

Молекулярная биология, 2019, T. 53, № 3, стр. 355-366
Участие клеточных систем репарации ДНК в репликации ВИЧ-1
А. Н. Анисенко a, b, *, М. Б. Готтих a, b
a Химический факультет Московского государственного университета им. М.В. Ломоносова,
119991 Москва, Россия
b Научно-исследовательский институт физико-химической биологии им. А.Н. Белозерского
Московского государственного университета им. М.В. Ломоносова
119991 Москва, Россия
* E-mail: a_anisenko@mail.ru
Поступила в редакцию 03.12.2018
После доработки 10.12.2018
Принята к публикации 12.12.2018
Аннотация
Серьезную проблему терапии ВИЧ-инфекции представляет возникновение лекарственно устойчивых форм вируса. Одним из перспективных подходов к решению этой проблемы считается разработка ингибиторов взаимодействия вирусных белков с клеточными белками-помощниками. Развитие этого подхода сдерживается недостаточно изученной ролью клеточных белков в патогенезе ВИЧ-инфекции. Так, известно, что интеграция вирусной ДНК в геном клетки приводит к появлению в клеточной ДНК ряда повреждений, репарация которых абсолютно необходима для успешной репликации вируса. Однако до сих пор не установлено точно, какие клеточные белки участвуют в репарации этих повреждений. В настоящем обзоре мы попытались суммировать данные о роли клеточных систем репарации в репликации ВИЧ-1, а также в репарации повреждений, возникающих в процессе интеграции вирусной ДНК в геном клетки.
ВВЕДЕНИЕ
Современные методы борьбы с ВИЧ-1 основаны на предупреждении передачи вируса и на медикаментозном подавлении ВИЧ-инфекции. В терапии ВИЧ в последние 20–30 лет проделана колоссальная работа, позволившая создать десятки уникальных препаратов и их комбинаций, применяемых в современной медицинской практике [1–3]. Это позволило существенно улучшить качество жизни ВИЧ-инфицированных и перевести это заболевание из разряда смертельно опасных в хронические. К сожалению, особенности жизненного цикла ВИЧ приводят к появлению в организме пациентов, получающих терапию, вирусных частиц, устойчивых к действию конкретных антиретровирусных агентов [4, 5]. Это связано, в первую очередь, с низкой точностью работы вирусной обратной транскриптазы (ОТ), приводящей к возникновению в вирусных белках мутаций, часть из которых вызывает резистентность к препаратам, мишенями которых эти белки являются. Таким образом, актуальным остается поиск новых мишеней для создания препаратов с минимальным риском развития резистентности.
Такими мишенями потенциально могут быть комплексы вирусных белков с клеточными белками, участвующими в репликации вируса. К соединениям, препятствующим формированию таких комплексов, не должна развиваться резистентность, поскольку поверхность взаимодействия двух белков чрезвычайно консервативна, и любая мутация в этом регионе негативно сказывается на стабильности комплекса [6, 7]. Однако развитие этого подхода тормозится из-за недостаточного понимания роли клеточных белков в патогенезе ВИЧ-инфекции. Важной стадия репликации ВИЧ-1 – интеграция ДНК вируса в геном клетки, катализируемая вирусной интегразой (ИН). В результате интеграции в геноме возникают повреждения, без репарации которых репликация вируса невозможна [8]. Однако до сих пор не установлено точно, какие клеточные белки вовлечены в репарацию и взаимодействуют ли они с ИН.
В настоящем обзоре суммированы сведения об участии клеточных систем репарации ДНК в репарации повреждений, возникающих в геноме клетки после интеграции в него вирусной ДНК.
ИНТЕГРАЦИЯ ВИЧ-1 КАК ИСТОЧНИК ПОВРЕЖДЕНИЙ ДНК
ВИЧ-1 относится к вирусам рода Lentivirus семейства Retroviridae. Генетический материал ВИЧ-1 представлен двумя идентичными молекулами одноцепочечной (+) РНК [9], ассоциированными с нуклеокапсидным белком p7, ОТ и ИН [10]. Последние два вирусных фермента осуществляют ключевые стадии жизненного цикла ВИЧ-1: обратную транскрипцию, т.е. синтез ДНК-копии вирусной геномной РНК, и интеграцию этой ДНК-копии в геном клетки. Процесс интеграции начинается еще в цитоплазме. С концами новосинтезированной ДНК связываются вирусные и клеточные белки, формируя прединтеграционный комплекс (ПИК), который транспортируется в ядро [11]. Основной компонент ПИК – ИН – катализирует реакцию 3'-процессинга, приводящую к отщеплению GT-динуклеотидов c обоих 3'-концов вирусной ДНК. После проникновения ПИК в ядро ИН связывает клеточную ДНК и катализирует реакцию переноса цепи, в результате которой вирусная ДНК встраивается в геном инфицированной клетки [12] (рис. 1).
Рис. 1.
Схема интеграции и постинтеграционной репарации повреждений ДНК, возникающих в ходе интеграции генома ВИЧ-1.
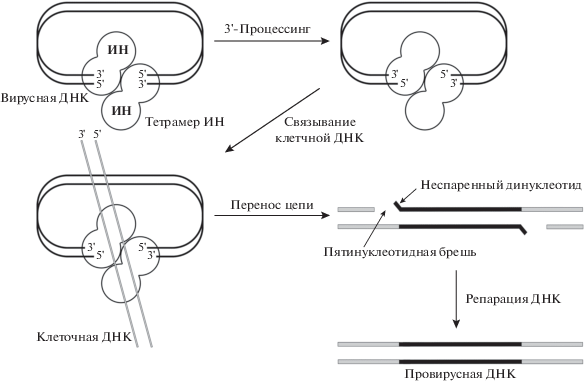
Интеграция ретровируса приводит к появлению множественных повреждений ДНК. Во-первых, как повреждение может быть воспринято нарушение структуры хроматина, вызванное встраиванием безгистоновой вирусной ДНК (около 10 000 п.н.). Во-вторых, оба конца вирусной ДНК встраиваются в комплементарные цепи клеточной ДНК на расстоянии 5 п.н. друг от друга (рис. 1). В результате в клеточной ДНК возникают одноцепочечные 5-нуклеотидные бреши, фланкирующие интегрированную ДНК вируса, а также неспаренные динуклеотиды CA на 5'-концах вирусной ДНК. Прохождение репликативной вилки через интегрированную, но нерепарированную ДНК в S-фазе клеточного цикла может приводить к преобразованию этих одноцепочечных разрывов в двухцепочечные, что представляет серьезную угрозу для стабильности генома [8].
Таким образом, интеграция ретровируса служит источником существенных повреждений клеточной ДНК, которые требуют незамедлительного восстановления для эффективной репликации вируса. В противном случае клетки с такими повреждениями могут быть элиминированы путем апоптоза, что негативно скажется на вирусной инфекции. Считается, что постинтеграционная репарация ДНК осуществляется клеточными белками [12, 13], поэтому рассмотрим коротко основные системы репарации повреждений в ДНК, действующие в клетках человека.
КЛЕТОЧНЫЕ СИСТЕМЫ ОТВЕТА НА ПОВРЕЖДЕНИЯ ДНК
Ежедневно в каждой клетке нашего организма возникают повреждения ДНК, число которых варьирует от десятков до тысяч событий в день [14]. Быстрое и точное устранение любых повреждений ДНК важно для поддержания стабильности генома [14], поэтому клетка использует широкий спектр механизмов обнаружения и исправления повреждений ДНК. Это высокоорганизованная и четко регулируемая белковая сеть, которая быстро активируется в ответ на повреждения. Такие сигнальные пути включают следующие обязательные компоненты: сенсоры повреждений, преобразователи сигнала, ряд переносчиков и финальные акцепторы сигнала. В зависимости от тяжести повреждений активация этих путей ведет к апоптозу, аресту клеточного цикла и/или привлечению белков, участвующих в репарации повреждений ДНК [15, 16].
В ответе на повреждения целостности цепей ДНК ключевую роль играют протеинкиназы из семейства PI3K: ATM (аtaxia telangiectasia mutated protein), реагирующая на двухцепочечные разрывы ДНК [17], ATR (ataxia-telangiectasia and Rad3-related protein), реагирующая на одноцепочечные разрывы ДНК и остановку репликативной вилки [17], и ДНК-зависимая протеинкиназа (DNA-PK – DNA-dependent protein kinase), участвующая в репарации двухцепочечных разрывов ДНК [17]), а также представители семейства поли(АDP-рибоза)-полимераз – PARP, узнающие одно- и двухцепочечные разрывы ДНК [18–20]. Все эти ферменты, за исключением DNA-PK, не участвуют непосредственно в классических путях репарации повреждений ДНК, а лишь регулируют процесс восстановления ее целостности. Так, например, ATM и ATR, связанные с поврежденной ДНК, запускают сигнальные каскады, приводящие к остановке клеточного цикла, а в некоторых случаях и к гибели клетки, а также к активации систем репарации ДНК [15]. Белки из семейства PARP привлекают факторы репарации ДНК к месту повреждения, а также регулируют их активность на транскрипционном уровне [21]. И только DNA-PK участвует в одном из ключевых компонентов репарационного пути – пути негомологичного объединения концов (NHEJ), в результате функционирования которого восстанавливается целостность ДНК, содержащей двухцепочечный разрыв [13, 15]. Рассмотрим подробнее пути репарации одно- и двухцепочечных повреждений ДНК.
Репарация двухцепочечных разрывов ДНК
Существуют два основных механизма репарации двухцепочечных разрывов ДНК, представляющих наиболее серьезную угрозу для жизни клетки: NHEJ и гомологичная рекомбинация (HR) [22, 23]. NHEJ может происходить на любой стадии клеточного цикла, в то время как HR может идти только в S- и G2-фазе клеточного цикла, поскольку для HR требуется неповрежденная сестринская хроматида [24].
В ходе NHEJ двухцепочечный разрыв узнается частью комплекса DNA-PK – гетеродимером Ku70/Ku80, который доставляет к месту повреждения каталитическую субъединицу DNA-PKcs, что приводит к ее автофосфорилированию и привлечению белков процессинга концов ДНК: Artemis и ДНК-лигазы (LigIV) в комплексе с XRCC4 и XLF. Этот путь не является высокоточным и часто приводит к возникновению мутаций [13, 23].
Устранение двухцепочечных разрывов ДНК по механизму HR проходит только в том случае, если рядом с поврежденной ДНК находится интактная копия ДНК, т.е. сестринская хроматида. Этот процесс возможен только в S- и G2-фазах клеточного цикла. HR начинается с процессинга концов двухцепочечного разрыва, в ходе которого образуются одноцепочечные 3'-концы, связанные с белком RPA. Затем RPA на одноцепочечных концах ДНК замещается рекомбиназой RAD51, которая привлекает в этот комплекс неповрежденную сестринскую хроматиду, используемую в качестве матрицы для достраивания поврежденных цепей. В результате HR восстанавливается целостность ДНК [25].
Репарация одноцепочечных повреждений ДНК
Наиболее часто повреждения появляются только в одной из цепей ДНК: одноцепочечные разрывы ДНК, модификации отдельных азотистых оснований или их потеря (AP-сайты), неспаренные нуклеотиды. Механизмы репарации этих повреждений предполагают использование второй цепи ДНК в качестве матрицы для исправления ошибок. Стоит отметить, что одноцепочечные повреждения ДНК менее опасны для клетки, чем двухцепочечные, но их восстановление должно происходить достаточно быстро, поскольку при прохождении репликативной вилки через одноцепочечный разрыв может образоваться двухцепочечный разрыв, опасный для стабильности генома. Существует несколько механизмов репарации повреждений, возникающих в одной цепи ДНК [26].
Прямая репарация – устранение модификаций нуклеотидов (УФ-индуцируемых пиримидиновых димеров и О6-алкилгуанина) за счет прямого устранения повреждения ферментами (фотолиазой и О6-метилгуанин-метилтрансферазой (MGMT) соответственно) [14].
Эксцизионная репарация оснований (ЭРО) восстанавливает повреждения в случае необратимых модификаций оснований, возникающих, например, в результате окислительного повреждения ДНК. ЭРО начинается с обнаружения поврежденного основания ДНК-гликозилазами, расщепляющими N-гликозидную связь в нуклеозиде, что приводит к появлению AP-сайта. AP-эндонуклеаза APE1 вносит разрыв в поврежденную цепь ДНК, приводя к новому типу повреждения – одноцепочечному разрыву. ДНК-полимеразы (Polβ или/и Polδ) замещают поврежденный фрагмент ДНК, после чего лигазы LigI или LigIII завершают процесс репарации, соединяя вновь синтезированный фрагмент ДНК с неповрежденной частью ДНК [27].
Эксцизионная репарация нуклеотидов (ЭРН) отвечает за репарацию крупных повреждений ДНК, таких как фотопродукты, аддукты ДНК с некоторыми химиотерапевтическими агентами. ЭРН может инициироваться по двум механизмам, которые отличаются способом узнавания повреждения: ассоциированному с транскрипцией и независимому от транскрипции. Транскрипционно-независимая ЭРН участвует в репарации повреждений в любом месте генома, в то время как транскрипционно-ассоциированная отвечает за репарацию в транскрибируемой цепи ДНК активных генов. Транскрипционно-независимая ЭРН начинается с узнавания сайта повреждения специфичным фактором XPC-RAD23B. Транскрипционно-зависимая ЭРН начинается в месте остановки РНК-полимеразы с привлечения факторов CSA, CSB и XAB2. После узнавания повреждения к этому месту привлекается транскрипционный фактор TFIIH, который индуцирует расплетание цепей ДНК примерно на 15 п.н. с обеих сторон от повреждения. С полученным комплексом связываются эндонуклеазы XPF/ERCC1 и XPG, которые вносят разрывы в ДНК с обеих сторон от повреждения. Полученная в ходе этого процесса брешь достраивается и лигируется с привлечением репликативных факторов (PCNA, RFC, Polδ, Polε, Polκ) и ДНК-лигаз (LigI, LigIII) [28].
Репарация неспаренных оснований – это путь, устраняющий ошибки репликации. Сразу после обнаружения неспаренных оснований с помощью комплекса MutSα (MSH2-MSH6) или MutSβ (MSH2-MSH3) к месту повреждения привлекается комплекс MutLα, состоящий из субъединиц PMS2 и MLH1. Белок PCNA, входящий в состав аппарата репликации, активирует MutLα. В результате этой активации MutLα разрезает вновь синтезированную цепь ДНК слева и справа от неспаренных нуклеотидов. Образующаяся “брешь” достраивается ДНК-полимеразой (Polδ или Polε) с последующим лигированием концов ДНК [29].
ПОСТИНТЕГРАЦИОННАЯ РЕПАРАЦИЯ ВИЧ-1
Как отмечалось выше, интеграция генома ВИЧ-1 в клеточную ДНК приводит к появлению повреждений ДНК: по обеим сторонам от вирусной ДНК в геномной ДНК находятся 5-нуклеотидные одноцепочечные бреши, а на 5'-концах вирусной ДНК расположены неспаренные динуклеотиды. Репарация этого интеграционного интермедиата приводит к образованию стабильно интегрированного провируса, с которого затем начинается синтез вирусных РНК.
Первоначально предполагалось, что репарация подобных повреждений может осуществляться непосредственно вирусными ферментами – ОТ и ИН [30]. Так in vitro показана способность очищенной ОТ ВИЧ-1 достраивать 5-нуклеотидную брешь в искусственных ДНК субстратах, имитирующих продукт интеграции вирусной ДНК [30]. Это не удивительно, поскольку ОТ ВИЧ-1 обладает активностью ДНК-зависимой ДНК-полимеразы. Интересно, что ни ИН, ни ОТ в индивидуальном состоянии не могли отщеплять неспаренный динуклеотид с 5'-концов вирусной ДНК, но в присутствии ОТ ИН смогла отщеплять этот динуклеотид и соединять концы ДНК по типу реакции дезинтеграции. Стоит отметить, однако, чрезвычайно низкую эффективность этого процесса (1–2%) [30]. Столь низкие значения эффективности репарации вирусными ферментами говорят о том, что вряд ли эти повреждения восстанавливаются в клетке таким способом. Более того, функциональный комплекс ИН–ПИК, выделенный из клеток, может осуществлять встраивание фрагмента вирусной ДНК в ДНК-субстрат in vitro, но образующийся при этом продукт содержит все типичные для интеграции повреждения ДНК [31]. Все эти данные показывают, что вирусные белки, входящие в состав ПИК, не способны завершить процесс интеграции, и ключевую роль в этом процессе должны играть клеточные системы репарации.
Роль пути негомологичного объединения концов и его компонентов в интеграции ВИЧ-1
Первые доказательства вовлеченности системы репарации NHEJ в репликацию ВИЧ-1 были получены René Daniel и соавт. в 1999 году [32], которые использовали предшественники B-лимфоцитов, выделенные из мышей с тяжелым комбинированным иммунодефицитом (scid-мыши), обусловленным делецией в гене DNA-PKcs. Заражение этих клеток VSV-G-псевдотипированным вектором на основе ВИЧ-1 (псевдовирусом) приводило к повышенной апоптотической гибели мутантных клеток по сравнению с контрольной клеточной линией. Помимо этого, в мутантных клетках зафиксирован пониженный уровень экспрессии репортерного белка – β-галактозидазы. Сниженная инфекционность псевдовируса на основе ВИЧ-1 показана также на других клеточных линиях, дефектных по DNA-PKcs [33]. Аналогичные результаты получены и для псевдовирусов на основе вируса саркомы Рауса (RSV) или вируса лейкоза мышей Молони (MMLV). Более того, экспрессия функционального варианта DNA-PKcs в клеточных линиях, дефектных по DNA-PKcs, предотвращала гибель клеток при их заражении псевдовирусом на основе ВИЧ-1 и восстанавливала его инфекционность [34].
Для доказательства того, что интеграция вирусной ДНК активирует гибель клеток, использовали псевдовирус на базе RSV, несущий неактивную мутантную форму ИН, неспособную осуществлять интеграцию. Гибель клеток в этом случае не наблюдалась [32]. Позднее аналогичный результат был получен и с псевдовирусом на основе ВИЧ-1 [34].
Как отмечалось выше, DNA-PKcs активируется при связывании с гетеродимером Ku70/Ku80, который узнает и связывает двухцепочечные разрывы в ДНК. Обнаружено увеличение активности DNA-PKcs в контрольных клетках с нормальным содержанием DNA-PKcs в ответ на инфицирование псевдовирусом с активной ИН, но не с ее мутантным вариантом [32]. Таким образом, гибель клеток, дефектных по главному компоненту NHEJ – DNA-PKcs, и увеличение каталитической активности DNA-PKcs в контрольных клетках в ответ на трансдукцию ретровирусными векторами обусловлена именно интеграцией, а не предшествующими этапами жизненного цикла вируса.
Наблюдаемые в работе [32] эффекты, вероятно, не универсальны для всех ретровирусов, поскольку инфицирование первичных клеток-предшественников B-лимфоцитов, мутантных по DNA-PKcs, вирусом лейкоза мышей Абельсона (A-MuLV) проходит так же, как и в контрольной клеточной линии [35]. Более того, цитопатический эффект ретровирусной трансдукции проявляется в клеточных линиях MEF, дефектных по DNA-PKcs, только при высоких значениях множественности инфекции (МИ > 1) [36]. МИ – это параметр, который отражает количество вирусных частиц, приходящихся на одну клетку. При низких значениях МИ (МИ < 0.1) трансдукция псевдовирусом на основе ВИЧ-1 протекает одинаково в клетках, дефектных по DNA-PKcs, и в контрольной клеточной линии; гибели клеток при этом не наблюдается. Надо отметить, что Daniel и соавт. воспроизвели условия, использованные в [36], но их результат существенно отличался: даже при низких значениях МИ (МИ = 0.1) наблюдалась сниженная трансдукция DNA-PKcs-дефектных клеток [34].
Как сказано выше, DNA-PKcs, а также ATM и ATR относятся к семейству фосфоинозитид-3-киназ (PI3K). Вортманнин – специфичный ингибитор PI3K. Эффективность трансдукции первичных клеток-предшественниц В-лимфоцитов, экспрессирующих нормальную DNA-PKcs, псевдовирусами на основе RSV или ВИЧ-1 в присутствии вортманнина снижена в 10 раз [37]. К сожалению, наблюдаемые эффекты можно объяснить ингибированием не только DNA-PKcs, но также и других PI3K.
Изучен также эффект другого компонента DNA-PK – белка Ku80 [38–44]. Как отмечено выше, гетеродимер Ku70/Ku80 первично узнает двухцепочечный разрыв в ДНК и привлекает к месту повреждения каталитическую субъединицу DNA-PKcs. Компоненты гетеродимера Ku70/Ku80 обнаружены в составе ПИК [38], показано также непосредственное взаимодействие Ku70 с ИН ВИЧ-1 [39–41]. Инфицирование клеток CEM4fx с подавленной экспрессией Ku80 штаммом NL4-3 ВИЧ-1 de novo приводит к снижению продукции новых вирусных частиц [42]. Уровень интеграции вирусной ДНК в таких клетках снижен в 3–4 раза по сравнению с контрольной клеточной линией, при этом обратная транскрипция и транспорт в ядро не затронуты. Негативное влияние снижения внутриклеточной концентрации Ku80 на трансдукцию псевдовирусами на основе ВИЧ-1 показано также с применением рибозимов и малых интерферирующих РНК (siРНК) к Ku80 [43]. Снижение инфекционности псевдовируса на основе ВИЧ-1 наблюдали также на клеточной линии человека НСТ с нокаутом одного из аллелей гена Ku80 [44], приводящим к снижению примерно на 50% количества как Ku80, так и Ku70, мало- стабильных в отсутствие своего партнера по гетеродимеризации. Однако снижение инфекционности объясняли влиянием Ku80 на транскрипцию с интегрированного провируса, а не на интеграцию, поскольку не обнаружено уменьшения количества интегрированной вирусной ДНК при понижении уровня Ku80 [44].
Необходимо отметить, что данные по влиянию компонентов DNA-PK на репликацию ВИЧ-1 достаточно противоречивы. Так, в крупномасштабном эксперименте по изучению роли белков-участников систем репарации повреждений ДНК в жизненном цикле ВИЧ-1 не обнаружено взаимосвязи между уровнем внутриклеточного Ku70 или DNA-PKcs и эффективностью инфекции [45]. Роль известных партнеров ИН, в том числе белка Ku70, в репликации ВИЧ-1 изучали также путем опосредованного CRISP-Cas9 нокаута генов [46]. Нокаут Ku70 не приводил к снижению инфекционности вируса, а наоборот, незначительно (в 1.5 раза) увеличивал ее. Подобные противоречия результатов, полученных в [45, 46] и в ранее опубликованных работах [32–34, 37, 42, 43], можно объяснить отсутствием проверки количества Ku70 и DNA-PKcs в клетках при крупномасштабных испытаниях [45, 46]. Гетеродимер Ku70/Ku80 отличается высокой стабильностью, количество его субъединиц практически не уменьшается даже через 24 ч после подавления трансляции циклогексимидом [47]. В работах [45, 46] клетки трансдуцировали векторами на основе ВИЧ-1 через 24 ч после добавления siРНК [45] или нокаута, опосредованного СRISP-Cas9 [46], что, вероятно, не позволяет существенно снизить концентрацию Ku70 или Ku80. В пользу этого предположения говорят данные, полученные с использованием клонов клеток НЕК 293Т с разным уровнем белка Ku70, которые убедительно показали, что снижение инфекционности псевдовируса на основе ВИЧ-1 прямо коррелирует со снижением уровня Ku70 [41].
Заканчивая обсуждение роли DNA-PK в репликации ВИЧ-1, необходимо отметить, что согласно [48] заражение первичных лимфоцитов вирусом или псевдовирусом может приводить к гибели клеток, причем в первую очередь гибнут клетки, содержащие вирусную кДНК, но не экспрессирующие вирусные белки. Гибель клеток, в которых наблюдается полноценная экспрессия вирусных белков, начинается только через 2 дня после инфицирования. Иными словами, заражение первичных лимфоцитов ВИЧ-1 служит сигналом к их последующей гибели. Особенно важно, что настоящим сигналом к гибели клеток является интеграция, поскольку использование вируса с неактивной ИН или ингибитора интеграции ралтегравира не приводит к гибели лимфоцитов. В ВИЧ-инфицированных первичных CD4 лимфоцитах и в клетках CEMX174 обнаружена повышенная активность DNA-PK и увеличение фосфорилирования р53 и гистона Н2АХ [32, 34, 48]. Однако самое удивительное то, что ингибирование фосфорилирующей активности DNA-PKcs специфичными низкомолекулярными ингибиторами NU7026 и NU7441 блокировало гибель клеток, не влияя при этом на уровень репликации ВИЧ-1 [48]. Этот результат полностью противоречит данным [34], согласно которым повышение уровня DNA-PKcs предотвращает гибель клеток. Результаты работы [48] вызвали оживленную дискуссию в научной среде [49, 50], однако, ее авторы настаивают, что предлагаемый механизм гибели ВИЧ-инфицированных Т-лимфоцитов за счет активации DNA-PKcs и фосфорилирования р53 необходимо учитывать при проведении антиретровирусной терапии [48].
Последний акцептор сигнала от комплекса DNA-PK в системе NHEJ–ДНК-лигаза (LigIV), которая осуществляет соединение концов ДНК. Установлено [38], что клетки, дефектные по этой лигазе, погибают от апоптоза в ответ на трансдукцию вектором на базе ВИЧ-1, причем этот эффект не связан с активной интеграцией вирусного генома. Перепроверка этих результатов [34] не подтвердила независимости гибели клеток, дефектных по LigIV, от интеграции: активация апоптоза происходила в ответ на трансдукцию псевдовирусом с активной ИН, но не псевдовирусом с неактивной ИН. Подобные отличия можно объяснить методологическими различиями в опытах по заражению клеток векторами на основе ВИЧ-1 – пассивной доставки вирусных частиц [34] и процедуры спинокуляции, которая заключается в длительном центрифугировании клеток с вирусом, что повышает эффективность трансдукции в 5–10 раз [38]. R. Daniel и соавт. повторили эксперимент с LigIV-дефектными клетками, но в условиях спинокуляции вируса, и установили, что при таком способе доставки гибель клеток действительно не зависит от интеграции вирусной ДНК и происходит и при использовании вектора с неактивной ИН [34]. Таким образом, доставка вируса методом спинокуляции может существенно маскировать эффекты, обусловленные интеграцией ретровируса.
Таким образом, большинство работ подтверждает вовлеченность компонентов системы репарации по пути NHEJ (Ku70, Ku80, DNA-PKcs и LigIV) в регуляцию интеграции ВИЧ-1 и выживание клеток в ответ на повреждения ДНК, возникающие в ходе этого процесса. Однако до сих пор не ясно, как именно компоненты этого пути участвуют в постинтеграционной репарации провирусной ДНК. В частности, как DNA-PK привлекается к месту повреждения ДНК, поскольку при интеграции не происходит разрыва обеих цепей и не образуются свободные концы ДНК, с которыми гетеродимер Ku связывается в процессе NHEJ.
Роль других факторов клеточного ответа на повреждения ДНК в репликации ВИЧ-1
Как уже сказано, в клетках, дефектных по DNA-PKcs, наблюдали снижение инфекционности псевдовируса на основе ВИЧ-1 на 80–90% по сравнению с контрольной клеточной линией [32]. Наличие остаточной инфекционности может объясняться существованием компенсаторных путей, регулирующих постинтеграционную репарацию. Возможно, в репарацию включаются другие PI3K, ATM и ATR, участвующие в ответе клетки на повреждения ДНК. На подобные мысли наталкивает наблюдение, что ретровирусная трансдукция в клетках, дефектных по DNA-PKcs, дополнительно снижается в присутствии вортманнина – неспецифичного ингибитора PI3K [37]. Известно, что в процессах первичного узнавания повреждений ДНК и запуска ответа на них принимает участие не только DNA-PK, но и ATM, ATR и белки из семейства PARP [51]. Рассмотрим свидетельства вовлеченности этих факторов клеточного ответа на повреждения ДНК в процесс интеграции ВИЧ-1.
ATM. Белок ATM активирует ряд участников репарации ДНК, а также задержку клеточного цикла в ответ на двухцепочечные разрывы ДНК. К месту повреждения эта протеинкиназа привлекается комплексом MRN, состоящим из белков Mre11, Rad50 и NBS1 [17]. В таком виде активированный белок ATM фосфорилирует белки p53 и CHK2, что приводит к остановке клеточного цикла, а также гистон H2AX, что позволяет привлечь к месту повреждения ДНК факторы репарации [15].
В ряде работ показано, что клетки, дефектные по ATM, хуже инфицируются псевдовирусами на основе ВИЧ-1 [33, 37, 52]. Однако снижение внутриклеточного уровня ATM в клетках дикого типа с помощью интерферирующих РНК не повлекло за собой снижения инфекции ВИЧ-1 [51]. По-видимому, остаточного уровня ATM может хватить для завершения процесса интеграции. Вместе с тем, зафиксирована гибель клеток, дефектных по ATM, в ответ на инфицирование ВИЧ-1 [51]. Как в случае клеток, дефектных по DNA-PKcs, апоптоз активировался лишь в ответ на интеграцию.
Специфичный ингибитор ATM (Ku-55933) эффективно подавляет инфекцию ВИЧ-1, снижая количество интегрированной ДНК и не влияя на общее количество суммарной вирусной ДНК [52]. Стоит, однако, отметить, что снижение количества интегрированной ДНК зависит от времени: через 24 ч после инфицирования уровень интегрированной ДНК одинаков в контрольных и обработанных Ku-55933 клетках, но через 48 ч количество интегрированной ДНК резко падает в клетках, обработанных ингибитором, и практически не изменяется в контрольной клеточной линии [52]. Вероятно, в период с 24 до 48 ч после инфицирования клетки, в которых невозможно репарировать постинтеграционные повреждения ДНК, погибают, что и вызывает наблюдаемые эффекты.
Как уже отмечалось, ATM привлекается к месту двухцепочечного разрыва комплексом MRN, состоящим из белков Mre11, Rad50 и NBS1 [17]. Методом иммунопреципитации хроматина показана ассоциация вирусной ДНК с NBS1 и ATM. Стоит отметить, что в клетках NBS1–/– ATM не взаимодействует с вирусной ДНК [53]. Соответственно, можно предположить, что в клетках, дефектных по NBS1, должна быть снижена постинтеграционная репарация. Однако данные по эффективности трансдукции клеток, дефектных по NBS1, а также по Mre11, псевдовирусами на основе ВИЧ-1 малочисленны и крайне противоречивы [33, 53]. Так, показано, что уменьшение числа компонентов комплекса MRN снижает эффективность трансдукции псевдовирусом на основе ВИЧ-1 [53], а в работе [33] эффективность трансдукция клеток, дефектных по NBS1 или Mre11, и контрольных клеточных линий была одинаковой. Тем не менее, секвенирование сайтов интеграции позволило выявить повышение частоты мутаций в местах интеграции в отсутствие NBS1 или Mre11, что говорит в пользу нарушения репарации в этих клетках [33].
ATR. Этот белок также относится к семейству PI3K. Он участвует в процессах регуляции ответа на повреждения ДНК. Спектр повреждений ДНК, в результате которых активируется ATR, достаточно широк и часто перекрывается со спектром ATM. ATR активируется преимущественно в ответ на одноцепочечные разрывы ДНК, связанные с репликативным белком A (RPA), и обычно находящиеся в непосредственной близости к двухцепочечным разрывам [54, 55]. Возможна также активация ATR фосфорилированной формой ATM [56].
Как только в клетке появляется одноцепочечный участок ДНК, с ним сразу связывается белок RPA, который защищает оцДНК от возможных повреждающих агентов. С помощью ATR-связывающего белка (ATRIP) RPA привлекает к месту повреждения ATR-киназу. С местом повреждения связывается также взаимодействующий с топоизомеразой белок TOPBP1, который активирует ATR. Активированная ATR фосфорилирует ряд эффекторов, например CHK1 и p53, которые блокируют клеточный цикл и активируют репарацию ДНК [15].
К сожалению, данные о вовлеченности ATR в репликацию ВИЧ-1 крайне скудны и противоречивы, в силу чего не удается дать взвешенной оценки результатов посвященных этому работ. В пользу участия ATR в интеграции или постинтеграционной репарации свидетельствуют данные работы [57]. При сверхэкспрессии доминантно-негативного мутанта ATR (укороченная версия ATR, узнающая повреждения ДНК, но не способная активировать ответ на повреждения ДНК) на фоне эндогенного ATR зафиксировано снижение количества клеток, стабильно трансдуцированных псевдовирусом на основе ВИЧ-1, а также количества интегрированной ДНК. В то же время, снижение уровня ATR при использовании соответствующих малых интерферирующих РНК не влияет на эффективность интеграции ретровируса [51, 58]. Обнаружено, однако, что ATR, как и АТМ [53], ассоциирован с вирусной ДНК, но в отличие от ATM его взаимодействие с ДНК не зависит от компонентов комплекса MRN. При нарушении взаимодействия между ATM и вирусной ДНК, комплекс ATR с вирусной ДНК сохраняется [53]. Таким образом, очевидно, что вопрос о роли ATR в интеграции ретровируса требует дальнейшего изучения.
PARP-1. PARP-1 относится к семейству поли-АDP-рибозилирующих белков (PARP), которые катализируют реакцию переноса АDP-рибозы. PARP-1 активируется в ответ на одно- и двухцепочечные повреждения ДНК, и поли-АDP-рибозилирует ряд молекул, вовлеченных в регуляцию ответа на повреждения ДНК [19]. Во-первых, аутополи-АDP-рибозилирование PARP-1 создает разветвленный каркас для привлечения к месту повреждения ДНК белков системы ответа на повреждения ДНК, например ATM, Mre11 и других. Во-вторых, PARP-1 привлекает и модифицирует белки-участники системы репарации: комплекс белков ЭРО, XPA (эксцизионная репарация нуклеотидов), MSH6 (репарация неспаренных оснований), DNA-PKcs (негомологичное объединение концов). Одновременно с этим PARP-1 регулируется компонентами путей репарации, например, гетеродимер Ku70/Ku80 увеличивает каталитическую активность PARP-1. В-третьих, PARP-1 может модулировать клеточный ответ на повреждения ДНК, регулируя транскрипцию генов системы репарации ДНК [21]. Таким образом, PARP-1, как и любой другой компонент системы ответа на повреждения ДНК, может самостоятельно запускать каскад реакций в ответ на повреждения или выступать в роли промежуточного звена, амплифицирующего сигнал.
Данные о роли PARP-1 в репликации ВИЧ-1 также противоречивы, как и данные о роли других участников систем репарации. Установлено, что PARP-1 играет важную роль в интеграции ВИЧ-1 [59–62]. Конкурентные ингибиторы PARP снижают эффективность ретровирусной трансдукции, а также количество интегрированной ДНК [61]. Аналогичные эффекты обнаружены на клетках мыши с нокаутом PARP-1, трансдуцированных псевдовирусами на основе ВИЧ-1 [62]. Установлено, что нокдаун PARP-1 с помощью siРНК приводит к подавлению экспрессии гена репортерного белка в составе вектора на основе ВИЧ-1 в нескольких линиях клеток человека [59].
Вместе с тем, важность PARP–1 в репликации ретровируса ставится под сомнение [36, 58, 63]. Так, конкурентный ингибитор PARP–1 3-метоксибензамид не влиял на трансдукцию клеток псевдовирусом на основе ВИЧ-1 [36], тогда как выявлено снижение трансдукции эмбриональных фибробластов мыши с нокаутом PARP-1. Однако на эмбриональных стволовых клетках мыши этот эффект не был воспроизведен. Снижение уровня PARP-1 в культуре клеток человека (HeLa) с помощью коротких шпилечных РНК (shРНК) не влияло на эффективность трансдукции псевдовирусом на базе ВИЧ-1, на основании чего предполагают, что PARP-1 не является ключевым фактором репликации ретровируса [36].
Противоположные результаты получены при использовании двух типов VSV-G-псевдотипированных векторов на основе ВИЧ-1: HIV-sinPPT и HIV-EGFPΔE [63]. Вектор HIV-EGFPΔE получен из природного изолята NL3-4 путем замены гена env на eGFP. Вектор HIV-sinPPT также построен на основе изолята NL3-4, он содержит делецию в длинном концевом повторе (LTR), препятствующую репликации вектора, а репортерный ген аналогичен гену в составе HIV-EGFPΔE. Уровни трансдукции и интеграции вектора HIV-EGFΔE в эмбриональные фибробласты мыши PARP-1–/– снижены, тогда как при использовании HIV-sinPPT такие эффекты не наблюдаются. На основании этих результатов сделан вывод о необязательности PARP-1 для ретровирусной репликации. В последующих работах показано, что PARP-1 принимает участие в репликации ВИЧ-1 на уровне репрессии транскрипции провируса путем эпигенетических модификаций хроматина, но не влияет на уровень интеграции [64, 65].
Путь эксцизионной репарации оснований и репликация ВИЧ-1
В ходе крупномасштабного скрининга факторов, участвующих в постинтеграционной репарации ДНК, проверено влияние нокдауна 232 генов, относящихся к разным системам репарации повреждений ДНК [45]. В рамках этого исследования установлено, что siРНК против 41 гена существенно снижают трансдукцию клеток псевдовирусами на основе ВИЧ-1. Все эти гены принадлежат к разным системам репарации повреждений ДНК, но наибольшее число генов из этого списка (13) относятся к системе ЭРО, что позволяет сделать вывод о значимости этого пути репарации для ранних этапов репликации ВИЧ-1.
Компоненты пути ЭРО – Polβ и FEN1, могут репарировать синтетические субстраты, напоминающие продукты интеграции вирусной ДНК в геном инфицированной клетки, при участии других факторов in vitro [66]. Одновременно с этим установлено, что эти ферменты не относятся к абсолютно необходимым для in vitro репарации. Их функцию могут выполнять и другие полимеразы и эндонуклеазы [66]. В ходе перепроверки полученных результатов на эмбриональных фибробластах мыши, дефектных по компонентам пути ЭРО (Neil–/–, Myh–/–, Ogg1–/–, Polβ–/–), подтверждена их вовлеченность в репликацию ретровируса на уровне интеграции или постинтеграционной репарации [45, 67]. Однако опосредованный CRISPR-Cas9 нокдаун Polβ в культуре моноцитов человека (THP-1) не повлиял на трансдукцию ВИЧ-1 [68]. В этом случае мы снова наблюдаем противоречивость данных, которую можно объяснить отсутствием прямых методов оценки эффективности постинтеграционной репарации в клетках или отличиями экспериментальных моделей ВИЧ-инфекции.
Однако, даже если принять во внимание, что в ряде экспериментальных систем нарушение функций компонентов пути ЭРО приводит к снижению ретровирусной трансдукции, остается неясным механизм, по которому этот путь влияет на репликацию ВИЧ-1. С одной стороны, возможно, что путь ЭРО вовлечен непосредственно в репарацию постинтеграционных повреждений ДНК. С другой стороны, компоненты ЭРО могут направлять интеграцию в наиболее предпочтительные участки генома. Так известно, что сайты интеграции обогащены G:C-парами, а ЭРО принимает участие в репарации окисленных форм гуанина. Изучение этого вопроса показало, что сверхэкспрессия мутантной каталитически неактивной формы Polβ в клетках Polβ–/– восстанавливает инфекционность псевдовируса на основе ВИЧ-1 [69]. Следовательно, полимеразная активность Polβ не важна для эффективной трансдукции ВИЧ-1. Вместе с тем, в этой же работе выявлено изменение паттерна сайтов интеграции в клетках Ogg–/– и MYH–/–, что свидетельствует в пользу вовлеченности компонентов ЭРО в процесс выбора сайта интеграции, а не постинтеграционной репарации провируса.
ЗАКЛЮЧЕНИЕ
Подводя итог, можно с уверенностью сказать, что существующие на сегодняшний день данные однозначно указывают на вовлеченность белков-участников систем репарации повреждений ДНК в жизненный цикл ВИЧ-1. Наибольшее число работ посвящено изучению компонентов пути негомологичного объединения концов. Существующие на сегодняшний день данные позволяют предположить, что вероятнее всего именно эти белки играют ключевую роль в постинтеграционной репарации повреждений ДНК. Однако остается непонятным, каким образом белки из этой системы репарации узнают повреждения, возникающие в ходе интеграции, ведь в этом случае не возникает свободных концов ДНК – характеристических повреждений, которые распознаются этой системой репарации.
Следует, однако, отметить, что ни в одной из рассмотренных работ не удалось однозначно установить, какие из этих белков участвуют в постинтеграционной репарации провируса, а какие влияют на другие этапы жизненного цикла ВИЧ-1. Основной причиной этой неопределенности является отсутствие методик прямой оценки эффективности постинтеграционной репарации ДНК. Только в 2018 году нам удалось разработать метод количественного определения уровня постинтеграционной репарации ДНК [70]. Можно надеяться, что использование этого метода позволит в будущем точно идентифицировать участников постинтеграционной репарации ДНК и предложить новые мишени для разработки анти-ВИЧ препаратов.
Работа выполнена за счет Российского научного фонда, грант № 17-14-01107 (оценка роли пути NHEJ в репликации ВИЧ-1), и гранта РФФИ_мк (№ 18-29-0812).
Авторы заявляют об отсутствии конфликтов интересов.
Список литературы
Laskey S.B., Siliciano R.F. (2014) A mechanistic theory to explain the efficacy of antiretroviral therapy. Nat. Rev. Microbiol. 12, 772–780.
Domingo P., Vidal F. (2011) Combination antiretroviral therapy. Expert. Opin. Pharmacother. 12, 995–998.
Cihlar T., Fordyce M. (2016) Current status and prospects of HIV treatment. Curr. Opin. Virol. 18, 50–56.
Solomon D.A., Sax P.E. (2015) Current state and limitations of daily oral therapy for treatment. Curr. Opin. HIV AIDS. 10, 219–225.
Iyidogan P., Anderson K.S. (2014) Current perspectives on HIV-1 antiretroviral drug resistance. Viruses. 6, 4095–4139.
Adamson C.S., Freed E.O. (2010) Novel approaches to inhibiting HIV-1 replication. Antiviral Res. 85, 119–141.
Tintori C., Brai A., Fallacara A.L., Fazi R., Schenone S., Botta M. (2014) Protein-protein interactions and human cellular cofactors as new targets for HIV therapy. Curr. Opin. Pharmacol. 18, 1–8.
Skalka A.M., Katz R.A. (2005) Retroviral DNA integration and the DNA damage response. Cell Death Differ. 12, 971–978.
Sükösd Z., Andersen E.S., Seemann S.E., Jensen M.K., Hansen M., Gorodkin J., Kjems J. (2015) Full-length RNA structure prediction of the HIV-1 genome reveals a conserved core domain. Nucl. Acids Res. 43, 10168–10179.
Muriaux D., Darlix J.L. (2010) Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol. 7, 744–753.
Engelman A. (2009) Isolation and analysis of HIV-1 preintegration complexes. Methods Mol. Biol. 485, 135–149.
Агапкина Ю.Ю., Приказчикова Т.А., Смолов М.А., Готтих М.Б. (2005) Структура и функции интегразы ВИЧ-1. Успехи биол. химии. 45, 87–122.
Княжанская Е.С., Шадрина О.А., Анисенко А.Н., Готтих М.Б. (2016) Роль ДНК-зависимой протеинкиназы в репликации ВИЧ-1. Молекуляр. биология. 50, 639–654.
Jackson S.P., Bartek J. (2009) The DNA-damage response in human biology and disease. Nature. 461, 1071–1078.
Ryan E.L., Hollingworth R., Grand R.J. (2016) Activation of the DNA damage response by RNA viruses. Biomolecules. 6, 2.
Norbury C.J., Zhivotovsky B. (2004) DNA damage-induced apoptosis. Oncogene. 23, 2797–2808.
Yang J., Yu Y., Hamrick H.E., Duerksen-Hughes P.J. (2003) ATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responses. Carcinogenesis. 24, 1571–1580.
Ciccia A., Elledge S.J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell. 40, 179–204.
Gagné J.P., Rouleau M., Poirier G.G. (2012) Structural biology. PARP-1 activation – bringing the pieces together. Science. 336, 678–679.
Ходырева С.Н., Лаврик О.И. (2016) Поли(ADP-рибоза)полимераза 1 – ключевой регулятор репарации ДНК. Молекуляр. биология. 50, 655–673.
Ko H.L., Ren E.C. (2012) Functional aspects of PARP1 in DNA repair and transcription. Biomolecules. 2, 524–548
Ceccaldi R., Rondinelli B., D’Andrea A.D. (2016) Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64.
Chang H.H., Pannunzio N.R., Adachi N., Lieber M.R. (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 18, 495–506.
Cannan W.J., Pederson D.S. (2016) Mechanisms and consequences of double-strand DNA break formation in chromatin. J. Cell Physiol. 231, 3–14.
Shinagawa H., Iwasaki H. (1996) Processing the holliday junction in homologous recombination. Trends Biochem. Sci. 21, 107–111.
Abbotts R., Wilson D.M. (2017) Coordination of DNA single strand break repair. Free Radic. Biol. Med. 107, 228–244.
Krokan H.E., Bjørås M. (2013) Base excision repair. Cold Spring Harb. Perspect. Biol. 5, a012583
Lans H., Marteijn J.A., Vermeulen W. (2012) ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin. 5, 4.
Kunkel T.A., Erie D.A. (2015) Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet. 49, 291–313.
Brin E., Yi J., Skalka A.M., Leis J. (2000) Modeling the late steps in HIV-1 retroviral integrase-catalyzed DNA integration. J. Biol. Chem. 275, 39287–39295.
Miller M.D., Wang B., Bushman F.D. (1995) Human immunodeficiency virus type 1 preintegration complexes containing discontinuous plus strands are competent to integrate in vitro. J. Virol. 69, 3938–3944.
Daniel R., Katz R.A., Skalka A.M. (1999) A role for DNA-PK in retroviral DNA integration. Science. 284, 644–647.
Sakurai Y., Komatsu K., Agematsu K., Matsuoka M. (2009) DNA double strand break repair enzymes function at multiple steps in retroviral infection. Retrovirology. 6, 114.
Daniel R., Greger J.G., Katz R.A., Taganov K.D., Wu X., Kappes J.C., Skalka A.M. (2004) Evidence that stable retroviral transduction and cell survival following DNA integration depend on components of the nonhomologous end joining repair pathway. J. Virol. 78, 8573–8581.
Coffin J.M., Rosenberg N. (1999) Retroviruses. Closing the joint. Nature. 399, 413–416.
Baekelandt V., Claeys A., Cherepanov P., De Clercq E., De Strooper B., Nuttin B., Debyser Z. (2000) DNA-Dependent protein kinase is not required for efficient lentivirus integration. J. Virol. 74, 11278–11285.
Daniel R., Katz R.A., Merkel G., Hittle J.C., Yen T.J., Skalka A.M. (2001) Wortmannin potentiates integrase-mediated killing of lymphocytes and reduces the efficiency of stable transduction by retroviruses. Mol. Cell Biol. 21, 1164–1172.
Li L., Olvera J.M., Yoder K.E., Mitchell R.S., Butler S.L., Lieber M., Martin S.L., Bushman F.D. (2001) Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 20, 3272–3281.
Studamire B., Goff S.P. (2008) Host proteins interacting with the Moloney murine leukemia virus integrase: multiple transcriptional regulators and chromatin binding factors. Retrovirology. 5, 48.
Zheng Y., Ao Z., Wang B., Jayappa K.D., Yao X. (2011) Host protein Ku70 binds and protects HIV-1 integrase from proteasomal degradation and is required for HIV replication. J. Biol. Chem. 286, 17722–17735.
Anisenko A.N., Knyazhanskaya E.S., Zalevsky A.O., Agapkina J.Y., Sizov A.I., Zatsepin T.S., Gottikh M.B. (2017) Characterization of HIV-1 integrase interaction with human Ku70 protein and initial implications for drug targeting. Sci. Rep. 7, 5649.
Jeanson L., Subra F., Vaganay S., Hervy M., Marangoni E., Bourhis J., Mouscadet J.F. (2002) Effect of Ku80 depletion on the preintegrative steps of HIV-1 replication in human cells. Virology. 300, 100–108.
Waninger S., Kuhen K., Hu X., Chatterton J.E., Wong-Staal F., Tang H. (2004) Identification of cellular cofactors for human immunodeficiency virus replication via a ribozyme-based genomics approach. J. Virol. 78, 12829–12837.
Manic G., Maurin-Marlin A., Laurent F., Vitale I., Thierry S., Delelis O., Dessen P., Vincendeau M., Leib-Mösch C., Hazan U., Mouscadet J.F., Bury-Moné S. (2013) Impact of the Ku complex on HIV-1 expression and latency. PLoS One. 8, e69691.
Espeseth A.S., Fishel R., Hazuda D., Huang Q., Xu M., Yoder K., Zhou H. (2011) siRNA screening of a targeted library of DNA repair factors in HIV infection reveals a role for base excision repair in HIV integration. PLoS One. 6, e17612.
Hultquist J.F., Schumann K., Woo J.M., Manganaro L., McGregor M.J., Doudna J., Simon V., Krogan N.J., Marson A. (2016) A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Rep. 17, 1438–1452.
Appelqvist H., Johansson A.C., Linderoth E., Johansson U., Antonsson B., Steinfeld R., Kågedal K., Ol-linger K. (2012) Lysosome-mediated apoptosis is associated with cathepsin D-specific processing of bid at Phe24, Trp48, and Phe183. Ann. Clin. Lab. Sci. 42, 231–242.
Cooper A., García M., Petrovas C., Yamamoto T., Koup R.A., Nabel G.J. (2013) HIV integration and T cell death: additional commentary. Retrovirology. 10, 150.
Skalka A.M. (2013) HIV: Integration triggers death. Nature. 498, 305-306.
Estaquier J., Zaunders J., Laforge M. (2013) HIV integrase and the swan song of the CD4 T cells? Retrovirology. 10, 149.
Dehart J.L., Andersen J.L., Zimmerman E.S., Ardon O., An D.S., Blackett J., Kim B., Planelles V. (2005) The ataxia telangiectasia-mutated and Rad3-related protein is dispensable for retroviral integration. J. Virol. 79, 1389–1396.
Lau A., Swinbank K.M., Ahmed P.S., Taylor D.L., Jackson S.P., Smith G.C., O’Connor M.J. (2005) Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nat. Cell Biol. 7, 493–500.
Smith J.A., Wang F.X., Zhang H., Wu K.J., Williams K.J., Daniel R. (2008) Evidence that the Nijmegen breakage syndrome protein, an early sensor of double-strand DNA breaks (DSB), is involved in HIV-1 post-integration repair by recruiting the ataxia telangiectasia-mutated kinase in a process similar to, but distinct from, cellular DSB repair. Virol. J. 5, 11.
Awasthi P., Foiani M., Kumar A. (2016) ATM and ATR signaling at a glance. J. Cell Sci. 129, 1285.
Yan S., Sorrell M., Berman Z. (2014) Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell Mol. Life Sci. 71, 3951–3967.
Durocher D., Jackson S.P. (2001) DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr. Opin. Cell Biol. 13, 225–231.
Daniel R., Kao G., Taganov K., Greger J.G., Favorova O., Merkel G., Yen T.J., Katz R.A., Skalka A.M. (2003) Evidence that the retroviral DNA integration process triggers an ATR-dependent DNA damage response. Proc. Natl. Acad. Sci. USA. 100, 4778–4783.
Ariumi Y., Turelli P., Masutani M., Trono D. (2005) DNA damage sensors ATM, ATR, DNA-PKcs, and PARP-1 are dispensable for human immunodeficiency virus type 1 integration. J. Virol. 79, 2973–2978.
Kameoka M., Nukuzuma S., Itaya A., Tanaka Y., Ota K., Ikuta K., Yoshihara K. (2004) RNA interference directed against poly(ADP-ribose) polymerase 1 efficiently suppresses human immunodeficiency virus type 1 replication in human cells. J. Virol. 78, 8931–8934.
Kameoka M., Nukuzuma S., Itaya A., Tanaka Y., Ota K., Inada Y., Ikuta K., Yoshihara K. (2005) Poly(ADP-ribose)polymerase-1 is required for integration of the human immunodeficiency virus type 1 genome near centromeric alphoid DNA in human and murine cells. Biochem. Biophys. Res. Commun. 334, 412–417.
Gäken J.A., Tavassoli M., Gan S.U., Vallian S., Giddings I., Darling D.C., Galea-Lauri J., Thomas M.G., Abedi H., Schreiber V., Ménissier-de Murcia J., Collins M.K., Shall S., Farzaneh F. (1996) Efficient retroviral infection of mammalian cells is blocked by inhibition of poly(ADP-ribose) polymerase activity. J. Virol. 70, 3992–4000.
Ha H.C., Juluri K., Zhou Y., Leung S., Hermankova M., Snyder S.H. (2001) Poly(ADP-ribose) polymerase-1 is required for efficient HIV-1 integration. Proc. Natl. Acad. Sci. USA. 98, 3364–3368.
Siva A.C., Bushman F. (2002) Poly(ADP-ribose) polymerase 1 is not strictly required for infection of murine cells by retroviruses. J. Virol. 76, 11904–11910.
Rom S., Reichenbach N.L., Dykstra H., Persidsky Y. (2015) The dual action of poly(ADP-ribose) polymerase-1 (PARP-1) inhibition in HIV-1 infection: HIV-1 LTR inhibition and diminution in Rho GTPase activity. Front. Microbiol. 6, 878.
Bueno M.T., Reyes D., Valdes L., Saheba A., Urias E., Mendoza C., Fregoso O.I., Llano M. (2013) Poly(ADP-ribose) polymerase 1 promotes transcriptional repression of integrated retroviruses. J. Virol. 87, 2496–2507.
Yoder K.E., Bushman F.D. (2000) Repair of gaps in retroviral DNA integration intermediates. J. Virol. 74, 11191–11200.
Yoder K.E., Espeseth A., Wang X.H., Fang Q., Russo M.T., Lloyd R.S., Hazuda D., Sobol R.W., Fishel R. (2011) The base excision repair pathway is required for efficient lentivirus integration. PLoS One. 6, e17862.
Goetze R.W., Kim D.H., Schinazi R.F., Kim B. (2017) A CRISPR/Cas9 approach reveals that the polymerase activity of DNA polymerase β is dispensable for HIV-1 infection in dividing and nondividing cells. J. Biol. Chem. 292, 14016–14025.
Bennett G.R., Peters R., Wang X.H., Hanne J., Sobol R.W., Bundschuh R., Fishel R., Yoder K.E. (2014) Repair of oxidative DNA base damage in the host genome influences the HIV integration site sequence preference. PLoS One. 9, e103164.
Anisenko A.N., Knyazhanskaya E.S., Isaguliants M.G., Gottikh M.B. (2018) A qPCR assay for measuring the post-integrational DNA repair in HIV-1 replication. J. Virol. Methods. 262, 12–19.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология