскопии и криоэлектронной микроскопии, представляют наиболее надежные источники структурной информации об РНК, такие эксперименты стоят дорого или технически сложны из-за физико-химических особенностей РНК. В результате предсказание строения РНК компьютерными методами – ценный альтернативный источник структурной информации. При исследованиях в этом направлении разработано много подходов, в том числе MC-Fold/MC-Sym [1], NAST [2], RNAbuilder [3], iFoldRNA [4], FARNA/Rosetta [5], ERNA-3D [6], RNA2D3D [7], RNAcomposer [8], 3dRNA [9, 10], созданный нами подход pk3D [11–14]. Осуществлено много превосходных работ по предсказанию вторичной структуры [15], эффективным методам формирования выборок [16, 17], правилам расчета свободной энергии [18–25] или в других тесно связанных областях [26–28]. Некоторые из этих методов представлены также в составе интернет-сервисов, таких как Vfold3D [29], iFoldRNA [4], RNACompposer [8], 3dRNA [9], SimRNAweb [30], MiRDB [28] и т.д. Пожалуйста, обратите внимание, что из-за ограниченности пространства и быстрого развития отрасли это далеко неполный перечень. Можно рекомендовать для дальнейшего чтения, например, один из обзоров литературы [31].
В целом, предсказание ab initio трехмерных структур РНК включает в себя два этапа. Первый состоит в создании структурных кандидатов различными методами, такими как сборка фрагментов [5], выборка методом Монте-Карло [22], последовательный рост Монте-Карло [11, 32, 33] и т.д. Второй этап – оценка этих сгенерированных кандидатов с помощью соответствующей функции свободной энергии или функции скоринга (подсчета баллов). Кандидата с наименьшей свободной энергией, или баллом, считают предсказанной структурой и затем его сравнивают с экспериментальным результатом для оценки эффективности подхода.
Традиционно функцию свободной энергии, или функцию скоринга, строят на основе обратного уравнения Больцмана, которое преобразует вероятности наблюдений в величины свободной энергии. Однако этот подход требует знания ключевых величин, которые в наибольшей степени вносят вклад в величины свободной энергии. Разные варианты выбора этих величин приведут к разным решениям и, следовательно, к разной эффективности. Кроме того, существенно влияет на эффективность и выбор эталонного состояния. Нет универсальной схемы для решения этих проблем.
В последнее время мы стали свидетелями поразительного прогресса машинного обучения как инструмента для обнаружения, характеристики, распознавания, классификации или генерации сложных данных и их быстрого применения в широком спектре областей: от классификации изображений, распознавания лиц, автоматического вождения, финансового анализа, диагностики заболеваний [34], игры в шахматы или компьютерные игры [35, 36] вплоть до квантовой физики [37–39]. Тем не менее, этот список далек от охвата всех уже известных областей применения, не говоря уже о потенциально ожидаемых в будущем. Поэтому очень интересно исследовать потенциал подхода, основанного на машинном обучении, для характеристики и классификации 3D-данных РНК, для предсказания структуры РНК компьютерными методами. Будем надеяться, что сделанная нами попытка привнесет новые идеи в эту важную физико-биологическую проблему.
В этой статье мы сообщаем о нашей работе по разработке новой функции скоринга (оценки) на основе машинного обучения. Центральная часть нашего подхода – многослойная нейронная сеть, являющаяся аппроксиматором по универсальной теореме аппроксимации. В частности, мы собрали большую базу данных структур РНК и разделили ее на три части: наборы для обучения, проверки и тестирования. Затем мы загрузили обучающий набор в многослойную нейронную сеть и обучили ее стандартным методом градиентного спуска. Каждой входной кандидатной структуре нейронная сеть давала оценку подобия, которая оценивала ее сходство с нативной. Мы протестировали эффективность этого подхода и сравнили его с недавно предложенным полноатомным потенциалом.
МЕТОДЫ
Вход и выход нейронных сетей. Мы построили две многослойные нейронные сети, одна из которых в качестве входных данных принимает крупнозернистые характеристики структуры РНК, а другая – полноатомные характеристики. Эти две сети были названы NET1 и NET2 и соответствующим образом обучены. Входные данные представлены в виде многомерных тензоров.
Чтобы войти в NET1, РНК-структуру S преобразовали в многомерный тензор следующим образом:
(1)
$\begin{gathered} P(S) = {{P}_{{base}}}[seqSep][cType][dType][bin] + \hfill \\ + \,\,{{P}_{{backbone}}}[seqSep][aaType][bin], \hfill \\ \end{gathered} $Второй член в уравнении (1) означает вероятность наблюдения пар атомов углерода в остове макромолекулы, которые расположены на расстоянии seqSep и относятся к типу aaType в пространственном интервале, обозначенном bin. На крупнозернистом уровне атомы основной цепи включают только атомы P и C4'. Тензор второго члена определяют аналогично первому, за исключением того, что индекс aaType обозначает три типа пар, включая P-P, P-C4', C4'-C4'.
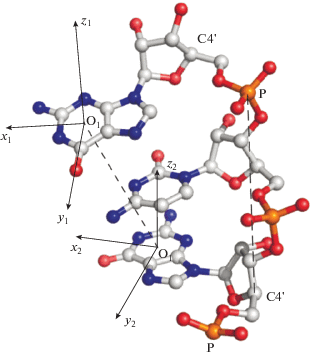
Рисунок 1 и следующие уравнения показывают, как рассчитать расстояния dxy и dz.
Рис. 1.
Фрагмент структуры РНК, показывающий, как вычислить структурные параметры. Атомы фосфора окрашены в оранжевый цвет, кислород в красный, углерод в серый, а азот – в синий. Локальные системы координат объяснены в тексте.
В дополнение к тензорам, приведенным в уравнении (1), число нуклеотидов целевой РНК и ее радиусов вращения вдоль трех основных осей также рассматривают как полезные функции и также вводят в нейронную сеть. В общей сложности для нейронной сети, работающей на крупнозернистом уровне, имеется 291 вход.
Чтобы войти в NET2, которая работает на полноатомном уровне, тензор, описывающий РНК-структуру S, определяют следующим образом:
что означает вероятности наблюдения пар атомов, которые имеют тип aaType и в пространственном интервале bin. Атомы классифицируются по 23 типам, как описано в литературе [18]. Соответственно, aaType может принимать 23 × 24/2 = 276 значений. Ширина шага расстояния равна 0.1 нм, а расстояние больше 2 нм игнорируется. Взяв в качестве примера стандартную А-спираль длиной 50 н., удалось установить, что общее число пар атомов составляет 908, и среди них 48 пар P-C4' находятся на расстоянии от 0.3 до 0.4 нм, тогда для Paa[P-C4'] [3] будет установлено значение 48/908. Аналогично вычисляют другие элементы P(S). Тензор P(S) вместе с числом нуклеотидов РНК и радиусами вращения вдоль трех основных осей вводят в нейронную сеть. Всего в нейронной сети на полноатомном уровне работает 5524 входа.
Нейронные сети разработаны для получения на выходе оценки подобия, которая оценивает подобие между предсказанной структурой и нативной. Математически сеть может быть описана функцией ${{y}_{i}} = f({{x}_{i}})$, где xi обозначает входной тензор для структуры, проиндексированной i, а yi обозначает результат на выходе или метку, как обычно называют. На этапе обучения метку yi принимали за среднеквадратическое отклонение (RMSD) структуры i от соответствующей нативной структуры.
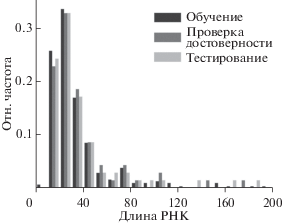
Наборы данных. Мы построили три набора данных для обучения сетей. В частности, загрузили неизбыточные РНК с разрешением более 3.5% с сайта банка NDB, удалили РНК, образующие комплексы с другими молекулами, а также убрали РНК с отсутствующими или нестандартными нуклеотидами. В итоге получен список, содержащий 462 РНК. Затем, чтобы генерировать сравнительные структуры, мы выполнили моделирование методом молекулярной динамики (MD) продолжительностью 40 нс для каждой РНК, при этом температуру постепенно увеличивали с 300 до 600 К. На этом этапе применяли программное обеспечение Gromacs (версия 4.5). С каждой траектории MD получали 300 структур, случайным образом и равномерно распределенных в диапазоне RMSD [0, RMSDmax], где RMSDmax = 1.0 нм для РНК длиной <100 н., 1.5 нм для РНК длиной в диапазоне [100, 200], и 2.0 нм для РНК длиной > > 200 н. Всего собрано 462 × 300 = 138 600 структур. 462 РНК случайным образом разделили на три набора данных: набор обучения, набор проверки и набор тестов; каждую нативную РНК-структуру связывали с 300 искусственными. Отношение количества структур между тремя наборами составило 322 : 70 : 70. Распределение по длинам последовательности РНК в трех наборах показано на рис. 2. Коды PDB для РНК приведены в дополнительных материалах (см. Приложение на сайте: http://www.molecbio.ru/downloads/2019/1/supp_ Wang_rus.pdf).
Рис. 2.
Распределение последовательностей по длине для трех наборов данных РНК. Количество РНК в трех наборах – 322, 70 и 70 соответственно. Самая левая гистограмма соответствует двум структурам шпилек длиной 8 н.
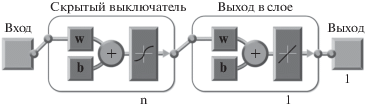
Архитектура многослойных нейронных сетей. Как NET1, так и NET2 содержат один скрытый слой, как показано на рис. 3. В сети NET1 имеется 291 узел во входном слое, 30 узлов в скрытом слое и 1 узел для вывода. В сети NET2 имеется 5524, 10 и 1 узлов в трех слоях соответственно. Мы также обучили сети с более значительными скрытыми слоями и обнаружили небольшое увеличение производительности. Мы протестировали для скрытого слоя разное число узлов: от 0 до 60. Результаты представлены в дополнительных материалах. Приведенные выше значения оптимизированы.
Рис. 3.
Архитектура нейронных сетей. Сеть содержит один входной слой, один скрытый слой и один выходной слой. Количество узлов в разных слоях подробно описано в тексте.
Функция активации нейронов в скрытом слое – это гиперболическая касательная функция, а для нейронов в выходном слое – линейная функция. Функция потерь представляет собой среднеквадратичную ошибку, скорректированную следующим образом:
где i – индекс образца, Yi и yi – RMSD для экспериментальной структуры и оценки сходства, заданного сетью, соответственно. Знаменатель ni – это число выборок в интервале RMSD, к которому принадлежит Yi; ширина интервала bin равна 0.1 нм. Экспоненциальный фактор применен для увеличения веса образцов с меньшими RMSD, поскольку тонкие структуры с минимальными уровнями энергии более важны, чем структуры с высокой энергией. Контролирующий фактор β в какой-то степени произволен и после нескольких раундов тестирования установлен на уровень 0.3. Согласно нашим тестам, использование экспоненциального коэффициента в функции потерь существенно повышает эффективность. Сеть обучена с помощью метода масштабированного сопряженного градиента, реализованного в пакете программ MATLAB как trainscg. Объем загрузки составлял 128.РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
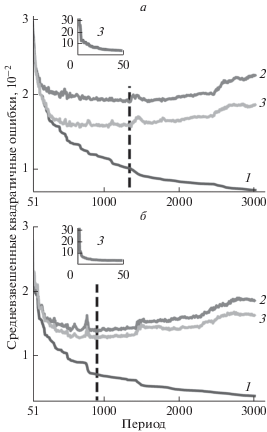
Значения потерь двух сетей в процессе обучения представлены на рис. 4. Как показано вертикальной линией, значение потерь для проверочного набора достигает минимума после 1335 периодов (epoch) для NET1 и после 906 – для NET2. Предполагаем, что сети на этом этапе обучения обладают максимальной способностью к обобщению. Дальнейшее обучение сетей продолжает снижать потери обучающего набора, но увеличивает потери проверочных и тестовых наборов и приводит к эффекту чрезмерного обучения. Поэтому параметры, полученные на этапе, обозначенном вертикальными линиями на рис. 4, используют для расчета приведенных ниже результатов.
Рис. 4.
Функции потерь при тренировках, проверках и тестировании, установленные во время обучения: (a) для NET1 и (б) для NET2. Вставки отражают первые 50 периодов. В каждом периоде для обучения использовали ~ 100 тыс. образцов. Вертикальные линии показывают, где минимизируется потеря проверочного набора. 1 – Тренировка; 2 – проверка достоверности; 3 – тестирование; --- минимизация потерь проверочного набора.
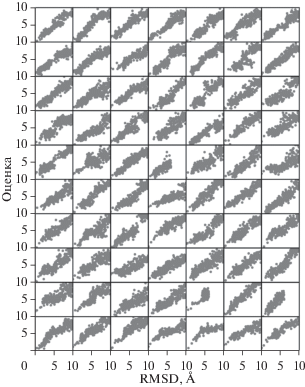
На рис. 5 и 6 показана корреляция между оценками подобия, предсказанными сетями, и фактическими значениями RMSD входных кандидатных структур, рассчитанными для тестового набора, содержащего 70 РНК с их производными. Обратите внимание, что на этапе тестирования входы в сеть были только кандидатными структурами, представленными уравнением (1). Сеть слепа к их RMSD, которые использовали только для оценки точности предсказания. В идеале, чем меньше RMSD, тем ниже оценка (больше подобия с нативной структурой), которую должна дать сеть, что обычно называют “функцией воронки”. На рис. 5 и 6 действительно показано, что корреляция между оценками и RMSD следует поведению воронки, а это указывает на хорошую производительность сетей.
Рис. 5.
Корреляция между оценками подобия, предсказанными сетью NET1, и фактическими RMSD, рассчитанными для 70 РНК в тестовом наборе. Каждая панель соответствует одной РНК и содержит 301 структуру.
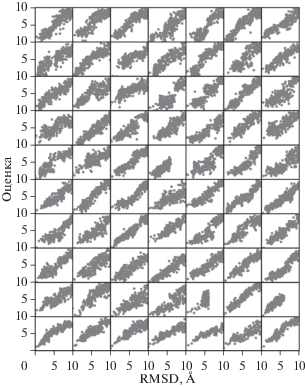
Рис. 6.
Корреляция между оценками подобия, предсказанными сетью NET2, и фактическими RMSD, рассчитанными для 70 РНК в тестовом наборе. Каждая панель соответствует одной РНК и содержит 301 структуру.
В табл. 1. показана способность NET1 и NET2 количественно распознавать нативную структуру среди искусственно созданных. Если мы выберем кандидата с наилучшим показателем подобия, предсказанного как нативный, то 39 из 70 РНК правильно оценены сетью NET1, а 49 из 70 – сетью NET2. Напротив, полноатомный статистический потенциал RASP [18] корректно предсказывает 26 из 70 РНК того же тестового набора. Если считать успешным, когда нативная структура находится в пределах 10 наилучших оценок (баллов), то 60 из 70 РНК правильно предсказаны сетью NET1, а 52 РНК – сетью NET2. Напротив, RASP идентифицирует всего лишь 31 из 70 образцов. Результаты показывают, что обученные сети дают существенно лучший результат, чем RASP.
Таблица 1.
Способность различных подходов к распознаванию нативных структур среди искусственных
| NET1 | NET2 | RASP | |
|---|---|---|---|
| Лучшая оценка | 39 | 49 | 26 |
| Лучшие 10 оценок | 60 | 52 | 31 |
Мы также вычислили показатели обогащения (ES) для сетей и сравнили с таковым для RASP. ES можно определить как
ES показывает степень перекрытия между верхними 10% оценок (${{E}_{{top10\% }}}$) и лучшими 10% RMSD (${{R}_{{top10\% }}}$). Идеальная линейность между оценкой и RMSD дает число 10, а совершенно случайное отношение дает 1. Мы вычислили ES для каждой из 70 РНК в тестовом наборе и нашли среднее значение 4.6 для NET1 и 5.3 для NET2. Среднее значение ES, для сравнения, было 4.4 для RASP. Оценки ES также показывают, что наши результаты лучше, чем полученные RASP.
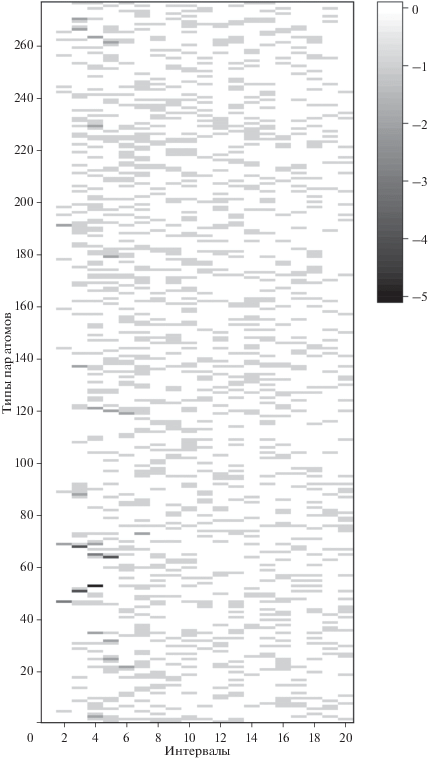
Наконец, мы провели анализ характеристик для NET2. Процедура состоит в следующем. В уравнении (2) для каждого элемента тензора проводят приравнивание к нулю, а затем проверку, насколько это воздействие влияет на производительность, измеряемую количеством нативных структур, правильно распознанных из искусственных. Расчет проводили так же, как в табл. 1, и для того же набора тестов. В общем случае приравнивание одного элемента к нулю приведет к ухудшению характеристик; и считают, что чем больше ухудшение, тем более важна функция, соответствующая этому элементу. Результаты показаны на рис. 7, где каждая точка данных соответствует одному элементу тензора, а его серый уровень указывает на величину ухудшения характеристик. Видно, что большинство изменений принадлежит диапазону от 1 до 2, это означает, что одну или менее двух нативных структур распознают из искусственно созданных для тестового набора из 70 РНК. Есть несколько больших изменений, близких к 5, в нижней левой части рисунка (темные блоки). Эти элементы соответствуют некоторым близким межмолекулярным взаимодействиям в диапазоне расстояний 0.3–0.5 нм. Однако мы обнаружили, что трудно получить интуитивное понимание этого наблюдения, связанное с трудностями при анализе результатов многослойных нейронных сетей. Мы также проецировали данные на рисунке на x-размерность, т.е. на расстояние. Обнаружено, что тензорные элементы в диапазоне расстояний 0.3–0.8 нм влияют на характеристики сильнее, чем другие. Этот факт понять легче с физической точки зрения, так как получается, что пространственно близкие атомы вносят более значительный вклад в стабильность молекулы. Анализ функций для NET1 дал аналогичные результаты и поэтому здесь не представлен.
Рис. 7.
Анализ характеристик для сети NET2. Оси x и y соответствуют первому и второму измерениям тензора в уравнении (2) соответственно. Ось x показывает пространственное расстояние между парами атомов, а ось y указывает разные типы пар атомов. Серый уровень данных указывает величину понижения производительности, когда соответствующий тензорный элемент установлен на ноль. Чем темнее элемент, тем больше понижение. Производительность можно измерить количеством нативных структур, распознанных из искусственно созданных, таких же, как в таблице. Величины понижающего диапазона варьируют от 0 до 5, в единицах количества РНК. Этот показатель рассчитан для 70 РНК в наборе тестов.
ВЫВОДЫ
В последнее время мы стали свидетелями удивительной способности методов машинного обучения в характеризации, классификации или генерации сложных данных в различных областях. Именно поэтому очень интересно исследовать потенциал машинного обучения в характеристике и классификации данных о структурах РНК. В этом исследовании мы разработали функцию скоринга, основанную на машинном обучении, для прогнозирования третичной структуры РНК. Мы построили три базы данных РНК и обучили две нейронные сети, NET1 и NET2, которые использовали грубозернистые и полноатомные структурные функции в качестве входных данных соответственно. Обе сети выдают оценку подобия между входной структурой и нативной. Что касается производительности, то корреляция между прогнозируемыми оценками и фактическими RMSD при изучении методом последовательного исключения следует поведению воронки, что указывает на хорошую производительность сетей. Количественно, если структуру с наилучшим результатом принимают как предсказанную, то NET1 правильно предсказывает 39 из 70 РНК, а NET2 – 49. Если считать успехом, когда нативная структура находится в пределах 10 наилучших баллов, то NET1 правильно предсказывает 60, а NET2 – 52 РНК. Оценка обогащения, которая характеризует перекрытие между наилучшим скорингом и лучшими RMSD, рассчитана как 4.6 и 5.3 в среднем для NET1 и NET2 соответственно. Сравнение приведенных выше результатов с результатами применения недавно предложенного полноатомного потенциала, показало, что наш подход существенно лучше.
Наш подход имеет много новых особенностей. По сравнению с традиционной функцией скоринга, основанной на обратном уравнении Больцмана, новая функция подсчета более гибкая, принимающая во внимание различные особенности, например, длину последовательности, радиус вращения и т.д. В принципе, все соответствующие функции могут быть легко включены в сеть, и их относительный вклад в окончательную оценку можно определить автоматически во время обучения. Более того, наш подход свободен от выбора эталонного состояния, что остается одной из сложных проблем традиционного способа вывода скоринг-функции.
Кроме того, мы хотели бы отметить, что проведенная работа предварительная и имеет большие возможности для улучшения. Во-первых, вход в нейронные сети был в некоторой степени традиционным – принято формирование структур по п.н. и стэкинг-взаимодействиям. Однако это необязательно для текущей схемы; и мы работаем над непосредственным вводом РНК-структур в сеть и тестированием производительности. Во-вторых, современные нейронные сети относительно просты. Напротив, как считают в сообществе исследователей машинного обучения, более крупная сеть обычно лучше при условии, что будет тщательно проработана проблема чрезмерного обучения. В ближайшем будущем мы будем пытаться создавать более сложные архитектуры, такие как глубокие сверхточные сети.
С появлением все новых и новых структур РНК методы машинного обучения будут все более полезными в плане проблемы прогнозирования структуры. Мы надеемся, что представленная нами работа окажется полезной и сможет стимулировать новые идеи в соответствующей области.
Авторы выражают благодарность Центру HPCC Университета Нанкина и Центру совместных инновационных разработок HPC Advanced Microstructures за их поддержку в компьютерной обработке данных.
Авторы выражают признательность Национальному фонду естественных наук Китая (№ 11774158, 11274157, 31671026, 11334004), Национальной программе фундаментальных исследований и развития Китая (2013CB834100) и проекту развития приоритетных академических программ (PAPD) высших учебных заведений провинции Цзянсу.
Список литературы
Parisien M., Major F. (2008) The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 452, 51‒55.
Jonikas M.A., Radmer R.J., Laederach A., Das R., Pearlman S., Herschlag D., Altman R.B. (2009) Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 15(2), 189‒199.
Flores S.C., Wan Y., Russell R., Altman R.B. (2010) Predicting RNA structure by multiple template homology modeling. Pac. Symp. Biocomput. 216‒227.
Sharma S., Ding F., Dokholyan N.V. (2008) iFoldRNA: three-dimensional RNA structure prediction and folding. Bioinformatics. 24, 1951‒1952.
Das R., Baker D. (2007) Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. USA. 104, 14664‒14669.
Zwieb C., Muller F. (1997) Three-dimensional comparative modeling of RNA. Nucleic Acids Symp. Ser. (36), 69‒71.
Martinez H.M., Maizel J.V. Jr., Shapiro B.A. (2008) RNA2D3D: a program for generating, viewing, and comparing 3-dimensional models of RNA. J. Biomol. Struct. Dyn. 25, 669‒683.
Popenda M., Szachniuk M., Antczak M., Purzycka K.J., Lukasiak P., Bartol N., Blazewicz J., Adamiak R.W. (2012) Automated 3D structure composition for large RNAs. Nucleic Acids Res. 40, e112. doi 10.1093/nar/ gks339
Zhao Y.J., Huang Y.Y., Gong Z., Wang Y., Man J., Xiao Y. (2012) Automated and fast building of three-dimensional RNA structures. Sci. Rep. 2, 734. doi 10.1038/ srep00734
Wang J., Mao K.K., Zhao Y.J., Zeng C., Xiang J., Zhang Y., Xiao Y. (2017) Optimization of RNA 3D structure prediction using evolutionary restraints of nucleotide-nucleotide interactions from direct coupling analysis. Nucleic Acids Res. 45(11), 6299‒6309.
Zhang J., Dundas J., Lin M., Chen R., Wang W., Liang J. (2009) Prediction of geometrically feasible three-dimensional structures of pseudoknotted RNA through free energy estimation. RNA. 15, 2248‒2263.
Zhang J., Zhang Y.J., Wang W. (2010) An RNA base discrete state model toward tertiary structure prediction. Chin. Phys. Lett. 27, 118702.
Zhang J., Bian Y.Q., Lin H., Wang W. (2012) RNA fragment modeling with a nucleobase discrete-state model. Phys. Rev. E. 85, 021909.
Li J., Zhang J., Wang J., Wang W. (2016) Structure prediction of RNA loops with a probabilistic approach. PLoS Comput. Biol. 12, e1005032.
Qasim R., Kauser N., Jilani T. (2011) Secondary structure prediction of RNA using machine learning method. Int. J. Comput. Appl. 10(6), 24‒28.
Frellsen J., Moltke I., Thiim M., Mardia K.V., Ferkinghoff-Borg J., Hamelryck T. (2009) A probabilistic model of RNA conformational space. PLoS Comput. Biol. 5, e1000406.
Wang Z., Xu J. (2011) A conditional random fields method for RNA sequence-structure relationship modeling and conformation sampling. Bioinformatics. 27, i102‒110.
Capriotti E., Norambuena T., Marti-Renom M.A., Melo F. (2011) All-atom knowledge-based potential for RNA structure prediction and assessment. Bioinformatics. 27, 1086‒1093.
Cao S., Chen S.J. (2006) Predicting RNA pseudoknot folding thermodynamics. Nucleic Acids Res. 34, 2634‒2652.
Tan Z.J., Chen S.J. (2011) Salt contribution to RNA tertiary structure folding stability. Biophys. J. 101, 176‒187.
Wu Y.Y., Zhang Z.L., Zhang J.S., Zhu X.L., Tan Z.J. (2015) Multivalent ion-mediated nucleic acid helix-helix interactions: RNA versus DNA. Nucleic Acids Res. 43, 6156‒6165.
Shi Y.Z., Wang F.H., Wu Y.Y., Tan Z.J. (2014) A coarse-grained model with implicit salt for RNAs: Predicting 3D structure, stability and salt effect. J. Chem. Phys. 141, 105102.
Shi Y.Z., Wu Y.Y., Wang F.H., et al. (2014) RNA structure prediction: Progress and perspective. Chinese Phys B. 23, 078701.
Gong S., Wang Y.J., Zhang W.B. (2015) The regulation mechanism of yitJ and metF riboswitches. J. Chem. Phys. 143, 045103.
Zhang W.B., Chen S.J. (2001) A three-dimensional statistical mechanical model of folding double-stranded chain molecules. J. Chem. Phys. 114, 7669‒7681.
Yang Y., Zhao H., Wang J., Zhou Y. (2014) SPOT-Seq-RNA: predicting protein-RNA complex structure and RNA-binding function by fold recognition and binding affinity prediction. Methods Mol. Biol. 1137, 119‒130.
Yang Y., Li X., Zhao H., Zhan J., Wang J., Zhou Y. (2017) Genome-scale characterization of RNA tertiary structures and their functional impact by RNA solvent accessibility prediction. RNA. 23, 14‒22.
Wang X., El Naqa I.M. (2008) Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 24, 325‒332.
Xu X., Zhao P., Chen S.J. (2014) Vfold: a web server for RNA structure and folding thermodynamics prediction. PLoS One. 9, e107504.
Magnus M., Boniecki M.J., Dawson W., Bujnicki J.M. (2016) SimRNAweb: a web server for RNA 3D structure modeling with optional restraints. Nucleic Acids Res. 44, W315‒319. doi 10.1093/nar/gkw279
Magnus M., Matelska D., Lach G., Chojnowski G., Boniecki M.J., Purta E., Dawson W., Dunin-Horka-wicz S., Bujnicki J.M. (2014) Computational modeling of RNA 3D structures, with the aid of experimental restraints. RNA Biol. 11, 522‒536.
Zhang J., Lin M., Chen R., Wang W., Liang J. (2008) Discrete state model and accurate estimation of loop entropy of RNA secondary structures. J. Chem. Phys. 128, 125107.
Tang K., Zhang J.F., Liang J. (2014) Fast protein loop sampling and structure prediction using distance-guided sequential chain-growth Monte Carlo Method. PLoS Comput. Biol. 10, e1003539.
Goodfellow I., Bengio Y., Courville A. (2016) Deep Learning. Cambridge, Massachusetts: The MIT Press.
Silver D., Huang A., Maddison C.J., Guez A., Sifre L., van den Driessche G., Schrittwieser J., Antonoglou I., Panneershelvam V., Lanctot M., Dieleman S., Grewe D., Nham J., Kalchbrenner N., Sutskever I., Lillicrap T., Leach M., Kavukcuoglu K., Graepel T., Hassabis D. (2016) Mastering the game of Go with deep neural networks and tree search. Nature. 529, 484‒489.
Silver D., Schrittwieser J., Simonyan K., Antonoglou I., Huang A., Guez A., Hubert T., Baker L., Lai M., Bolton A., Chen Y., Lillicrap T., Hui F., Sifre L., van den Driessche G., Graepel T., Hassabis D. (2017) Mastering the game of Go without human knowledge. Nature. 550, 354‒359.
Carleo G., Troyer M. (2017) Solving the quantum many-body problem with artificial neural networks. Science. 355, 602‒605.
Carrasquilla J., Melko R.G. (2017) Machine learning phases of matter. Nat. Phys. 13, 431‒434.
van Nieuwenburg E.P.L., Liu Y.H., Huber S.D. (2017) Learning phase transitions by confusion. Nat. Phys. 13, 435‒439.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология