

Молекулярная биология, 2019, T. 53, № 2, стр. 240-255
Факторы рестрикции вируса иммунодефицита человека и их неоднозначная роль в инфекции
А. А. Зотова a, b, *, А. А. Атемасова a, А. В. Филатов c, Д. В. Мазуров b, c
a Биологический факультет Московского государственного университета им. М.В. Ломоносова
119991 Москва, Россия
b Институт биологии гена Российской академии наук
119334 Москва, Россия
c Государственный научный центр “Институт иммунологии”
Федерального медико-биологического агентства России
115478 Москва, Россия
* E-mail: ashunaeva@gmail.com
Поступила в редакцию 02.11.2018
После доработки 18.11.2018
Принята к публикации 19.11.2018
Аннотация
Вирусом иммунодефицита человека (ВИЧ) инфицировано более 37 млн человек по всему миру. Антиретровирусная терапия позволяет контролировать инфекцию, однако не способна полностью удалить вирус из организма пациента. Понимание того, как клетка отвечает на ВИЧ-инфекцию и какие клеточные белки в этом участвуют, может способствовать созданию новых препаратов с антиретровирусной активностью. Обзор посвящен описанию известных на сегодняшний день факторов рестрикции ВИЧ, обсуждению их неоднозначной роли в инфекции и взаимодействию с акцессорными белками ВИЧ.
Вирус иммунодефицита человека (ВИЧ) принадлежит к семейству Retroviridae, роду Lentivirus с длительным инкубационным периодом. ВИЧ включает следующие виды: ВИЧ-1, наиболее распространенный и патогенный, и ВИЧ-2, отличающийся от ВИЧ-1 по строению и патогенному действию. Различают четыре группы ВИЧ-1: M (major, основная), N (non-M, non-O – ни M, ни O), O (outlier, непохожий) и P (putative, предполагаемый). В настоящее время считается, что ВИЧ-1 группы М произошел от вируса иммунодефицита (SIV) шимпанзе в начале ХХ века [1]. Более 70 млн человек инфицировано ВИЧ-1 группы М и для 30 млн это заболевание уже привело к летальному исходу. ВИЧ-1 других групп (N, O, P) также произошел от SIV (шимпанзе в случае группы N и гориллы в случае групп O и P), но гораздо реже встречается у людей. ВИЧ-1 группы О найден примерно у 100 тыс. жителей Камеруна и близлежащих стран [2], вирус N и P групп обнаружен только у нескольких человек [3–5].
После расшифровки нуклеотидной последовательности ВИЧ-1 стало очевидно, что его геном устроен намного сложнее, чем многих ретровирусов животных. Геном последних включает гены gag, pol и env, которые кодируют только структурные белки (например, геном вируса лейкоза мышей, MLV). В 3'-области генома ВИЧ-1 обнаружены открытые рамки считывания, не характерные для других представителей семейства. Здесь закодированы регуляторные белки Tat и Rev и вспомогательные белки Vif, Vpr, Vpu и Nef (у вируса ВИЧ-2 к ним также относится Vpx). Tat и Rev необходимы ВИЧ-1 для репликации во всех клетках хозяина: они запускают транскрипцию генов с провирусной ДНК и обеспечивают транспорт РНК вируса из ядра в цитоплазму. Вспомогательные, или акцессорные, белки необходимы для репликации лишь в некоторых типах клеток, хотя их основной функцией принято считать противодействие клеточным механизмам противовирусной защиты. Потребовались годы, чтобы найти мишени вспомогательных белков ВИЧ – факторы рестрикции (рис. 1). Хотя литературы по рестрикционным факторам много (см., например, обзоры [6–8]), их список постоянно расширяется, а роль некоторых так до конца не выяснена или пересматривается, поэтому требуется периодически обобщать вновь появляющиеся данные.
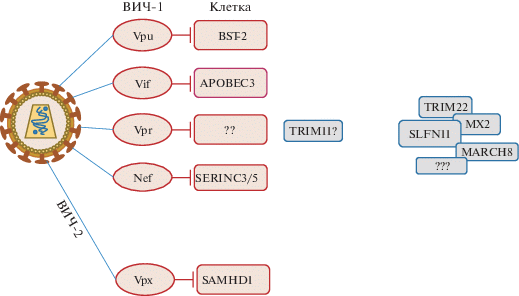
К наиболее известным рестрикционным факторам, механизмы действия которых установлены, можно отнести IFITM1–3 [9], APOBEC3 [10–12], TRIM5α [13–15], Mx2 [16–18], Schlaffen11 [19], SAMHD1 [20–22] и BST-2 (также известный как Tetherin) [23, 24].
Далее мы подробно рассмотрим клеточные белки, вовлеченные в рестрикцию ВИЧ, в той последовательности, в которой они воздействуют на различные этапы цикла репликации ВИЧ, – от ранних до поздних.
IFITM
Проникновение ВИЧ-1 в клетку вызывает активацию паттернраспознающих рецепторов (PRR), в том числе, Toll-подобного рецептора 7 (TLR7), и продукцию интерферонов (IFN) I типа [25]. За последние годы обнаружено множество генов факторов рестрикции различных вирусов, экспрессия которых зависит от продукции IFN I типа. Некоторые из них (MxB, BST-2, APOBEC3) детально изучены в контексте конкретных вирусных инфекций, в частности, ретровирусной. Среди таких интерферонстимулируемых генов (ISGs, IFN stimulated genes) идентифицировано семейство IFITM (interferon-induced transmembrane gene family) с широкой противовирусной активностью.
Семейство белков IFITM было описано 30 лет назад и тогда же выявлена индукция их экспрессии IFN I и II типов [26]. Противовирусная функция белков IFITM1, IFITM2 и IFITM3 обнаружена не так давно, в 2009 году, – в результате скрининга малых интерферирующих РНК (siРНК) [9]. Оказалось, что эти белки выполняют функцию факторов рестрикции на ранних этапах заражения клетки вирусом гриппа А подтипа H1N1, а также флавивирусами – вирусом лихорадки Денге и лихорадки Западного Нила [9]. В 2011 году выявлено противовирусное действие белков IFITM в отношении ВИЧ-1 – нокдаун IFITM1, IFITM2 и IFITM3 приводил к усилению инфекции [27].
Известно, что IFITM1 локализуется на плазматической мембране клетки, тогда как IFITM2 и IFITM3 – в поздних эндосомах и лизосомах [28, 29]. Белки содержат два трансмембранных домена и один крайне консервативный внутриклеточный домен, что интересным образом зеркально отражает структуру другого фактора рестрикции – BST-2, действующего на этапе высвобождения частиц от зараженной клетки [24] (см. ниже). Принцип рестрикции проникновения вируса в клетку белками IFITM связывают с двумя аспектами: регулированием уровня холестерина на мембране клетки и ингибированием процесса слияния вируса с клеткой [30–32]. Кроме того, белки IFITM способны инкорпорироваться в вирусные частицы, что приводит к снижению их инфекционности [33]. Оказалось, что у таких вирионов нарушен процесс слияния с клеточной мембраной. На T-клетках человека SupT1 с Tet-On-регулируемой экспрессией разных IFITM показано, что все три белка ингибируют инфекцию свободным вирусом (cell-free) ВИЧ-1 [27]. Однако IFITM3 проявляет низкую эффективность в рестрикции ВИЧ-1 при межклеточной трансмиссии (инфекция от-клетки-к-клетке) [33].
Недавно, при исследовании IFITM-опосредованной рестрикции ВИЧ-1 в зависимости от тропности вируса (для проникновения в клетку вирус использует в качестве корецепторов CCR5 или CXCR4), показано, что Х4-тропный вирус эффективно подвергается рестрикции белками IFITM2 и IFITM3 в эндосомах и лизосомах, тогда как R5-тропный вирус – рестрикции IFITM1, который экспрессируется на плазматической мембране клетки [34]. Эта избирательность обусловлена тем, что проникновение вириона ВИЧ-1 в клетку происходит путем слияния с мембраной эндосомы в случае использования в качестве корецептора CXCR4 и с плазматической мембраной – для CCR5-тропных вирусов [34].
Рестрикция белками IFITM происходит до интеграции провируса в геном клетки, поэтому борьба ВИЧ-1 с IFITM-рестрикцией может идти в следующих направлениях: либо изменение сайта проникновения частиц в клетку, либо наличие в вирионах белков, противодействующих фактору рестрикции. Действительно, показано, что со временем накопление мутаций в белке Env позволяет вирусу избежать рестрикции [35].
Таким образом, белки IFITM играют важную роль в процессе слияния мембран вируса и клетки на этапе внедрения вириона в хозяйскую клетку, а также при межклеточной трансмиссии. Известно, что в этом задействован белок оболочки ВИЧ-1, однако точный механизм пока неясен.
TRIM
Белки семейства TRIM, будучи E3-лигазами, участвуют в регуляции клеточного цикла, аутофагии и реакциях врожденного иммунитета. TRIM-белки задействованы в сигнальных путях NF-κB и IFN I типа и опосредованно влияют на ВИЧ-инфекцию. Однако несколько белков этого семейства напрямую ингибируют вирусную инфекцию на разных этапах репликации вируса, в том числе обеспечивая протеасомную деградацию вирусных белков [36–41].
Семейство TRIM впервые охарактеризовали еще в 2001 году – как группу белков с консервативным трехкомпонентным (TRIpartite Motif) N-концевым доменом RBCC (Really Interesting New Gene (RING) E3 ligase domain, B boxes and Сoiled-Сoil domains) [42]. RBCC-домен обеспечивает Е3-лигазную функцию белка, а также олигомеризацию, что необходимо для его функциональной активности [43, 44].
С-концевой домен, напротив, вариабельный и ответственен за взаимодействие TRIM с белками-партнерами. По типу С-концевого домена TRIM белки делят на 11 подсемейств. Мотив PRY/SPRY (SPla and the RYanodine Receptor) встречается на С-конце более чем у 30 белков TRIM и ассоциируется с противовирусной активностью белков TRIM5, TRIM22, TRIM11 и TRIM15 соответствующего подсемейства [40, 45, 46].
Белок TRIM5α семейства TRIM считается ключевой молекулой, обеспечивающей устойчивость обезьян Старого Света (африканских зеленых мартышек, макак-резусов и др.) к инфекции ВИЧ-1 [13], тогда как у обезьян Нового Света (например, у ночных обезьян) эту функцию выполняет TRIM5, слитый с циклофилином А (TRIMCyp) [14, 47].
Предложено несколько моделей, описывающих механизм рестрикции ВИЧ-1 белком TRIM5α, каждая из которых основана на взаимодействии SPRY-домена с капсидом вируса, в результате чего нарушается процесс разборки вируса – происходит его преждевременное “раздевание”. Однако точный механизм вмешательства TRIM5α в распаковку вириона до сих пор не выяснен. Известно, что для связывания с капсидом TRIM5α должен образовать димеры и тримеры в антипараллельной ориентации за счет суперспирализованных (coiled-coil) доменов. Затем TRIM5α формирует гексагонподобную сеть, охватывающую капсид [48–51]. Интересно, что формирование гексогональной сети справедливо и для TRIM-Cyp, из чего можно предположить существование единого механизма рестрикции ВИЧ разными белками TRIM [52].
Еще одно следствие формирования гексагональных структур молекулой TRIM5α на капсиде вируса – запуск противовирусного клеточного ответа [53] (рис. 2). В результате димеризации RING-доменов TRIM5α на поверхности капсида усиливается Е3-лигазная активность TRIM5α и активизируется синтез полиубиквитиновых цепей, связанных с боковой цепью Lys63. Эти цепи убиквитина активируют киназный комплекс TAK1 путем аутофосфорилирования, что, в свою очередь, приводит к транслокации факторов транскрипции AP-1 и NF-κB в ядро и последующей экспрессии IFN I типа [51].
Рис. 2.
Роль белков TRIM в репликации ВИЧ. Ub – убиквитин; K48-, K63-polyUb – цепочки убиквитина, связанные через Lys48 и Lys63 соответственно; Р – фосфорилирование.
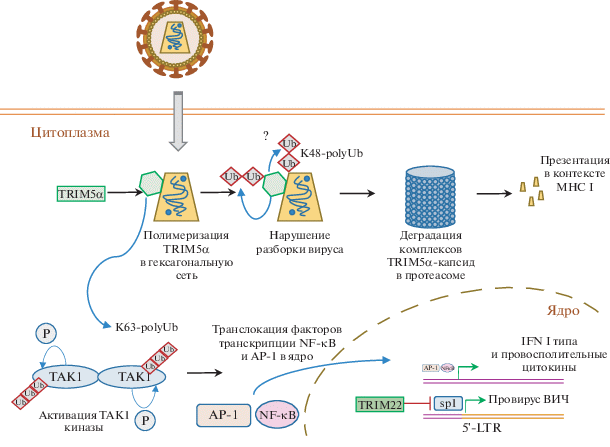
Еще одна успешная противовирусная стратегия реализуется TRIM-белками за счет направления вирусных компонентов на протеасомную деградацию. Но до сих пор остается открытым вопрос, выполняет ли эту функцию TRIM5α в отношении капсида ВИЧ-1. На данный момент не обнаружено сайтов убиквитинирования на белке капсида ВИЧ-1 [54], но известна способность TRIM5α к самоубиквитинированию с последующей деградацией в протеасоме – куда он, вероятно, увлекает и вирусные компоненты [55] (рис. 2). Кроме того, обнаружено, что компоненты нуклеокапсида вируса, в том числе интеграза, подвергаются эффективной деградации в протеасоме – и не без участия TRIM5α [56].
Интересно, что гомолог TRIM5α у человека не обладает выраженной противовирусной активностью. Однако единственная аминокислотная замена в домене PRY-SPRY в TRIM5α человека способна заметно реабилитировать его как фактор рестрикции ВИЧ-1 [57]. Другой белок семейства – TRIM22, будучи паралогом TRIM5α у человека, вовлечен в ингибирование репликации ВИЧ-1 посредством IFN I типа [58, 59]. Кроме того, TRIM22, независимо от присущей ему лигазной функции, ингибирует Tat- и NF-κB-независимую транскрипцию вирусных генов с LTR-промотора ВИЧ-1 путем прямого взаимодействия с фактором Sp1 [60, 61]. Эти наблюдения заслуживают внимания и клиницистов – обсуждается возможность контролировать транскрипцию вирусного генома в латентном периоде [62].
Как именно ВИЧ-1 противодействует клеточной рестрикции, опосредованной TRIM-белками, на данный момент неясно. Известно, что уровень экспрессии TRIM11 в клетке регулируется вспомогательным вирусным белком Vpr, причем его при низкой концентрации в клетке [63]; но этот регуляторный механизм тоже еще далек от понимания.
SAMHD1
Роль SAMHD1 (sterile alpha motif and histidine-aspartate domain containing protein 1) в рестрикции ВИЧ была независимо определена двумя исследовательскими группами в 2011 году. Белок идентифицировали масс-спектрометрией как партнер вспомогательного белка Vpx вируса ВИЧ-2 (у ВИЧ-1 Vpx отсутствует) [20, 64]. SAMHD1 экспрессируется на высоком уровне в миелоидных клетках и резидентных CD4+ Т-лимфоцитах и представляет собой дезоксинуклеозидтрифосфатазу, ответственную за регуляцию пула dNTPs и ингибирование обратной транскрипции ВИЧ [20, 22, 64, 65].
Гистидин/аспартатный (HD) каталитический домен белка SAMHD1 гидролизует dNTP с образованием дезоксирибонуклеозида и трифосфата, удаляя тем самым строительные блоки для синтеза вирусной кДНК [22]. Активность фермента определяется его тетрамеризацией и аллостерически контролируется GTP и всеми четырьмя dNTPs [66–70].
Недавно показано, что SAMHD1 обладает также металлзависимой 3′→5′ экзонуклеазной активностью в отношении одноцепочечных ДНК и РНК in vitro, из чего можно предполагать существование еще одного присущего этому ферменту механизма рестрикции ВИЧ – связывание и деградация вирусной РНК [71]. Важно отметить, что как dNTP-трифосфатгидролазная, так и 3′→5′ экзонуклеазная активность SAMHD1 выполняется HD-доменом. Ryoo и др. [72] на точечных мутантах SAMHD1 с заменами D137N или Q548A продемонстрировали способность SAMHD1 с отсутствием dNTP-трифосфатгидролазной активности и сохраненной 3′→5′ экзонуклеазной ингибировать инфекцию ВИЧ-1. Однако ингибирующее влияние SAMHD1 не наблюдалось в противоположной ситуации. Так авторы пришли к выводу, что именно 3′→5′ экзонуклеазная активность SAMHD1 вносит основной вклад в рестрикцию ВИЧ-1 [72]. Хотя эти результаты впоследствии не получили подтверждения [73, 74], Ryoo и соавт. объяснили это различиями в постановке экспериментов [75]. На сегодня вопрос о значимости 3′→5′ экзонуклеазной активности SAMHD1 в рестрикции ВИЧ по-прежнему остается открытым.
Вспомогательный белок ВИЧ-2 Vpx связывается с С-концом SAMHD1 и адаптерной молекулой DCAF1 и инициирует образование Е3-убиквитинлигазного комплекса с последующей деградацией SAMHD1 в протеасоме [20, 64, 76, 77]. Механизм этого противостояния ВИЧ-1 SAMHD1-рестрикции пока тоже до конца не изучен. Есть данные о том, что Vpr содержит тот же мотив, что и Vpx, необходимый для связывания с адаптером DCAF1 и ареста клеточного цикла в фазе G2 [77]. Кроме того, недавно Kyei с соавт. [78] обнаружили способность ВИЧ-1 использовать регулятор клеточного цикла циклин L2 для нейтрализации SAMHD1 в макрофагах. Важная роль SAMHD1 в рестрикции ВИЧ-1 показана на примере макрофагов, выделенных от пациентов с синдромом Айкарди–Гутьерес, вызванным мутациями в SAMHD1. Макрофаги таких пациентов обладают значительно более высокой чувствительностью к заражению ВИЧ-1 в сравнении с макрофагами здоровых людей [21].
APOBEC3 (CEM15)
APOBEC3 (apolipoprotein B editing complex 3) – семейство цитидиновых дезаминаз, которое у приматов включает семь белков: APOBEC3A (A3A), A3C, A3H с одним каталитическим доменом и A3B, A3D, A3F, A3G с двумя [79, 80]. Белки APOBEC3, в особенности A3G, экспрессируются на высоком уровне во многих типах клеток, включая CD4+ Т-лимфоциты, дендритные клетки и макрофаги, [81]. Кроме того, экспрессия APOBEC3 стимулируется IFN I типа [81]. Первоначально A3G уделялось наибольшее внимание среди прочих белков семейства, но впоследствии стало ясно, что A3H, A3F, A3D, A3B также активны в отношении ВИЧ, а вирусный белок Vif противодействует каждому из них [82].
Противовирусная роль белков APOBEC3 проявляется после того, как они попадают в вирусные частицы, а значит, только в следующем цикле репликации вируса [83, 84]. Рестрикция осуществляется путем индуцированного дезаминазами преобразования C→U в (–)-цепи ДНК ВИЧ. Урацил распознается полимеразой как тимин, что приводит к G→A мутации при синтезе (+)-цепи ДНК [83]. Гипермутагенез при обратной транскрипции вирусной РНК в ДНК имеет два важных последствия: во-первых, аберрантные последовательности узнаются клеткой и уничтожаются; во-вторых, происходит интеграция мутированной ДНК провируса в геном клетки, но из-за большого количества образовавшихся преждевременных стоп-кодонов (например, после замены TGG→TAG) и прочих мутаций инфекционные вирусные частицы не продуцируются [10, 83]. Замечено, что APOBEC3-зависимый мутагенез происходит с большей частотой на 3'-конце вирусной последовательности. Это объясняется тем, что в отличие от (–)-цепи вирусной ДНК, которая синтезируется с одного сайта (primary binding site) в области 5'-LTR, синтез (+)-цепи ДНК инициируется с двух полипуриновых трактов – центрального (cPPT) и на 3'-конце (3'-PPT). Наличие двух сайтов инициации синтеза (+)-цепи ДНК и параллельного расщепления РНК-матрицы создает предпосылки для неравновесного и более длительного пребывания (–)-цепи ДНК в одноцепочечном состоянии (а это субстрат APOBEC3) в 3'‑области генома ВИЧ [85, 86].
Белки семейства APOBEC3 ингибируют лентивирусную инфекцию, используя альтернативный механизм – не связанный с дезаминазной активностью. Показано, что А3-дезаминазы нарушают обратную транскрипцию, препятствуя синтезу (–)-цепи, а также связыванию тРНК с вирусной мРНК [10, 87]. Однако в ряде работ [88, 89] эта гипотеза не получила подтверждения, так как выявлена неспособность белков APOBEC3 к рестрикции ВИЧ при отсутствии каталитического дезаминазного домена.
В качестве инструмента противодействия APOBEC3-опосредованной рестрикции ВИЧ использует вспомогательный белок Vif, который индуцирует деградацию дезаминаз до того, как они успевают инкорпорироваться в вирусные частицы. Происходит связывание Vif с молекулой APOBEC3 в зараженной клетке и привлечение Е3-убиквитинлигазного комплекса, в состав которого входят элогин В, элонгин С и RBX1, с последующей деградацией APOBEC3 [84, 90, 91]. Однако Vif не способен полностью элиминировать APOBEC3 в клетке, продуцирующей вирус, что было продемонстрировано наличием множественных мутаций в провирусах клеток пациентов с острой и хронической ВИЧ-инфекцией, а также при вертикальной передаче ВИЧ новорожденным [82, 92]. Возможно, при индукции IFN I типа баланс APOBEC3–Vif сдвигается в сторону факторов рестрикции, и они успевают попасть в вирусные частицы до связывания с Vif.
По-видимому, при неполном сдерживании APOBEC3-опосредованной рестрикции вирус получает даже дополнительные преимущества. Дезаминазы А3 могут не справиться с тем, чтобы полностью подавить репликацию вируса, но обеспечивают высокий уровень мутагенеза, а значит более высокий риск появления так называемых escape-мутантов с устойчивостью к антиретровирусным препаратам. Низкую эффективность антиретровирусной терапии у некоторых пациентов уже связывают с дефектным Vif [93]. Итак, APOBEC3 можно рассматривать как пример фактора рестрикции, действие которого вирусу удалось обратить в свою пользу.
MxB
Белки MxA и MxB (Mx1 и Mx2 у мышей) относятся к семейству GTPаз, экспрессия обоих белков индуцируется IFN I типа. Известно, что MxA обладает широкой противовирусной активностью в отношении как ДНК-, так и РНК-содержащих вирусов, но не ретровирусов [94, 95]. Недавно три независимые исследовательские группы обнаружили, что MxB вовлечен в рестрикцию ВИЧ-1 – не влияя на обратную транскрипцию вирусной РНК, он дестабилизирует преинтеграционный комплекс и тем самым снижает интеграцию провирусной ДНК в геном клетки [16–18, 96].
В отличие от MxA, у MxB, помимо GTPазного домена, есть удлиненный N-концевой домен, необходимый для рестрикции ВИЧ-1 [16, 96, 97]. Этот домен несет сигнал ядерной локализации (NLS) и может связываться с капсидом вируса после гомодимеризации (2 молекулы MxB связываются между собой в антипараллельном направлении). Показано, что Arg-богатый мотив этого домена напрямую связывается с капсидом ВИЧ-1 и определяет способность MxB к рестрикции вируса [98]. Предполагают, что, как и при TRIM5α-рестрикции, это приводит к ингибированию “раздевания” вируса [99].
Таким образом, MxB воздействует на репликацию ВИЧ-1 в двух клеточных компартментах и, соответственно, на следующих стадиях цикла репликации вируса: “раздевания” в цитоплазме, импорта преинтеграционного комплекса в ядро и интеграции провируса в геном клетки. Важно заметить, что у клинических изолятов ВИЧ-1, несущих мутации H87Q и G116A в белке капсида, снижена чувствительность к MxB-опосредованной рестрикции и повышена репликативная активность по сравнению с другими циркулирующими штаммами [100, 101]. Эти данные можно рассматривать как подтверждение гипотезы о давлении фактора MxB на ВИЧ-1, причем в условиях in vivo.
Schlafen11 (SLFN11)
SLFN11 принадлежит к семейству Schlafen – ISG-белков, регулирующих пролиферацию клетки, иммунный ответ и репликацию вирусов [102]. Не так давно обнаружено участие SLFN11 в рестрикции ВИЧ [19]. Показано, что SLFN11 не влияет на ранние стадии цикла инфекции ретровируса, включая обратную транскрипцию, интеграцию и транскрипцию. SLFN11 действует на поздней стадии репликации вируса, селективно ингибируя трансляцию вирусных белков. SLFN11 связывает тРНК и противодействует изменениям в клеточном пуле тРНК, вызванном наличием ВИЧ. Это новый противовирусный механизм врожденного иммунного ответа, в котором SLFN11 селективно ингибирует синтез вирусных белков в ВИЧ-инфицированных клетках.
Интересно, что за 18 лет до этого открытия, еще в 1994 году, было замечено снижение уровня трансляции вирусных белков при стимуляции IFN I типа, но механизм регуляции тогда не исследовали [103]. А недавно показано, что SLFN11 при стимуляции IFN I типа ингибирует трансляцию не только вирусных, но и других кодоннеоптимизированных транскриптов в клетке [104] – так что этот белок, по-видимому, не стоит рассматривать как специфический фактор рестрикции ВИЧ. Скорее всего, SLFN11 вовлечен в общий противовирусный ответ клетки.
MARCH8 (c-MIR)
Недавно обнаружено участие белка MARCH8 (membrane-associated RING-CH8) в рестрикции ВИЧ-1 [105]. MARCH8 представляет собой Е3-убиквитинлигазу с высокой экспрессией в дифференцированных миелодных клетках – макрофагах и дендритных клетках. К моменту открытия противовирусной функции белка было известно, что MARCH8 снижает экспрессию некоторых клеточных трансмембранных белков, в частности, MHC II класса и TRAIL-R1 (TNF-related apoptosis-inducing ligand receptor 1) [106, 107]. Предполагается, что MARCH8 узнает трехмерную структуру трансмембранных доменов белков, а не специфические мотивы; однако достоверных экспериментальных доказательств этому пока нет.
Как показано Tada и соавт. [105], эктопическая экспрессия MARCH8 в клетках-продуцентах вируса не влияет на уровень продукции вируса, однако снижает инфекционность вирусных частиц. MARCH8 блокирует попадание белка оболочки вируса в вирионы путем снижения экспрессии Env на поверхности клетки, вероятно, путем взаимодействия с ним. В результате, происходит существенное снижение эффективности слияния вируса с клеткой. На данный момент точная стратегия MARCH8-опосредованной рестрикции ВИЧ неизвестна, но показано, что вспомогательные белки Vpr, Vpu и Nef не интерферируют с MARCH8 [105].
SERINC3 и SERINC5
Долгое время не было понимания того, каким образом вирусный белок Nef повышает инфекционность вирусных частиц [106, 107]. Usami и соавт. [108] предположили, что Nef противодействует некоторому фактору рестрикции, снижая его плотность на поверхности клетки и препятствуя инкорпорации в вирионы. В результате проведенного протеомного анализа вирионов Nef+ ВИЧ-1 и Nef– ВИЧ-1 в последних обнаружены мембранные белки SERINC3 и SERINC5 (SERin INCorporator). Показано, что внедрение SERINC3 и SERINC5 в вирионы ВИЧ, действительно, снижает их способность инфицировать хозяйские клетки [108].
Семейство SERINC состоит из пяти белков, характеризующихся наличием 10–11 трансмембранных доменов. Белки SERINC участвуют в биосинтезе сфинголипидов и фосфатидилсерина [109]. Однако только SERINC3 и SERINC5 считаются факторами рестрикции ВИЧ-1, причем SERINC5 обладает более выраженным противовирусным действием [110, 111]. Предполагаемый механизм действия SERINC5 заключается в формировании олигомеров на мембране вирусных частиц, что приводит к снижению эффективности слияния вирусной и клеточной мембран [112]. Кроме того, попадание SERINC5 в вирусные частицы приводит к повышению их чувствительности к широконейтрализующим антителам, узнающим консервативный домен gp41 белка оболочки [112, 113].
Вспомогательный белок ВИЧ-1 Nef эффективно удаляет с поверхности клетки SERINC3 и SERINC5, препятствуя их попаданию в вирионы [108, 111, 114]. Механизм отрицательной регуляции SERINC3 и SERINC5 аналогичен снижению экспрессии CD4 на поверхности клетки: в каждом случае Nef использует клеточную транспортную систему для обеспечения деградации таргетных клеточных белков в эндолизосомах. Недавно показано, что чувствительность SERINC5 к Nef-опосредованной деградации определяется структурой его внутриклеточного домена ICL4 (intracellular loop 4). Так, при замене чувствительного к Nef варианта ICL4 на устойчивый белок SERINC5 становится резистентным к действию Nef, который уже не препятствует SERINC5-индуцируемой рестрикции ВИЧ [115].
BST-2 (CD317, Tetherin)
Одной из причин, по которой ВИЧ-1, принадлежащий группе М, широко распространился среди людей и стал пандемичным, стало приобретение высокой резистентности к клеточному фактору BST-2 за счет эволюции и адаптации вирусного белка Vpu к BST-2-индуцируемой рестрикции [116]. Другие группы ВИЧ-1, а также ВИЧ-2 и SIV, не эволюционировали в этом направлении и, предположительно, защищаются от действия белка BST-2 более древним и менее эффективным способом – при помощи акцессорного белка Nef [117–120]. Вспомогательный вирусный белок Vpu закодирован в геноме вируса ВИЧ-1, но отсутствует у вируса ВИЧ-2 и большинства штаммов SIV. Дефектный по гену vpu вирус, Δvpu-ВИЧ-1, обладает определенными характерными чертами, отличающими его от вируса дикого типа: меньше “шипов” на поверхности вириона и скопление “кластеров” вирусных частиц у поверхности клетки [121–123]. Ранее показано, что снижение содержания шипов на вирусной оболочке обусловлено взаимодействием молекулы CD4 с вирусным белком оболочки gp160 в аппарате Гольджи. Vpu связывается с CD4, освобождая тем самым белок оболочки, который беспрепятственно может транспортироваться к плазматической мембране клетки [124].
Труднее было понять, чем вызвана кластеризация вирусных частиц Δvpu-ВИЧ-1 у поверхности клетки. C помощью технологии сайт-направленного мутагенеза в гене vpu показано, что в высвобождении gp160 и кластеризации вирионов у поверхности клетки задействованы разные домены Vpu. Так, скопление вирионов Δvpu-ВИЧ-1 наблюдалось только в определенных типах клеток и могло быть усилено действием IFN I типа [125, 126]. Vpu-дефектный вирус оказался также более чувствителен к подавлению репликации под действием IFN-α. Кроме того, такие вирусные частицы оказались зрелыми и могли быть удалены с поверхности зараженной клетки при обработке протеазой. Следовательно, есть клеточный белок, способный удерживать вирионы Δvpu-ВИЧ-1 на поверхности клетки и тем самым препятствующий их отпочковыванию. По данным электронной микроскопии, вирусные частицы Δvpu-ВИЧ-1 не только прикрепляются к поверхности зараженной клетки, но и “слипаются” друг к другу [23]. Значит, молекула, задействованная в этом процессе, может быть инкорпорирована в оболочку вируса и предотвращать распространение вируса путем удержания вирусных частиц на поверхности зараженной клетки. Все эти наблюдения дают основания считать, что белок Vpu появился и закрепился в ходе коэволюции ВИЧ-1 и человека как мера противодействия IFN-опосредованной защите клетки.
Neil с соавт. [23] предложили название “tetherin” (от англ. tether – привязывать, ограничивать) для поверхностного белка клетки, ответственного за этот процесс. Для идентификации конкретной молекулы, выполняющей эту роль, с помощью микрочипов проанализировали транскриптом клеток до и после стимуляции IFN-α. Среди кандидатов оказался белок BST-2 (bone marrow stromal antigen 2) и три IFN-индуцируемых трансмембранных белка: IFITM1, IFITM2 и IFITM3. Способность BST-2 “привязывать” вирус к мембране подтвердили экспериментально. Важно, что BST-2 никак не влиял на экспрессию и процессинг вирусного белка Gag, ингибируя только высвобождение сформированных вирусных частиц из клетки.
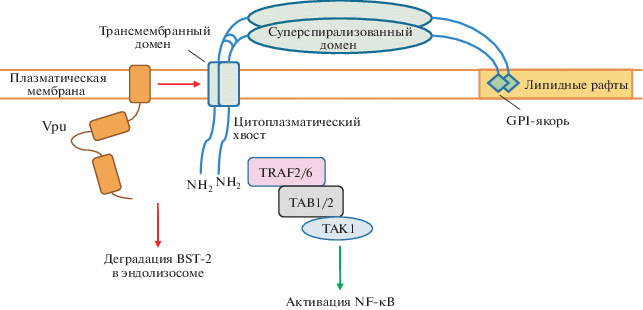
BST-2 обладает уникальной структурой: N-конец с цитоплазматическим доменом переходит в трансмембранный домен, затем располагается внешний суперспирализованный домен и С-концевой гликозилфосфатидилинозитольный якорь (GPI-якорь), часто называемый вторым трансмембранным доменом [127, 128] (рис. 3). BST-2 локализован в липидных рафтах плазматической мембраны, в транс-Гольджи сети и в эндосомах [127, 129, 130]. Два ассоциированных с мембраной домена определяют способность белка удерживать вирусные частицы у мембраны и связывать их друг с другом: при почковании вирионов один из доменов остается на мембране, тогда как другой оказывается встроенным в липидную оболочку вируса [131–134] (рис. 4). Показано, что BST-2 функционирует в качестве гомодимеров и гомотетрамеров, ковалентно связанных через остатки цистеинов, расположенных в суперспирализованных внешних доменах BST-2 [129, 131, 135, 136] (рис. 3).
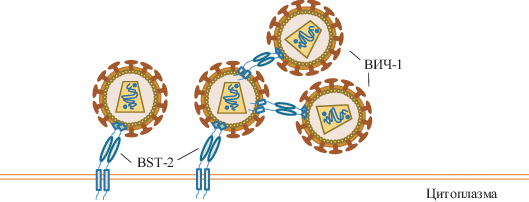
Прикрепление вируса к мембране зараженной клетки снижает инфицирование “свободным” вирусом. Недавно стало известно, что ВИЧ-1 более эффективно передается через межклеточные контакты, в особенности через вирусологический синапс, характеризующийся привлечением в область контакта клеток молекул адгезии, вирусных белков и клеточных рецепторов [137, 138]. Интересным и по-прежнему открытым остается вопрос о роли BST-2 в межклеточной передаче ВИЧ. На данный момент имеется несколько противоречащих друг другу данных. С одной стороны, показано ингибирующее влияние BST-2 на передачу инфекции от клетки к клетке [139–142]; с другой стороны, известно, что при межклеточной трансмиссии вируса BST-2 не только не работает как рестрикционный фактор ВИЧ, но, напротив, способствует более эффективному заражению соседних клеток за счет того, что удерживает в области контакта инфицированной/неинфицированной клеток вирусные частицы [143–145]. Разработка методов количественного измерения межклеточной инфекции, позволяющих отделить клетки-продуценты от зараженных в единой культуре, может разрешить это противоречие и выявить роль BST-2 в межклеточной передаче ВИЧ-1 [146, 147].
Цитоплазматический хвост BST-2 определяет участие белка во внутриклеточном сигналинге. Ранее BST-2 обнаружили среди белков, активирующих сигнальный путь NF-κB [148]. Удерживание вируса у поверхности и активация сигнального пути NF-κB – две самостоятельные функции белка, выполняемые разными доменами; при этом связывание вириона и димеризация BST-2 индуцируют запуск NF-κB-сигналинга [149, 150]. К предполагаемому сайту связывания TRAF на молекуле BST-2 привлекаются TRAF2 и TRAF6, что ведет к активации TAK1 и, соответственно, запуску канонического пути NF-κB-сигналинга с последующей экспрессией провоспалительных цитокинов IL-6, CXCL10 и IFN-β.
Вспомогательный белок Vpu вируса ВИЧ-1 связывается с трансмембранным доменом BST-2 [151] и привлекает SCF E3-убиквитинлигазу, что приводит к убиквитинированию и деградации BST-2 в эндолизосоме [151, 152]. Интересно, что у обезьян белок Nef SIV эффективно противодействует BST-2 [116, 117], тогда как у человека Nef HIV не считается фактором, интерферирующим с BST-2.
СПОСОБЫ ВЫЯВЛЕНИЯ НОВЫХ ФАКТОРОВ РЕСТРИКЦИИ ВИЧ
Большинство известных на сегодняшний день факторов рестрикции ВИЧ (IFITM, BST-2, APOBEC3) были найдены среди ISGs. В инфицированной клетке, благодаря вирусным сенсорам (TLR, RLR и т.п.), запускаются сигнальные пути IFN и NF-κB, что приводит к экспрессии белков клеточной защиты от вирусной инфекции. Вполне логично, что среди них обнаружили факторы рестрикции ВИЧ.
Еще одна эффективная стратегия исследования факторов рестрикции заключается в поиске белков-партнеров вспомогательных белков вируса. ВИЧ способен инфицировать клетки без экспрессии Vif, Vpr, Vpu и Nef (Vpx у ВИЧ-2), однако их присутствие в геноме вируса неслучайно. Главной функцией вспомогательных белков считается противодействие клеточной защите. Идентификация факторов рестрикции SAMHD1 и SERINC3/5 как мишеней соответственно Vpx и Nef подтверждает это предположение.
С развитием технологий высокопроизводительного секвенирования появились новые возможности для поиска факторов рестрикции. Уже сегодня накоплены огромные массивы данных, полученные в результате siРНК-, shРНК- и CRISPR/Cas9-скринингов по поиску факторов рестрикции [153] и факторов репликации ВИЧ-1 [154–158]. Эти массивы данных еще “ждут своего часа”: систематизации [159] и интерпретации в соответствии с результатами экспериментальных исследований, – но уже сейчас их использование стимулирует поиск и позволяет быстрее находить новые, интересные с клинической точки зрения, клеточные белки, вовлеченные в процесс ВИЧ-инфекции.
Работа выполнена при поддержке гранта РНФ “Поиск новых факторов рестрикции ВИЧ-1, ограничивающих репликацию вируса в условиях его межклеточной трансмиссии, путем скрининга библиотеки нокаутов GeCKO” (проект № 15-15-00135) и гранта РФФИ “Изучение роли KPNA1, CD82 и ряда других белков в репликации HTLV-1, выявленных с помощью скрининга библиотеки нокаутов GeCKO” (проект № 18-34-00712).
Список литературы
Sharp P.M., Hahn B.H. (2011) Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 1(1), a006841. doi 10.1101/cshperspect.a006841
Mourez T., Simon F., Plantier J.-C. (2013) Non-M variants of human immunodeficiency virus type 1. Clin. Microbiol. Rev. 26(3), 448–461. doi 10.1128/CMR.00012-13
Vallari A., Holzmayer V., Harris B., Yamaguchi J., Ngansop C., Makamche F., Mbanya D., Kaptué L., Ndembi N., Gürtler L., Devare S., Brennan C.A. (2011) Confirmation of putative HIV-1 group P in Cameroon. J. Virol. 85(3), 1403–1407. doi 10.1128/ JVI.02005-10
Vallari A., Bodelle P., Ngansop C., Makamche F., Ndembi N., Mbanya D., Kaptué L., Gürtler L.G., McArthur C.P., Devare S.G., Simon F. (2010) Four new HIV-1 group N isolates from Cameroon: prevalence continues to be low. AIDS Res. Hum. Retroviruses 26(9806), 109–115. doi 10.1016/S0140-6736(11)61457-8
Plantier J.C., Leoz M., Dickerson J.E., De Oliveira F., Cordonnier F., Lemée V., Damond F., Robertson D.L., Simon F. (2009) A new human immunodeficiency virus derived from gorillas. Nat. Med. 15(8), 871–872. doi 10.1038/nm.2016
Soliman M., Srikrishna G., Balagopal A. (2017) Mechanisms of HIV-1 control. Curr. HIV/AIDS Rep. 14(3), 101–109. doi 10.1007/s11904-017-0357-9
Карамов Э.В., Петров Р.В. (2011) Суверенный иммунитет. Часть 2. Клеточные факторы антиретровирусной защиты: тетерин, семейство APOBEC3, клеточные микроРНК. CRISPR/Cas системы прокариот. Физиология и патология иммунной системы. 15 (4), 3–23.
Карамов Э.В., Петров Э.В. (2011) Суверенный иммунитет. Часть 1. Особенности антиретровирусного иммунного ответа. Клеточные факторы защиты, взаимодействующие с капсидными белками ретровирусов: TRIM5, циклофилин. Физиология и патология иммунной системы 15 (3), 3–22.
Brass A.L., Huang I.-C., Benita Y., John S.P., Krishnan M.N., Feeley E.M., Ryan B.J., Weyer J.L., van der Weyden L., Fikrig E., Adams D.J., Xavier R.J., Farzan M., Elledge S.J. (2009) The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 139(7), 1243–1254. doi 10.1016/j.cell.2009.12.017
Bishop K.N., Verma M., Kim E.-Y., Wolinsky S.M., Malim M.H. (2008) APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 4(12), e1000231. doi 10.1371/journal.ppat.1000231
Sheehy A.M., Gaddis N.C., Choi J.D., Malim M.H. (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 418(6898), 646–650. doi 10.1038/nature00939
Harris R.S., Bishop K.N., Sheehy A.M., Craig H.M., Petersen-Mahrt S.K., Watt I.N., Neuberger M.S., Malim M.H. (2003) DNA deamination mediates innate immunity to retroviral infection. Cell. 113(6), 803–809. http://www.ncbi.nlm.nih.gov/pubmed/12809610.
Stremlau M., Owens C.M., Perron M.J., Kiessling M., Autissier P., Sodroski J. (2004) The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 427(6977), 848–853. doi 10.1038/nature02343
Sayah D.M., Sokolskaja E., Berthoux L., Luban J. (2004) Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 430(6999), 569–573. doi 10.1038/nature02777
Stremlau M., Perron M., Lee M., Li Y., Song B., Javanbakht H., Diaz-Griffero F., Anderson D.J., Sundquist W.I., Sodroski J. (2006) Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. USA. 103(14), 5514–5519. doi 10.1073/pnas.0509996103
Kane M., Yadav S.S., Bitzegeio J., Kutluay S.B., Zang T., Wilson S.J., Schoggins J.W., Rice C.M., Yamashita M., Hatziioannou T., Bieniasz P.D. (2013) MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 502(7472), 563–566. doi 10.1038/nature12653
Goujon C., Moncorgé O., Bauby H., Doyle T., Ward C.C., Schaller T., Hué S., Barclay W.S., Schulz R., Malim M.H. (2013) Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature. 502(7472), 559–562. doi 10.1038/nature12542
Liu Z., Pan Q., Ding S., Qian J., Xu F., Zhou J., Cen S., Guo F., Liang C. (2013) The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe. 14(4), 398–410. doi 10.1016/j.chom.2013.08.015
Li M., Kao E., Gao X., Sandig H., Limmer K., Pavon-Eternod M., Jones T.E., Landry S., Pan T., Weitzman M.D., David M. (2012) Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature. 491(7422), 125–128. doi 10.1038/ nature11433
Hrecka K., Hao C., Gierszewska M., Swanson S.K., Kesik-Brodacka M., Srivastava S., Florens L., Washburn M.P., Skowronski J. (2011) Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 474(7353), 658–661. doi 10.1038/nature10195
Berger A., Sommer A.F.R., Zwarg J., Hamdorf M., Welzel K., Esly N., Panitz S., Reuter A., Ramos I., Jatiani A., Mulder L.C.F., Fernandez-Sesma A., Rutsch F., Simon V., König R., Flory E. (2011) SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutières syndrome are highly susceptible to HIV-1 infection. PLoS Pathog. 7(12), e1002425. doi 10.1371/journal.ppat.1002425
Goldstone D.C., Ennis-Adeniran V., Hedden J.J., Groom H.C.T., Rice G.I., Christodoulou E., Walker P.A., Kelly G., Haire L.F., Yap M.W., de Carvalho L.P.S., Stoye J.P., Crow Y.J., Taylor I.A., Webb M. (2011) HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 480(7377), 379–382. doi 10.1038/nature10623
Neil S.J.D., Zang T., Bieniasz P.D. (2008) Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 451(7177), 425–430. doi 10.1038/ nature06553
Sauter D., Specht A., Kirchhoff F. (2010) Tetherin: holding on and letting go. Cell. 141(3), 392–398. doi 10.1016/j.cell.2010.04.022
Francis M.L., Meltzer M.S. (1993) Induction of IFN-alpha by HIV-1 in monocyte-enriched PBMC requires gp120-CD4 interaction but not virus replication. J. Immunol. 151(4), 2208–2216. http://www.ncbi. nlm.nih.gov/pubmed/8345204.
Jaffe E.A., Armellino D., Lam G., Cordon-Cardo C., Murray H.W., Evans R.L. (1989) IFN-gamma and IFN-alpha induce the expression and synthesis of Leu 13 antigen by cultured human endothelial cells. J. Immunol. 143(12), 3961–3966. http://www.ncbi.nlm.nih.gov/ pubmed/2512344.
Lu J., Pan Q., Rong L., He W., Liu S.-L., Liang C. (2011) The IFITM proteins inhibit HIV-1 infection. J. Virol. 85(5), 2126–2137. doi 10.1128/JVI.01531-10
Weston S., Czieso S., White I.J., Smith S.E., Wash R.S., Diaz-Soria C., Kellam P., Marsh M. (2016) Alphavirus restriction by IFITM proteins. Traffic. 17(9), 997–1013. doi 10.1111/tra.12416
Feeley E.M., Sims J.S., John S.P., Chin C.R., Pertel T., Chen L.-M., Gaiha G.D., Ryan B.J., Donis R.O., Elledge S.J., Brass A.L. (2011) IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 7(10), e1002337. doi 10.1371/journal.ppat.1002337
Li K., Markosyan R.M., Zheng Y.-M., Golfetto O., Bungart B., Li M., Ding S., He Y., Liang C., Lee J.C., Gratton E., Cohen F.S., Liu S.-L. (2013) IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 9(1), e1003124. doi 10.1371/journal.ppat.1003124
Desai T.M., Marin M., Chin C.R., Savidis G., Brass A.L., Melikyan G.B. (2014) IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 10(4), e1004048. doi 10.1371/journal.ppat.1004048
Amini-Bavil-Olyaee S., Choi Y.J., Lee J.H., Shi M., Huang I.-C., Farzan M., Jung J.U. (2013) The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe. 13(4), 452–464. doi 10.1016/j.chom.2013.03.006
Compton A.A., Bruel T., Porrot F., Mallet A., Sachse M., Euvrard M., Liang C., Casartelli N., Schwartz O. (2014) IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe. 16(6), 736–747. doi 10.1016/j.chom.2014.11.001
Foster T.L., Wilson H., Iyer S.S., Coss K., Doores K., Smith S., Kellam P., Finzi A., Borrow P., Hahn B.H., Neil S.J.D. (2016) Resistance of transmitted founder HIV-1 to IFITM-mediated restriction. Cell Host Microbe. 20(4), 429–442. doi 10.1016/j.chom.2016.08.006
Yu J., Li M., Wilkins J., Ding S., Swartz T.H., Esposito A.M., Zheng Y.-M., Freed E.O., Liang C., Chen B.K., Liu S.-L. (2015) IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep. 13(1), 145–156. doi 10.1016/j.celrep.2015.08.055
Rajsbaum R., García-Sastre A., Versteeg G.A. (2014) TRIMmunity: the roles of the TRIM E3-Ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 426(6), 1265–1284. doi 10.1016/j.jmb.2013.12.005
Versteeg G.A., Benke S., García-Sastre A., Rajsbaum R. (2014) InTRIMsic immunity: positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev. 25(5), 563–576. doi 10.1016/j.cytogfr.2014.08.001
Ozato K., Shin D.-M., Chang T.-H., Morse H.C. (2008) TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8(11), 849–860. doi 10.1038/nri2413
Uchil P.D., Hinz A., Siegel S., Coenen-Stass A., Pertel T., Luban J., Mothes W. (2013) TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 87(1), 257–272. doi 10.1128/JVI.01804-12
Uchil P.D., Quinlan B.D., Chan W.-T., Luna J.M., Mothes W. (2008) TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 4(2), e16. doi 10.1371/journal.ppat.0040016
Versteeg G.A., Rajsbaum R., Sánchez-Aparicio M.T., Maestre A.M., Valdiviezo J., Shi M., Inn K.-S., Fernandez-Sesma A., Jung J., García-Sastre A. (2013) The E3-Ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity. 38(2), 384–398. doi 10.1016/j.immuni.2012.11.013
Reymond A., Meroni G., Fantozzi A., Merla G., Cairo S., Luzi L., Riganelli D., Zanaria E., Messali S., Cainarca S., Guffanti A., Minucci S., Pelicci P.G., Ballabio A. (2001) The tripartite motif family identifies cell compartments. EMBO J. 20(9), 2140–2151. doi 10.1093/emboj/20.9.2140
Esposito D., Koliopoulos M.G., Rittinger K. (2017) Structural determinants of TRIM protein function. Biochem. Soc. Trans. 45(1), 183–191. doi 10.1042/ BST20160325
Napolitano L.M., Meroni G. (2012) TRIM family: pleiotropy and diversification through homomultimer and heteromultimer formation. IUBMB Life. 64(1), 64–71. doi 10.1002/iub.580
Sardiello M., Cairo S., Fontanella B., Ballabio A., Meroni G. (2008) Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 8(1), 225. doi 10.1186/1471-2148-8-225
Han K., Lou D.I., Sawyer S.L. (2011) Identification of a genomic reservoir for new TRIM genes in primate genomes. PLoS Genet. 7(12), e1002388. doi 10.1371/ journal.pgen.1002388
Nisole S., Lynch C., Stoye J.P., Yap M.W. (2004) A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. USA. 101(36), 13324–13328. doi 10.1073/ pnas.0404640101
Lamichhane R., Mukherjee S., Smolin N., Pauszek R.F., Bradley M., Sastri J., Robia S.L., Millar D., Campbell E.M. (2017) Dynamic conformational changes in the rhesus TRIM5α dimer dictate the potency of HIV-1 restriction. Virology. 500, 161–168. doi 10.1016/j.virol.2016.10.003
Li Y.-L., Chandrasekaran V., Carter S.D., Woodward C.L., Christensen D.E., Dryden K.A., Pornil-los O., Yeager M., Ganser-Pornillos B.K., Jensen G.J., Sundquist W.I. (2016) Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. Elife. 5. doi 10.7554/eLife.16269
Sastri J., Campbell E.M. (2011) Recent insights into the mechanism and consequences of TRIM5α retroviral restriction. AIDS Res. Hum. Retroviruses. 27(3), 231–238. doi 10.1089/AID.2010.0367
Pertel T., Hausmann S., Morger D., Züger S., Guerra J., Lascano J., Reinhard C., Santoni F.A., Uchil P.D., Chatel L., Bisiaux A., Albert M.L., Strambio-De-Castillia C., Mothes W., Pizzato M., Grütter M.G., Luban J. (2011) TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 472(7343), 361–365. doi 10.1038/nature09976
Wagner J.M., Christensen D.E., Bhattacharya A., Dawidziak D.M., Roganowicz M.D., Wan Y., Pumroy R.A., Demeler B., Ivanov D.N., Ganser-Pornillos B.K., Sundquist W.I., Pornillos O. (2018) General model for retroviral capsid pattern recognition by TRIM5 proteins. J. Virol. 92(4), e01563-17. doi 10.1128/JVI.01563-17
Yudina Z., Roa A., Johnson R., Biris N., de Souza Aranha Vieira D.A., Tsiperson V., Reszka N., Taylor A.B., Hart P.J., Demeler B., Diaz-Griffero F., Ivanov D.N. (2015) RING dimerization links higher-order assembly of TRIM5α to synthesis of K63-linked polyubiquitin. Cell Rep. 12(5), 788–797. doi 10.1016/j.celrep.2015.06.072
van Tol S., Hage A., Giraldo M., Bharaj P., Rajsbaum R. (2017) The TRIMendous role of TRIMs in virus–host interactions. Vaccines. 5(3), e23. doi 10.3390/ vaccines5030023
Rold C.J., Aiken C. (2008) Proteasomal degradation of TRIM5α during retrovirus restriction. PLoS Pathog. 4(5), e1000074. doi 10.1371/journal.ppat.1000074
Kutluay S.B., Perez-Caballero D., Bieniasz P.D. (2013) Fates of retroviral core components during unrestricted and TRIM5-restricted infection. PLoS Pathog. 9(3), e1003214. doi 10.1371/journal.ppat.1003214
Yap M.W., Nisole S., Stoye J.P. (2005) A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol. 15(1), 73–78. doi 10.1016/j.cub.2004.12.042
Barr S.D., Smiley J.R., Bushman F.D. (2008) The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 4(2), e1000007. doi 10.1371/journal.ppat.1000007
Singh R., Gaiha G., Werner L., McKim K., Mlisana K., Luban J., Walker B.D., Karim S.S.A., Brass A.L., Ndung’u T., CAPRISA Acute Infection Study Team (2011) Association of TRIM22 with the type 1 interferon response and viral control during primary HIV-1 infection. J. Virol. 85(1), 208–216. doi 10.1128/ JVI.01810-10
Kajaste-Rudnitski A., Marelli S.S., Pultrone C., Pertel T., Uchil P.D., Mechti N., Mothes W., Poli G., Luban J., Vicenzi E. (2011) TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappaB-responsive long terminal repeat elements. J. Virol. 85(10), 5183–5196. doi 10.1128/JVI.02302-10
Turrini F., Marelli S., Kajaste-Rudnitski A., Lusic M., Van Lint C., Das A.T., Harwig A., Berkhout B., Vicenzi E. (2015) HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology. 12(1), 104. doi 10.1186/s12977-015-0230-0
Deeks S.G., Lewin S.R., Ross A.L., Ananworanich J., Benkirane M., Cannon P., Chomont N., Douek D., Lifson J.D., Lo Y.-R., Kuritzkes D., Margolis D., Mellors J., Persaud D., Tucker J.D., Barre-Sinoussi F., International AIDS Society Towards a Cure Working Group (2016) International AIDS Society global scientific strategy: towards an HIV cure 2016. Nat. Med. 22(8), 839–850. doi 10.1038/nm.4108
Yuan T., Yao W., Huang F., Sun B., Yang R. (2014) The human antiviral factor TRIM11 is under the regulation of HIV-1 Vpr. PLoS One. 9(8), e104269. doi 10.1371/journal.pone.0104269
Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Ségéral E., Yatim A., Emiliani S., Schwartz O., Benkirane M. (2011) SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 474(7353), 654–657. doi 10.1038/nature10117
Descours B., Cribier A., Chable-Bessia C., Ayinde D., Rice G., Crow Y., Yatim A., Schwartz O., Laguette N., Benkirane M. (2012) SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+ T-cells. Retrovirology. 9(1), 87. doi 10.1186/1742-4690-9-87
Ji X., Tang C., Zhao Q., Wang W., Xiong Y. (2014) Structural basis of cellular dNTP regulation by SAMHD1. Proc. Natl. Acad. Sci. USA. 111(41), E4305–E4314. doi 10.1073/pnas.1412289111
Ji X., Wu Y., Yan J., Mehrens J., Yang H., DeLucia M., Hao C., Gronenborn A.M., Skowronski J., Ahn J., Xiong Y. (2013) Mechanism of allosteric activation of SAMHD1 by dGTP. Nat. Struct. Mol. Biol. 20(11), 1304–1309. doi 10.1038/nsmb.2692
Hansen E.C., Seamon K.J., Cravens S.L., Stivers J.T. (2014) GTP activator and dNTP substrates of HIV-1 restriction factor SAMHD1 generate a long-lived activated state. Proc. Natl. Acad. Sci. USA. 111(18), E1843–E1851. doi 10.1073/pnas.1401706111
Amie S.M., Bambara R.A., Kim B. (2013) GTP is the primary activator of the anti-HIV restriction factor SAMHD1. J. Biol. Chem. 288(35), 25001–25006. doi 10.1074/jbc.C113.493619
Yan J., Kaur S., DeLucia M., Hao C., Mehrens J., Wang C., Golczak M., Palczewski K., Gronenborn A.M., Ahn J., Skowronski J. (2013) Tetramerization of SAMHD1 is required for biological activity and inhibition of HIV infection. J. Biol. Chem. 288(15), 10406–10417. doi 10.1074/jbc.M112.443796
Beloglazova N., Flick R., Tchigvintsev A., Brown G., Popovic A., Nocek B., Yakunin A.F. (2013) Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 288(12), 8101–8110. doi 10.1074/ jbc.M112.431148
Ryoo J., Choi J., Oh C., Kim S., Seo M., Kim S.-Y., Seo D., Kim J., White T.E., Brandariz-Nuñez A., Diaz-Griffero F., Yun C.-H., Hollenbaugh J.A., Kim B., Baek D., Ahn K. (2014) The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 20(8), 936–941. doi 10.1038/nm.3626
Seamon K.J., Sun Z., Shlyakhtenko L.S., Lyubchenko Y.L., Stivers J.T. (2015) SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res. 43(13), 6486–6499. doi 10.1093/nar/gkv633
Antonucci J.M., St. Gelais C., de Silva S., Yount J.S., Tang C., Ji X., Shepard C., Xiong Y., Kim B., Wu L. (2016) SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 22(10), 1072–1074. doi 10.1038/nm.4163
Ryoo J., Hwang S.-Y., Choi J., Oh C., Ahn K. (2016) Reply to SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 22(10), 1074–1075. doi 10.1038/nm.4164
Ahn J., Hao C., Yan J., DeLucia M., Mehrens J., Wang C., Gronenborn A.M., Skowronski J. (2012) HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 287(15), 12550–12558. doi 10.1074/jbc.M112.340711
Wei W., Guo H., Han X., Liu X., Zhou X., Zhang W., Yu X.-F. (2012) A novel DCAF1-binding motif required for Vpx-mediated degradation of nuclear SAMHD1 and Vpr-induced G2 arrest. Cell. Microbiol. 14(11), 1745–1756. doi 10.1111/j.1462-5822.2012.01835.x
Kyei G.B., Cheng X., Ramani R., Ratner L. (2015) Cyclin L2 is a critical HIV dependency factor in macrophages that controls SAMHD1 abundance. Cell Host Microbe. 17(1), 98–106. doi 10.1016/j.chom.2014.11.009
Jarmuz A., Chester A., Bayliss J., Gisbourne J., Dunham I., Scott J., Navaratnam N. (2002) An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 79(3), 285–296. doi 10.1006/geno.2002.6718
Harris R.S., Petersen-Mahrt S.K., Neuberger M.S. (2002) RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell. 10(5), 1247–1253. http://www.ncbi.nlm.nih.gov/pubmed/ 12453430.
Refsland E.W., Stenglein M.D., Shindo K., Albin J.S., Brown W.L., Harris R.S. (2010) Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38(13), 4274–4284. doi 10.1093/ nar/gkq174
Desimmie B.A., Delviks-Frankenberrry K.A., Burdick R.C., Qi D., Izumi T., Pathak V.K. (2014) Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 426(6), 1220–1245. doi 10.1016/j.jmb.2013.10.033
Mangeat B., Turelli P., Caron G., Friedli M., Perrin L., Trono D. (2003) Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 424(6944), 99–103. doi 10.1038/nature01709
Mariani R., Chen D., Schröfelbauer B., Navarro F., König R., Bollman B., Münk C., Nymark-McMahon H., Landau N.R. (2003) Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 114(1), 21–31. http://www.ncbi.nlm.nih.gov/ pubmed/12859895.
Suspène R., Rusniok C., Vartanian J.-P., Wain-Hobson S. (2006) Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 34(17), 4677–4684. doi 10.1093/nar/gkl555
Yu Q., König R., Pillai S., Chiles K., Kearney M., Palmer S., Richman D., Coffin J.M., Landau N.R. (2004) Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11(5), 435–442. doi 10.1038/nsmb758
Holmes R.K., Malim M.H., Bishop K.N. (2007) APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 32(3), 118–128. doi 10.1016/j.tibs.2007.01.004
Albin J.S., Brown W.L., Harris R.S. (2014) Catalytic activity of APOBEC3F is required for efficient restriction of Vif-deficient human immunodeficiency virus. Virology. 450–451, 49–54. doi 10.1016/j.virol.2013.11.041
Browne E.P., Allers C., Landau N.R. (2009) Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology. 387(2), 313–321. doi 10.1016/j.virol.2009.02.026
Yu X., Yu Y., Liu B., Luo K., Kong W., Mao P., Yu X.-F. (2003) Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 302(5647), 1056–1060. doi 10.1126/science.1089591
Sheehy A.M., Gaddis N.C., Malim M.H. (2003) The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9(11), 1404–1407. doi 10.1038/nm945
Kim E.-Y., Lorenzo-Redondo R., Little S.J., Chung Y.-S., Phalora P.K., Maljkovic Berry I., Archer J., Penugonda S., Fischer W., Richman D.D., Bhattacharya T., Malim M.H., Wolinsky S.M. (2014) Human APOBEC3 induced mutation of human immunodeficiency virus type-1 contributes to adaptation and evolution in natural infection. PLoS Pathog. 10(7), e1004281. doi 10.1371/journal.ppat.1004281
Fourati S., Malet I., Binka M., Boukobza S., Wirden M., Sayon S., Simon A., Katlama C., Simon V., Calvez V., Marcelin A.-G. (2010) Partially active HIV-1 Vif alleles facilitate viral escape from specific antiretrovirals. AIDS. 24(15), 2313–2321. doi 10.1097/ QAD.0b013e32833e515a
Dicks M.D.J., Goujon C., Pollpeter D., Betancor G., Apolonia L., Bergeron J.R.C., Malim M.H. (2016) Oligomerization requirements for MX2-mediated suppression of HIV-1 infection. J. Virol. 90(1), 22–32. doi 10.1128/JVI.02247-15
Haller O., Kochs G. (2011) Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J. Interf. Cytokine Res. 31(1), 79–87. doi 10.1089/jir.2010.0076
Matreyek K.A., Wang W., Serrao E., Singh P.K., Levin H.L., Engelman A. (2014) Host and viral determinants for MxB restriction of HIV-1 infection. Retrovirology. 11(1), 90. doi 10.1186/s12977-014-0090-z
Fribourgh J.L., Nguyen H.C., Matreyek K.A., Alvarez F.J.D., Summers B.J., Dewdney T.G., Aiken C., Zhang P., Engelman A., Xiong Y. (2014) Structural insight into HIV-1 restriction by MxB. Cell Host Microbe. 16(5), 627–638. doi 10.1016/j.chom.2014.09.021
Goujon C., Greenbury R.A., Papaioannou S., Doyle T., Malim M.H. (2015) A triple-arginine motif in the amino-terminal domain and oligomerization are required for HIV-1 inhibition by human MX2. J. Virol. 89(8), 4676–4680. doi 10.1128/JVI.00169-15
Fricke T., White T.E., Schulte B., de Souza Aranha Vieira D.A., Dharan A., Campbell E.M., Brandariz-Nuñez A., Diaz-Griffero F. (2014) MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology. 11(1), 68. doi 10.1186/s12977-014-0068-x
Nakayama E.E., Saito A., Sultana T., Jin Z., Nohata K., Shibata M., Hosoi M., Motomura K., Shioda T., Sangkitporn S., Loket R., Saeng-Aroon S. (2018) Naturally occurring mutations in HIV-1 CRF01_AE capsid affect viral sensitivity to restriction factors. AIDS Res. Hum. Retroviruses. 34(4), 382–392. doi 10.1089/AID.2017.0212
Wei W., Guo H., Ma M., Markham R., Yu X.-F. (2016) Accumulation of MxB/Mx2-resistant HIV-1 capsid variants during expansion of the HIV-1 epidemic in human populations. EBioMedicine. 8, 230–236. doi 10.1016/j.ebiom.2016.04.020
Mavrommatis E., Fish E.N., Platanias L.C. (2013) The schlafen family of proteins and their regulation by interferons. J. Interferon Cytokine Res. 33(4), 206–210. doi 10.1089/jir.2012.0133
Coccia E.M., Krust B., Hovanessian A.G. (1994) Specific inhibition of viral protein synthesis in HIV-infected cells in response to interferon treatment. J. Biol. Chem. 269(37), 23087–23094. http://www.ncbi.nlm.nih.gov/pubmed/7521875.
Stabell A.C., Hawkins J., Li M., Gao X., David M., Press W.H., Sawyer S.L. (2016) Non-human primate schlafen11 inhibits production of both host and viral proteins. PLOS Pathog. 12(12), e1006066. doi 10.1371/journal.ppat.1006066
Tada T., Zhang Y., Koyama T., Tobiume M., Tsunetsugu-Yokota Y., Yamaoka S., Fujita H., Tokunaga K. (2015) MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat. Med. 21(12), 1502–1507. doi 10.1038/nm.3956
Ohmura-Hoshino M., Matsuki Y., Aoki M., Goto E., Mito M., Uematsu M., Kakiuchi T., Hotta H., Ishido S. (2006) Inhibition of MHC class II expression and immune responses by c-MIR. J. Immunol. 177(1), 341–354. http://www.ncbi.nlm.nih.gov/pubmed/16785530.
van de Kooij B., Verbrugge I., de Vries E., Gijsen M., Montserrat V., Maas C., Neefjes J., Borst J. (2013) Ubiquitination by the membrane-associated RING-CH-8 (MARCH-8) ligase controls steady-state cell surface expression of tumor necrosis factor-related apoptosis inducing ligand (TRAIL) receptor 1. J. Biol. Chem. 288(9), 6617–6628. doi 10.1074/jbc.M112.448209
Usami Y., Wu Y., Göttlinger H.G. (2015) SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature. 526(7572), 218–223. doi 10.1038/nature15400
Inuzuka M., Hayakawa M., Ingi T. (2005) Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 280(42), 35776–35783. doi 10.1074/jbc.M505712200
Zhang X., Zhou T., Yang J., Lin Y., Shi J., Zhang X., Frabutt D.A., Zeng X., Li S., Venta P.J., Zheng Y.-H. (2017) Identification of SERINC5-001 as the predominant spliced isoform for HIV-1 restriction. J. Virol. 91(10), e00137-17. doi 10.1128/JVI.00137-17
Rosa A., Chande A., Ziglio S., De Sanctis V., Bertorelli R., Goh S.L., McCauley S.M., Nowosielska A., Antonarakis S.E., Luban J., Santoni F.A., Pizzato M. (2015) HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature. 526(7572), 212–217. doi 10.1038/nature15399
Sood C., Marin M., Chande A., Pizzato M., Melikyan G.B. (2017) SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 292(14), 6014–6026. doi 10.1074/jbc.M117.777714
Beitari S., Ding S., Pan Q., Finzi A., Liang C. (2017) Effect of HIV-1 Env on SERINC5 antagonism. J. Virol. 91(4), e02214-16. doi 10.1128/JVI.02214-16
Matheson N.J., Sumner J., Wals K., Rapiteanu R., Weekes M.P., Vigan R., Weinelt J., Schindler M., Antrobus R., Costa A.S.H., Frezza C., Clish C.B., Neil S.J.D., Lehner P.J. (2015) Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe. 18(4), 409–423. doi 10.1016/j.chom.2015.09.003
Dai W., Usami Y., Wu Y., Göttlinger H. (2018) A long cytoplasmic loop governs the sensitivity of the anti-viral host protein SERINC5 to HIV-1 Nef. Cell Rep. 22(4), 869–875. doi 10.1016/j.celrep.2017.12.082
Sauter D., Schindler M., Specht A., Landford W.N., Münch J., Kim K.-A., Votteler J., Schubert U., Bibollet-Ruche F., Keele B.F., Takehisa J., Ogando Y., Ochsenbauer C., Kappes J.C., Ayouba A., Peeters M., Learn G.H., Shaw G., Sharp P.M., Bieniasz P., Hahn B.H., Hatziioannou T., Kirchhoff F. (2009) Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe. 6(5), 409–421. doi 10.1016/j.chom.2009.10.004
Jia B., Serra-Moreno R., Neidermyer W., Rahmberg A., Mackey J., Fofana I. Ben, Johnson W.E., Westmoreland S., Evans D.T. (2009) Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by Tetherin/BST2. PLoS Pathog. 5(5), e1000429. doi 10.1371/journal.ppat.1000429
Sauter D., Hué S., Petit S.J., Plantier J.-C., Towers G.J., Kirchhoff F., Gupta R.K. (2011) HIV-1 group P is unable to antagonize human tetherin by Vpu, Env or Nef. Retrovirology. 8(1), 103. doi 10.1186/1742-4690-8-103
Sauter D., Unterweger D., Vogl M., Usmani S.M., Heigele A., Kluge S.F., Hermkes E., Moll M., Barker E., Peeters M., Learn G.H., Bibollet-Ruche F., Fritz J.V., Fackler O.T., Hahn B.H., Kirchhoff F. (2012) Human tetherin exerts strong selection pressure on the HIV-1 group N Vpu protein. PLoS Pathog. 8(12), e1003093. doi 10.1371/journal.ppat.1003093
Zhang F., Wilson S.J., Landford W.C., Virgen B., Gregory D., Johnson M.C., Munch J., Kirchhoff F., Bieniasz P.D., Hatziioannou T. (2009) Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. 6(1), 54–67. doi 10.1016/ j.chom.2009.05.008
Strebel K., Klimkait T., Maldarelli F., Martin M.A. (1989) Molecular and biochemical analyses of human immunodeficiency virus type 1 vpu protein. J. Virol. 63(9), 3784–3791. http://www.ncbi.nlm.nih.gov/ pubmed/2788224.
Terwilliger E.F., Cohen E.A., Lu Y.C., Sodroski J.G., Haseltine W.A. (1989) Functional role of human immunodeficiency virus type 1 vpu. Proc. Natl. Acad. Sci. USA. 86(13), 5163–5167. http://www.ncbi.nlm.nih.gov/ pubmed/2472639.
Klimkait T., Strebel K., Hoggan M.D., Martin M.A., Orenstein J.M. (1990) The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 64(2), 621–629. http://www.ncbi.nlm.nih.gov/pubmed/2404139.
Willey R.L., Maldarelli F., Martin M.A., Strebel K. (1992) Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J. Virol. 66(1), 226–234. http:// www.ncbi.nlm.nih.gov/pubmed/1727486.
Neil S.J.D., Eastman S.W., Jouvenet N., Bieniasz P.D. (2006) HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2(5), 354–367. doi 10.1371/journal.ppat.0020039
Sakai H., Tokunaga K., Kawamura M., Adachi A. (1995) Function of human immunodeficiency virus type 1 Vpu protein in various cell types. J. Gen. Virol. 76(11), 2717–2722. doi 10.1099/0022-1317-76-11-2717
Kupzig S., Korolchuk V., Rollason R., Sugden A., Wilde A., Banting G. (2003) Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 4(10), 694–709. http://www.ncbi. nlm.nih.gov/pubmed/12956872.
Sauter D. (2014) Counteraction of the multifunctional restriction factor tetherin. Front. Microbiol. 5(APR), 1–14. doi 10.3389/fmicb.2014.00163
Goto T., Kennel S.J., Abe M., Takishita M., Kosaka M., Solomon A., Saito S. (1994) A novel membrane antigen selectively expressed on terminally differentiated human B cells. Blood. 84(6), 1922–1930. http:// www.ncbi.nlm.nih.gov/pubmed/8080996.
Masuyama N., Kuronita T., Tanaka R., Muto T., Hirota Y., Takigawa A., Fujita H., Aso Y., Amano J., Tanaka Y. (2009) HM1.24 is internalized from lipid rafts by clathrin-mediated endocytosis through interaction with α-adaptin. J. Biol. Chem. 284(23), 15927–15941. doi 10.1074/jbc.M109.005124
Perez-Caballero D., Zang T., Ebrahimi A., McNatt M.W., Gregory D.A., Johnson M.C., Bieniasz P.D. (2009) Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 139(3), 499–511. doi 10.1016/j.cell.2009.08.039
Hammonds J., Spearman P. (2009) Tetherin is as tetherin does. Cell. 139(3), 456–457. doi 10.1016/ j.cell.2009.10.011
Venkatesh S., Bieniasz P.D. (2013) Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog. 9(7), e1003483. doi 10.1371/journal.ppat.1003483
Bieniasz P.D. (2009) The cell biology of HIV-1 virion genesis. Cell Host Microbe. 5(6), 550–558. doi 10.1016/j.chom.2009.05.015
Andrew A.J., Miyagi E., Kao S., Strebel K. (2009) The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology. 6(1), 80. doi 10.1186/1742-4690-6-80
Ohtomo T., Sugamata Y., Ozaki Y., Ono K., Yoshimura Y., Kawai S., Koishihara Y., Ozaki S., Kosaka M., Hirano T., Tsuchiya M. (1999) Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem. Biophys. Res. Commun. 258(3), 583–591. doi 10.1006/ bbrc.1999.0683
Jolly C., Kashefi K., Hollinshead M., Sattentau Q.J. (2004) HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199(2), 283–293. doi 10.1084/jem.20030648
Sattentau Q.J. (2011) The direct passage of animal viruses between cells. Curr. Opin. Virol. 1(5), 396–402. doi 10.1016/j.coviro.2011.09.004
Casartelli N., Sourisseau M., Feldmann J., Guivel-Benhassine F., Mallet A., Marcelin A.-G., Guatelli J., Schwartz O. (2010) Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 6(6), e1000955. doi 10.1371/journal.ppat.1000955
Kuhl B.D., Sloan R.D., Donahue D.A., Bar-Magen T., Liang C., Wainberg M.A. (2010) Tetherin restricts direct cell-to-cell infection of HIV-1. Retrovirology. 7(1), 115. doi 10.1186/1742-4690-7-115
Blanchet F.P., Stalder R., Czubala M., Lehmann M., Rio L., Mangeat B., Piguet V. (2013) TLR-4 engagement of dendritic cells confers a BST-2/tetherin-mediated restriction of HIV-1 infection to CD4+ T cells across the virological synapse. Retrovirology. 10(1), 6. doi 10.1186/1742-4690-10-6
Giese S., Marsh M. (2014) Tetherin can restrict cell-free and cell-cell transmission of HIV from primary macrophages to T cells. PLoS Pathog. 10(7). doi 10.1371/journal.ppat.1004189
Jolly C., Booth N.J., Neil S.J.D. (2010) Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J. Virol. 84(23), 12185–12199. doi 10.1128/JVI.01447-10
Coleman C.M., Spearman P., Wu L. (2011) Tetherin does not significantly restrict dendritic cell-mediated HIV-1 transmission and its expression is upregulated by newly synthesized HIV-1 Nef. Retrovirology. 8(1), 26. doi 10.1186/1742-4690-8-26
Zhong P., Agosto L.M., Ilinskaya A., Dorjbal B., Truong R., Derse D., Uchil P.D., Heidecker G., Mothes W. (2013) Cell-to-cell transmission can overcome multiple donor and target cell barriers imposed on cell-free HIV. PLoS One. 8(1), e53138. doi 10.1371/journal.pone.0053138
Mazurov D., Ilinskaya A., Heidecker G., Lloyd P., Derse D. (2010) Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 6(2), e1000788. doi 10.1371/journal.ppat.1000788
Shunaeva A., Potashnikova D., Pichugin A., Mishina A., Filatov A., Nikolaitchik O., Hu W.-S., Mazurov D. (2015) Improvement of HIV-1 and human T cell lymphotropic virus type 1 replication-dependent vectors via optimization of reporter gene reconstitution and modification with intronic short hairpin RNA. J. Virol. 89(20), 10591–10601. doi 10.1128/JVI.01940-15
Matsuda A., Suzuki Y., Honda G., Muramatsu S., Matsuzaki O., Nagano Y., Doi T., Shimotohno K., Harada T., Nishida E., Hayashi H., Sugano S. (2003) Large-scale identification and characterization of human genes that activate NF-κB and MAPK signaling pathways. Oncogene. 22(21), 3307–3318. doi 10.1038/sj.onc.1206406
Galão R.P., Le Tortorec A., Pickering S., Kueck T., Neil S.J.D. (2012) Innate sensing of HIV-1 assembly by tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe. 12(5), 633–644. doi 10.1016/j.chom.2012.10.007
Tokarev A., Suarez M., Kwan W., Fitzpatrick K., Singh R., Guatelli J. (2013) Stimulation of NF-κB activity by the HIV restriction factor BST2. J. Virol. 87(4), 2046–2057. doi 10.1128/JVI.02272-12
Kobayashi T., Ode H., Yoshida T., Sato K., Gee P., Yamamoto S.P., Ebina H., Strebel K., Sato H., Koyanagi Y. (2011) Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J. Virol. 85(2), 932–945. doi 10.1128/JVI.01668-10
Douglas J.L., Viswanathan K., McCarroll M.N., Gustin J.K., Fruh K., Moses A.V. (2009) Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a TrCP-dependent mechanism. J. Virol. 83(16), 7931–7947. doi 10.1128/JVI.00242-09
Liu L., Oliveira N.M., Cheney K.M., Pade C., Dreja H., Bergin A.-M.H., Borgdorff V., Beach D.H., Bishop C.L., Dittmar M.T., McKnight Á. (2011) A whole genome screen for HIV restriction factors. Retrovirology. 8(1), 94. doi 10.1186/1742-4690-8-94
Zhou H., Xu M., Huang Q., Gates A.T., Zhang X.D., Castle J.C., Stec E., Ferrer M., Strulovici B., Hazuda D.J., Espeseth A.S. (2008) Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 4(5), 495–504. doi 10.1016/ j.chom.2008.10.004
Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J., Lieberman J., Elledge S.J. (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science. 319(5865), 921–926. doi 10.1126/science.1152725
Park R.J., Wang T., Koundakjian D., Hultquist J.F., Lamothe-Molina P., Monel B., Schumann K., Yu H., Krupzcak K.M., Garcia-Beltran W., Piechocka-Trocha A., Krogan N.J., Marson A., Sabatini D.M., Lander E.S., Hacohen N., Walker B.D. (2017) A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 49(2), 193–203. doi 10.1038/ng.3741
König R., Zhou Y., Elleder D., Diamond T.L., Bonamy G.M.C., Irelan J.T., Chiang C.-Y., Tu B.P., De Jesus P.D., Lilley C.E., Seidel S., Opaluch A.M., Caldwell J.S., Weitzman M.D., Kuhen K.L., Bandyopadhyay S., Ideker T., Orth A.P., Miraglia L.J., Bushman F.D., Young J.A., Chanda S.K. (2008) Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 135(1), 49–60. doi 10.1016/j.cell.2008.07.032
Nguyen D.G., Yin H., Zhou Y., Wolff K.C., Kuhen K.L., Caldwell J.S. (2007) Identification of novel therapeutic targets for HIV infection through functional genomic cDNA screening. Virology. 362(1), 16–25. doi 10.1016/J.VIROL.2006.11.036
Gélinas J.-F., Gill D.R., Hyde S.C. (2018) Multiple inhibitory factors act in the late phase of HIV-1 replication: a systematic review of the literature. Microbiol. Mol. Biol. Rev. 82(1), e00051-17. doi 10.1128/ MMBR.00051-17
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология