

Молекулярная биология, 2019, T. 53, № 4, стр. 531-540
Молекулярные механизмы ненаследуемой толерантности к антибиотикам у бактерий и архей
Т. М. Хлебодарова a, *, В. А. Лихошвай a
a Институт цитологии и генетики Сибирского отделения Российской академии наук
630090 Новосибирск, Россия
* E-mail: tamara@bionet.nsc.ru
Поступила в редакцию 22.01.2019
После доработки 22.01.2019
Принята к публикации 04.02.2019
Аннотация
Феномен бактериальной персистенции, известный также как ненаследуемая толерантность части популяции бактерий к антибиотикам, описан более 70 лет назад. Этот тип толерантности вносит вклад в хронизацию инфекционных заболеваний, включая туберкулез. В настоящее время появление персистентных клеток в популяциях бактерий связывают с функционированием некоторых индуцируемых стрессом молекулярных триггеров, включая системы токсин-антитоксин. В представленном обзоре рассмотрены генетические и метаболические особенности персистентных клеток, обсуждаются механизмы их возникновения. Предложена гипотеза происхождения клеток-персистеров на основе бистабильности, возникающей благодаря нелинейным свойствам системы сопряженной транскрипции-трансляции. В рамках этой гипотезы феномен бактериальной персистенции современных клеток рассматривается как результат генетического закрепления фенотипической множественности, возникшей у примитивных клеток в процессе нейтрально-сопряженной коэволюции – генетического дрейфа множественных нейтрально-сопряженных мутаций. Наша гипотеза объясняет свойства клеток-персистеров, а также их происхождение и неистребимость.
ВВЕДЕНИЕ
Феномен бактериальной персистенции – ненаследуемой толерантности части микробной популяции к антибиотикам, привлекает все большее внимание [1–11]. Это явление считается одной из причин хронического течения многих инфекционных болезней, в том числе туберкулеза и заболеваний, связанных со стафилококковой инфекцией [2–4, 6, 10–12]. Суть этого феномена состоит в том, что чувствительная к антибиотику популяция бактериальных клеток практически всегда, даже после длительной обработки антибиотиком, содержит некоторое число (10–3–10–6) толерантных клеток, способных восстановить популяцию после прекращения воздействия. Однако свойство толерантности эти клетки своим потомкам не передают. Воссозданная популяция клеток бактерий так же чувствительна к воздействию антибиотика, как и исходная.
Феномен персистенции был открыт более 70 лет назад [13], но до начала нынешнего столетия систематические экспериментальные и теоретические исследования ненаследуемой толерантности бактерий к антибиотикам практически не проводились. Основные усилия вплоть до конца 20-го века были направлены на изучение генетических механизмов формирования устойчивости бактерий к антибиотикам, по-видимому, в связи с тем, что вскоре после открытия Bigger J.W. [13] феномена, впоследствии названного бактериальной персистенцией, был разработан метод “отпечатков”. Благодаря этому методу установлено предсуществование в колониях бактерий клеток, устойчивых к антибиотикам, и причиной этой устойчивости были мутации генов [14]. Интерес к этому феномену возник только в начале 2000-х годов после описания мутаций, которые существенно повышали частоту персистентных клеток в культурах Escherichia coli [15], и после появления методов идентификации и выделения этих клеток [16–18]. Оказалось, что персистентные клетки постоянно возникающие в экспоненциально растущей культуре E. coli, генетически идентичны основной популяции клеток, но отличаются низким уровнем метаболизма, медленным ростом, небольшими размерами и устойчивостью к антибиотикам [16–19].
В настоящее время персистентные клетки и клетки, получившие устойчивость к антибиотикам в результате мутаций (резистентные), различают по способности их потомков расти в присутствии антибиотиков – потомки резистентных клеток в отличие от потомков клеток-персистеров растут в присутствии антибиотиков (см. обзор [12]).
Количество персистентных клеток в популяции существенно возрастает в присутствии различных стрессовых факторов, а именно, в условиях голодания, при действии агентов, повреждающих ДНК, при окислительном стрессе, а также при переходе в стационарную фазу роста или при переходе от планктонной формы к пленочной культуре [10, 17, 20–25]. Эти данные позволяют предположить, что бактериальная персистенция может быть отражением более общей стратегии адаптации клеток к внешним воздействиям, нежели только к антибиотикам.
В настоящее время персистентные клетки обнаружены не только у разных видов бактерий [17, 26], но и у архей [27].
ТРИГГЕР, КАК ОДИН ИЗ МЕХАНИЗМОВ ПЕРЕКЛЮЧЕНИЯ КЛЕТОК В ПЕРСИСТЕНТНОЕ СОСТОЯНИЕ
Считается, что персистентные клетки – это альтернативное физиологическое состояние [18], которое возникает в результате переключения клетки с одной генетической программы на другую [16] и отражает феномен фенотипической множественности. В современной научной литературе механизмы возникновения в генетически однородной популяции клеток с различными фенотипами обсуждаются достаточно широко. Наиболее вероятными считаются регуляторные механизмы по типу обратной связи, создающие внутренние условия для возникновения бистабильности, и стохастическая экспрессия генов [28–41].
По данным транскриптомного анализа по функциональной активности персистентные клетки E. coli отличаются от клеток и в экспоненциальной, и в стационарной фазе роста. В персистентных клетках выявлена дифференциальная экспрессия более 200 генов, включая гены, активируемые различными стрессовыми воздействиями, в том числе и гены токсин-антитоксиновых (ТА) систем [16, 17, 42]. Исследование клеточных популяций с мутациями генов различных ТА-систем также подтверждало участие последних в формировании клеток-персистеров [16, 42–44]. Первая обнаруженная мутация, существенно повышающая частоту персистентных клеток в популяции [15], относится именно к ТА-системе HipBA.
ТА-системы – это двухкомпонентные регуляторные системы стрессового ответа, широко распространенные у прокариот (см. обзоры [45–47]). ТА-системы относятся к системам триггерного типа, которые организованы по принципу обратной связи. Этот механизм создает внутренние условия для формирования и поддержания бистабильности – двух состояний, при которых клетка может стабильно существовать [28–34].
Математическое моделирование механизмов регуляции ТА-системы HipBA E. coli показало, что триггерных механизмов в сочетании со стохастичностью процессов, протекающих в клетке, и/или со структурными особенностями регуляторных областей ТА-систем достаточно для формирования фенотипической гетерогенности микробных популяций [48–50]. Возможность формирования бистабильности показана также на общих моделях функционирования ТА-систем [51, 52]. При этом отмечено, что потенциал бистабильности существенно возрастает при учете стохастических флуктуаций, изменений условий роста культуры и увеличения числа ТА-модулей в геноме [51]. Последний фактор – большое число ТА-модулей в геноме – характерен для бактерий (см. обзор [53]).
Экспериментальные данные в целом соответствуют идее о том, что какая-то доля клеток-персистеров, по крайней мере у E. coli, может возникать благодаря особенностям функционирования именно ТА-систем [16, 42–45, 54–56]. Однако отмечено, что мутации отдельных генов ТА-систем и их регуляторов, даже нескольких, хотя и снижают частоту возникновения клеток-персистеров, но не удаляют их из популяции [16, 42, 43, 56, 57]. У других видов бактерий, в частности у Staphylococcus aureus, делеции отдельных ТА-модулей могут не влиять на частоту возникновения клеток-персистеров [10]. Результаты этих исследований свидетельствуют о том, что ТА-системы не единственные участники формирования феномена персистенции.
Установлено, что триггерные механизмы, организованные по типу обратной связи, не уникальны для ТА-систем. Они характерны для многих индуцируемых стрессом систем, обеспечивающих адекватную реакцию клетки на изменение внешних условий, в том числе для окислительного стресса и SOS-ответа на повреждение ДНК. Поэтому тот факт, что частота персистентных клеток возрастает при действии стрессов [21, 24, 58, 59], не только подтверждает участие этих систем в формировании персистенции, но и позволяет предположить, что триггерный механизм можно рассматривать как один из достаточно распространенных механизмов переключения бактериальной клетки в состояние персистенции, но, возможно, как мы отмечали выше, не единственный. Об этом свидетельствуют также представленные ниже данные.
МЕТАБОЛИЧЕСКИЕ АСПЕКТЫ ВОЗНИКНОВЕНИЯ ПЕРСИСТЕНТНЫХ КЛЕТОК
Бактериальные клетки, находящиеся в персистентном состоянии, отличаются низким уровнем метаболизма, о чем свидетельствует их медленный рост, небольшие размеры и низкий уровень синтеза белков [16–19]. Поэтому не удивительно, что голодание, недостаток какого-либо субстрата или необходимость адаптации к новому субстрату оказались факторами, инициирующими переход клеток в персистентное состояние [19, 24, 60–63]. Снижение уровня метаболизма при ограничении питательных ресурсов – это естественная реакция клеток.
Прямым отражением энергетического состояния клетки является уровень внутриклеточного АТР. Действительно, у E. coli [64], S. aureus [10] и Pseudomonas aeruginosa [65] снижение уровня как АТР, так и сАМР [59], производного АТР, приводит к увеличению числа клеток-персистеров в популяции. Механизмы и пути реализации действия этих факторов при формировании фенотипической гетерогенности различны, они могут протекать как с участием различных ТА-систем [36, 60, 61, 66], так и без них [10].
Анализ мутантных клеток показал, что кроме ТА-систем на формирование персистеров в популяциях бактерий действительно влияют мутации генов клеточного метаболизма, например, мутации генов, кодирующих ферменты цикла Кребса, и гена phoU E. coli и его гомологов у других видов бактерий, в том числе у Mycobacterium tuberculosis [63, 67–73]. Выявлено также влияние мутаций генов глобальных регуляторов транскрипции и трансляции, в том числе генов факторов транскрипции fis, hns, hnr и dksA E. coli [74].
Все это свидетельствует о множественных механизмах перехода клеток к персистентному фенотипу. В пользу этой возможности свидетельствуют также данные количественного анализа клеток-персистеров из различных природных изолятов E. coli [75].
КЛЮЧЕВАЯ РОЛЬ СИСТЕМЫ ТРАНСЛЯЦИИ В ФОРМИРОВАНИИ ФЕНОТИПИЧЕСКОЙ МНОЖЕСТВЕННОСТИ
Необходимо отметить несколько фактов, которые позволяют выделить систему трансляции среди систем, участвующих в реализации персистентного фенотипа у бактерий. Во-первых, ее крайне низкая активность, наиболее характерная для персистентной клетки [16–19].
Во-вторых, наибольшее влияние на частоту возникновения клеток-персистеров оказывает неспецифическое блокирование процессов транскрипции и трансляции [76].
В-третьих, именно компоненты системы трансляции, включая мРНК, рРНК и тРНК, являются мишенью токсинов большей части хорошо изученных ТА-систем у бактерий. Взаимодействие токсинов с различными типами РНК ведет к их деградации и ингибированию трансляции (см. обзор [53]), что, по-видимому, можно рассматривать как основной фактор перехода клетки в персистентное состояние при участии ТА-систем.
И, в-четвертых, система трансляции сама обладает внутренним свойством реализации сложных динамических режимов функционирования [77]. Она относится к автокаталитическим системам, поскольку синтезирует не только белки собственного аппарата, но и ферменты, деградирующие их, что имитирует наличие петель обратной регуляции, характерных для триггерных систем.
Все перечисленные факты указывают на систему трансляции, сопряженную у бактерий с транскрипцией, как на необходимый компонент феномена бактериальной персистенции, возникающей при адаптации клеток к внешним условиям.
В связи с этим возникает несколько вопросов, в частности, какие свойства сопряженной системы транскрипции‒трансляции играют ключевую роль в формировании фенотипической множественности, и можно ли считать бистабильность и стохастичность механизмов регуляции экспрессии генов необходимыми условиями проявления феномена бактериальной персистенции [36, 49, 50, 78].
Ответ на последний вопрос получен в результате анализа динамики функционирования простейшей детерминированной модели одиночного цикла размножения клетки с синхронным типом инициации репликации и равномерным делением, в которой также отсутствуют какие-либо регуляторные контуры, создающие внутренние условия для возникновения бистабильности [79]. Основной результат, полученный с использованием этой модели, состоял в том, что, действительно, без учета стохастичности внутриклеточных процессов и в отсутствие каких-либо регуляторных воздействий возможны два различных устойчивых сценария реализации клеточного цикла. Первый сценарий назван R-фенотипом. Он характеризуется коротким периодом одиночного цикла размножения, большим объемом новорожденной “клетки” (здесь и далее кавычками подчеркнуто, что речь идет о моделируемой, а не о природной клетке), интенсивной репликацией и активным метаболизмом. “Клетки”, которые развивались по второму сценарию, обозначены как S-фенотип [79]. Эти клетки имели при рождении относительно малые размеры, характеризовались медленным ростом, низким уровнем метаболизма и репликации. Характеристики R-фенотипа были такими же, как у клеток E. coli, Bacillus subtilis и Salmonella typhimurium, растущих на богатой среде [80], а S‑фенотип был удивительно похож на так называемый персистентный фенотип природных клеток [16, 19]. Т.е. оказалось, что для объяснения возникновения у бактерий фенотипической множественности нет необходимости привлекать какие-либо специальные механизмы регуляции молекулярно-генетических и биохимических процессов, в том числе и стохастические флуктуации экспрессии генов, контролирующих рост и деление клетки.
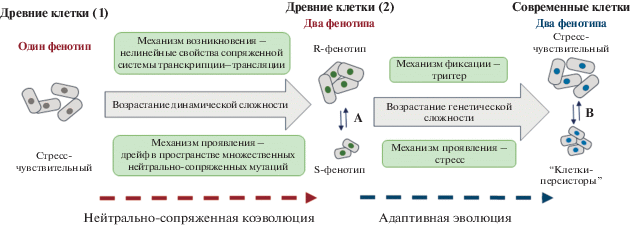
Рис. 1.
Эволюционный сценарий возникновения персистентных клеток. Переход (А) между R- и S-фенотипами происходит благодаря флуктуациям концентраций внутриклеточных компонентов во время деления клетки, переход (В) – при стрессе.
В этой связи необходимо отметить, что флуктуации активного и неактивного состояния гена (генов) в индивидуальной клетке, обусловленные стохастичностью взаимодействия регуляторных факторов с промоторами генов-мишеней, могут быть механизмом формирования на популяционном уровне бимодального распределения клеток по уровню экспрессии генов [41] и даже, возможно, механизмом возникновения клеток-персистеров [78]. Однако бистабильность, возникающая в результате стохастичности экспрессии генов, не является ни достаточной, ни необходимой для формирования бимодального распределения клеток в популяции [39].
В дальнейшем оказалось, что в основе наблюдаемой фенотипической множественности клеточных циклов лежат нелинейные свойства сопряженной системы транскрипции‒трансляции, универсальные для всех типов клеток [81, 82].
Таким образом, молекулярные предпосылки формирования феномена бистабильности имманентно присутствуют в одноклеточном организме. Общих принципов хранения и передачи наследственной информации, в основе которых лежат фундаментальные биохимические законы функционирования живых организмов, достаточно для возникновения гетерогенности фенотипических параметров клеточных циклов.
ПРОИСХОЖДЕНИЕ ПЕРСИСТЕНТНЫХ КЛЕТОК
На основании полученных данных [79, 81] высказана гипотеза, согласно которой персистентные клетки, способные в условиях стресса переключаться из состояния активного развития в неактивное, произошли от клеток, которые приобрели способность осуществлять одиночный цикл размножения нескольким альтернативными способами.
Анализ условий формирования фенотипической бистабильности показал, что возможность реализации формулы размножения “один генотип → два фенотипа” обеспечивается процессом нейтрально-сопряженной коэволюции – дрейфом популяции в пространстве множественных нейтрально-сопряженных мутаций в условиях относительного недостатка питательных ресурсов [82].
Этот тип эволюции по сути относится к явлению нейтральной эволюции, открытой и исследованной в работах М. Кimura и ряда других исследователей [83–87], значение которой в эволюционном процессе высоко оценивается и сейчас [88]. Однако в своей основе нейтрально-сопряженная коэволюция кардинально отличается от классической нейтральной эволюции Кimura [83, 84]. Основное различие состоит в том, что эта эволюция протекает в пространстве не одиночных, а множественных мутаций, сгруппированных по принципу нейтральной согласованности. Каждая отдельная мутация в такой группе (минимальное число мутаций в группе равно двум) может быть повреждающей, но их совместное воздействие на динамику биохимического процесса (в данном случае на процесс сопряженной транскрипции-трансляции) на физиологическом уровне компенсирует вредное действие друг друга. Т.е., по отношению к эволюционному отбору на уровне приспособленности организма эти группы мутаций выглядят, как нейтральные.
Возможность компенсации “вредных” мутаций легко просматривается в каталитических свойствах обратимых биохимических реакций, которые лежат в основе всех клеточных процессов, и ранее была проиллюстрирована М.В. Волькенштейном [89, 90] на примере простейшей ферментативной реакции, описываемой уравнением Михаэлиса–Ментен [91, 92]. В более общем виде идея существования компенсаторных механизмов минимизации фенотипических проявлений “вредных” мутаций высказывалась И.И. Шмальгаузеном еще в 1946 году (цит. по [93]). Возможная роль компенсаторных мутаций в эволюции отмечалась и Кimura [94]. Такой тип компенсации – через множественные сопряженные мутации, показан на примере эволюции последовательностей стартовых кодонов и сайтов инициации трансляции [95].
Возможность существования компенсаторных мутаций, связанных с функционированием ферментов, показана, например, в областях аминоацил-тРНК-синтетазы, вовлеченных в распознавание тРНК [96], а также в сопряженно действующих ферментах, таких как топоизомеразы и гиразы, контролирующие суперспирализацию ДНК [97–99]. С помощью геномного анализа выявлено большое число компенсаторных мутаций в районах мРНК с высокой стабильностью локальных вторичных структур [100]. Эти данные свидетельствуют о том, что компенсаторные мутации достаточно широко распространены в геномных последовательностях. Они возникают намного реже одиночных, однако вероятность их фиксации, как показано Кimura [94], достаточно высока.
Уникальность данного типа эволюции, названного нейтрально-сопряженной коэволюцией [82, 101], состоит в том, что при определенных условиях нейтральный дрейф обеспечивал фактически безальтернативное появление из модельных “клеток”, изначально способных осуществлять одиночный цикл только одним способом (стратегия развития один генотип → один фенотип), потомков, которые приобретали способность реализовывать одиночный цикл не одним, а несколькими альтернативными (стратегия развития один генотип → несколько фенотипов). Ранее аналогичное свойство наблюдали на модели стохастического эволюционного дрейфа популяции простейших самовоспроизводящихся особей [102], однако в этой работе отсутствовала привязка к конкретной биологической системе.
Вполне естественно предположить, что свойство фенотипической множественности оказалось достаточно полезным для выживания популяций, периодически попадающих в условия, неблагоприятные для выживания активно растущих клеток – R-фенотип, согласно [79], и некритичных для клеток с низким уровнем метаболизма – S-фенотип, согласно [79]. Поэтому свойство бистабильности закрепилось как эволюционно выгодное приобретение путем формирования в клетках специальных молекулярно-генетических механизмов, в том числе, ТА молекулярных триггеров, переводящих в условиях стресса клетки из чувствительного к стрессу фенотипа в устойчивый, что мы и наблюдаем у современных видов бактерий [16, 42–44, 103].
Исходя из полученных данных, предложен представленный на рис. 1 сценарий эволюционных молекулярно-генетических механизмов возникновения современных персистентных клеток [82].
Этот сценарий показывает как в результате нейтральной коэволюции и благодаря нелинейным свойствам сопряженной системы транскрипции-трансляции из условной популяции “древних клеток” (1), состоящей из клеток, способных осуществлять одиночный цикл исключительно единственным способом, возникает условная популяция “древних клеток” (2). В этой популяции наряду с клетками первого типа появляются клетки качественно нового типа – способные осуществлять одиночный цикл развития несколькими альтернативными способами (R- и S-фенотипы). Условная популяция “современных клеток” возникает из клеток второго типа “древней популяции” в результате формирования на генетическом уровне регуляторных контуров триггерного типа, которые включают оба фенотипа в норму реакции. Чувствительные и персистентные клетки в популяциях современных видов бактерий представлены генетически идентичными клетками, осуществляющими клеточный цикл альтернативными способами [16–19].
Этот сценарий не отрицает возможности того, что персистентные клетки могли и не быть прямыми потомками бистабильных клеток, а возникли какими-то другими способами [62, 67, 104, 105], в том числе, путем прямого формирования в изначально монофенотипических клетках специальных триггерных механизмов регуляции конкретных молекулярно-генетических систем, обеспечивающих условия для формирования и поддержания бистабильности [49, 51, 52, 103].
Однако сценарий, представленный на рис. 1, в отличие от ранее предложенных, обладает очень важным свойством – он позволяет объяснить такие горячие проблемы происхождения клеток-персистеров, как их неистребимость, постоянное присутствие в популяции даже в отсутствие стрессовых воздействий, а также не наследуемость толерантности к стрессовым условиям при восстановлении численности популяции из клеток-персистеров после снятия стресса.
РОЛЬ НЕЙТРАЛЬНО-СОПРЯЖЕННОЙ КОЭВОЛЮЦИИ В ВОЗНИКНОВЕНИИ СЛОЖНОСТИ И РАЗНООБРАЗИЯ ОДНОКЛЕТОЧНЫХ ОРГАНИЗМОВ (ГИПОТЕЗА)
Теоретически показано, что при определенных условиях эволюционный дрейф в пространстве множественных нейтрально-сопряженных мутаций обеспечивает фактически безальтернативное возникновение в генетически однородной популяции двух клеточных форм, отличных друг от друга по уровню метаболизма, длине клеточного цикла и размерам “клетки” [81, 82, 101]. Этот процесс протекает в условиях относительного недостатка питательных ресурсов [82, 101], нередкого для бактерий, живущих в естественных условиях, и в его основе лежат нелинейные свойства системы сопряженной транскрипции‒трансляции [81, 82], сходной в основных чертах у всех живых организмов.
Эта универсальность позволяет предположить, что способностью осуществлять одиночный цикл размножения нескольким альтернативными способами (свойство фенотипической множественности) может обладать практически любой одноклеточный организм. Условия для появления фенотипической множественности могли возникать многократно, начиная с самых ранних этапов эволюции одноклеточных организмов. Возможно, даже раньше, чем были сформированы основные принципы хранения и передачи наследственной информации (система сопряженной транскрипции‒трансляции), поскольку феномен фенотипической множественности можно рассматривать как проявление автокаталитических свойств биохимических систем и их существенной нелинейности. В пользу этого предположения косвенно свидетельствует существование персистентных клеток в разных доменах (царствах) живых организмов: и у бактерий, и у архей [16, 26, 27].
Согласно этой точке зрения, феномен бактериальной персистенции современных клеток – ненаследуемой толерантности части микробной популяции к антибиотикам, отражает более общую стратегию адаптации клетки к внешним воздействиям и является результатом генетического закрепления фенотипической множественности, возникшей у примитивных клеток в процессе нейтрально-сопряженной коэволюции.
Необходимо отметить более общее значение способности клеток с самых ранних этапов своей эволюции реализовывать одиночный цикл не одним, а несколькими альтернативными фенотипами, обладающими различной устойчивостью к внешним воздействиям. В изменяющихся условиях внешней среды стратегия развития “один генотип → несколько фенотипов” создает условия для независимой эволюции чувствительных и устойчивых к стрессу клеточных фенотипов, каждый из которых в процессе адаптации может достичь стадии максимальной приспособленности. А это означает, что их дальнейшая эволюция снова будет протекать в рамках нейтрально-сопряженной коэволюции [82], ведущей к возникновению альтернативных клеточных фенотипов, но уже на новой генетической основе. О возможности многократного повторения данного сценария свидетельствует существование клеток-персистеров у разных видов бактерий, относящихся к разным классам [17, 26, 67]. О возможности независимой эволюции альтернативных клеточных форм косвенно свидетельствуют данные геномного и филогенетического анализа бактерий и архей. Так, в пределах рода Methanococcus архей есть и мезофильные (M. maripaludis, M. vannielii, M. voltae), и гипертермофильные виды (M. jannaschii, M. igneus), оптимальные условия роста которых различаются на 50оС, а геномное содержание G+C практически идентично [106]. Аналогичный пример можно привести и для Deinococcus – экстремофильных бактерий, которые в целом характеризуются высокой устойчивостью к различным видам облучения и высыханию, однако, среди них есть как мезофильные, так и термофильные виды [107]. Из анализа ортологичных групп белков также следует, что сестринскими видами являются гипертермофильная бактерия Thermus thermophilus и мезофильная бактерия D. radiodurans [108]. Более того, на филогенетических деревьях, построенных на основе последовательностей высококонсервативных генов, мезофилы часто группируются в одном кластере с термофилами (цит. по [109]).
Здесь также можно отметить некоторые общие черты фенотипа архей, которые роднят их с предполагаемым S-фенотипом “древних клеток” [79] и позволяют предположить возможность их независимой эволюции именно из этой альтернативной клеточной формы: археи не только в основной своей массе являются экстремофилами, но и обладают в среднем более длинным клеточным циклом [110] и более мелкими размерами, чем бактерии (см. базу данных: BioNumbers, http://www.bionumbers. hms.harvard.edu).
С этой точки зрения можно также посмотреть на явление спорообразования – генетический механизм адаптации клеток к крайне неблагоприятным внешним воздействиям. Спорообразование, характерное для некоторых классов бактерий (Bacilli, Clostridia), проявляется способностью формирования спор, которые обладают чрезвычайной устойчивостью ко многим стрессовым факторам, что обусловлено не только дополнительными внешними оболочками, но и практически полным отключением метаболизма (см. обзор [111]). Не исключено, что это один из путей эволюционного использования устойчивости к стрессу S-фенотипа “древних клеток”, связанной с его крайне низкой метаболической активностью.
Таким образом, не исключено, что эволюционные сценарии возникновения не только феномена бактериальной персистенции, но и способности различных классов одноклеточных организмов жить в экстремальных условиях или пережидать их, отражают независимую адаптивную эволюцию предполагаемого S-фенотипа “древних клеток” [82, 101], которая на разных этапах и в разное время привела к формированию генетически различных механизмов закрепления устойчивости к стрессу или способности клетки переходить в это состояние в неблагоприятных условиях внешней среды.
Более того, не видно принципиальных запретов к тому, чтобы нейтрально-сопряженная коэволюция в определенные периоды прошлого выступила в качестве дизайнера и драйвера усложнения живых систем по описанному выше сценарию: возрастание динамической сложности поведения популяции живых организмов → включение динамической сложности в норму реакции путем формирования специализированных генетических программ → возрастание сложности строения организма [82, 101].
Этот уникальный потенциал нейтрально-сопряженной коэволюции является особенно интригующим, так как показывает направление поиска решения проблемы эволюционного усложнения строения живых систем – одной из самых актуальных и сложных проблем теории эволюции, которая не только не решена до сих пор, но и, по мнению Е.В. Кунина [109], для решения которой до сих пор даже не предложено подходов.
Работа выполнена при финансовой поддержке программы фундаментальных исследований СО РАН (проект № 0324-2019-0040).
Настоящая работа не содержит исследований, выполненных с использованием животных.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Dhar N., McKinney J.D. (2007) Microbial phenotypic heterogeneity and antibiotic tolerance. Curr. Opin. Microbiol. 10, 30–38.
Lewis K. (2007) Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5, 48–56.
Lewis K. (2008) Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 322, 107–131.
Lewis K. (2012) Persister cells: molecular mechanisms related to antibiotic tolerance. Handb. Exp. Pharmacol. 211, 121–133.
Jayaraman R. (2008) Bacterial persistence: some new insights into an old phenomenon. J. Biosci. 33, 795–805.
Kint C.I., Verstraeten N., Fauvart M., Michiels J. (2012) New-found fundamentals of bacterial persistence. Trends Microbiol. 20, 577–585.
Amato S.M., Fazen C.H., Henry T.C., Mok W.W., Orman M.A., Sandvik E.L., Volzing K.G., Brynildsen M.P. (2014) The role of metabolism in bacterial persistence. Front. Microbiol. 5, 70.
Kester J.C., Fortune S.M. (2014) Persisters and beyond: mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit. Rev. Biochem. Mol. Biol. 49, 91–101.
Gerdes K., Semsey S. (2016) Pumping persisters. Nature. 534, 41–42.
Conlon B.P., Rowe S.E., Gandt A.B., Nuxoll A.S., Donegan N.P., Zalis E.A., Clair G., Adkins J.N., Cheung A.L., Lewis K. (2016) Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 1, 16051.
Defraine V., Fauvart M., Michiels J. (2018) Fighting bacterial persistence: Current and emerging anti-persister strategies and therapeutics. Drug Resist. Update. 38, 12–26.
Wood T.K., Knabel S.J., Kwan B.W. (2013) Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 79, 7116–7121.
Bigger J.W. (1944) Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet. 244, 497–500.
Lederberg J., Lederberg E.M. (1952) Replica plating and indirect selection of bacterial mutants. J. Bacteriol. 63, 399–406.
Moyed H.S., Bertrand K.P. (1983) hipA, a newly recognized gene of Escherichia coli K12 that affects the frequency of persisters after inhibition of murein synthesis. J. Bacteriol. 155, 768–775.
Balaban N.Q., Merrin J., Chait R., Kowalik L., Leibler S. (2004) Bacterial persistence as a phenotypic switch. Science. 305, 1622–1625.
Keren I., Shah D., Spoering A., Kaldalu N., Lewis K. (2004) Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186, 8172–8180.
Shah D., Zhang Z., Khodursky A., Kaldalu N., Kurg K., Lewis K. (2006) Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6, 53.
Radzikowski J.L., Vedelaar S., Siegel D., Ortega Á.D., Schmidt A., Heinemann M. (2016) Bacterial persistence is an active σS stress response to metabolic flux limitation. Mol. Syst. Biol. 12, 882.
Nguyen D., Joshi-Datar A., Lepine F., Bauerle E., Olakanmi O., Beer K., McKay G., Siehnel R., Schafhauser J., Wang Y., Britigan B.E., Singh P.K. (2011) Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science. 334, 982–986.
Grant S.S., Hung DT (2013) Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence. 4, 273–283.
Grant S.S., Kaufmann B.B., Chand N.S., Haseley N., Hung D.T. (2012) Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc. Natl. Acad. Sci. USA. 109, 12147–12152.
Shapiro J.A., Nguyen V.L., Chamberlain N.R. (2011) Evidence for persisters in Staphylococcus epidermidis RP62a planktonic cultures and biofilms. J. Med. Microbiol. 60, 950–960.
Bernier S.P., Lebeaux D., DeFrancesco A.S., Valomon A., Soubigou G., Coppée J.Y., Ghigo J.M., Beloin C. (2013) Starvation, together with the SOS response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 9, e1003144.
Verstraeten N., Knapen W.J., Kint C.I., Liebens V., Van den Bergh B., Dewachter L., Michiels J.E., Fu Q., David C.C., Fierro A.C., Marchal K., Beirlant J., Versйes W., Hofkens J., Jansen M., Fauvart M., Michiels J. (2015) Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell. 59, 9–21.
Grassi L., Di Luca M., Maisetta G., Rinaldi A.C., Esin S., Trampuz A., Batoni G. (2017) Generation of persister cells of Pseudomonas aeruginosa and Staphylococcus aureus by chemical treatment and evaluation of their susceptibility to membrane-targeting agents. Front. Microbiol. 8, 1917.
Megaw J., Gilmore B.F. (2017) Archaeal persisters: persister cell formation as a stress response in Haloferax volcanii. Front. Microbiol. 8, 1589.
Ferrell J.E. Jr. (2002) Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr. Opin. Cell. Biol. 14, 140–148.
Angeli D., Ferrell J.E. Jr., Sontag E.D. (2004) Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feedback systems. Proc. Natl. Acad. Sci. USA. 101, 1822–1827.
Ozbudak E.M., Thattai M., Lim H.N., Shraiman B.I., Van Oudenaarden A. (2004) Multistability in the lactose utilization network of Escherichia coli. Nature. 427, 737–740.
Smits W.K., Kuipers O.P., Veening J.W. (2006) Phenotypic variation in bacteria: the role of feedback regulation. Nat. Rev. Microbiol. 4, 259–271.
Dubnau D., Losick R. (2006) Bistability in bacteria. Mol. Microbiol. 61, 564–572.
Piggot P. (2010) Epigenetic switching: bacteria hedge bets about staying or moving. Curr. Biol. 20, R480–R482.
Avendaño M.S., Leidy C., Pedraza J.M. (2013) Tuning the range and stability of multiple phenotypic states with coupled positive-negative feedback loops. Nat. Commun. 4, 2605.
Kaern M., Elston T.C., Blake W.J., Collins J.J. (2005) Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6, 451–464.
Sureka K., Ghosh B., Dasgupta A., Basu J., Kundu M., Bose I. (2008) Positive feedback and noise activate the stringent response regulator rel in mycobacteria. PLoS One. 3, e1771.
To T.L., Maheshri N. (2010) Noise can induce bimodality in positive transcriptional feedback loops without bistability. Science. 327, 1142–1145.
Zheng X.D., Yang X.Q., Tao Y. (2011) Bistability, probability transition rate and first-passage time in an autoactivating positive-feedback loop. PLoS One. 6, e17104.
Shu C.C., Chatterjee A., Dunny G., Hu W.S., Ramkrishna D. (2011) Bistability versus bimodal distributions in gene regulatory processes from population balance. PLoS Comput. Biol. 7, e1002140.
Ghosh S., Banerjee S., Bose I. (2012) Emergent bistability: effects of additive and multiplicative noise. Eur. Phys. J. E. Soft. Matter. 35, 11.
Thomas P., Popović N., Grima R. (2014) Phenotypic switching in gene regulatory networks. Proc. Natl. Acad. Sci. USA. 111, 6994–6999.
Dörr T., Vulić M., Lewis K. (2010) Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8, e1000317.
Tripathi A., Dewan P.C., Siddique S.A., Varadarajan R. (2014) MazF-induced growth inhibition and persister generation in Escherichia coli. J. Biol. Chem. 289, 4191–4205.
Schumacher M.A., Balani P., Min J., Chinnam N.B., Hansen S., Vulić M., Lewis K., Brennan R.G. (2015) HipBA-promoter structures reveal the basis of heritable multidrug tolerance. Nature. 524(7563), 59–64.
Gerdes K., Christensen S.K., Løbner-Olesen A. (2005) Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 3, 371–382.
Otsuka Y. Prokaryotic toxin-antitoxin systems: novel regulations of the toxins. (2016) Curr. Genet. 62, 379–382.
Soo V.W., Cheng H.Y., Kwan B.W., Wood T.K. (2014) de novo synthesis of a bacterial toxin/antitoxin system. Sci. Rep. 4, 4807.
Lou C., Li Z., Ouyang Q. (2008) A molecular model for persister in E. coli. J. Theor. Biol. 255, 205–209.
Koh R.S., Dunlop M.J. (2012) Modeling suggests that gene circuit architecture controls phenotypic variability in a bacterial persistence network. BMC Syst. Biol. 6, 47.
Feng J., Kessler D.A., Ben-Jacob E., Levine H. (2014) Growth feedback as a basis for persister bistability. Proc. Natl. Acad. Sci. USA. 111, 544–549.
Fasani R.A., Savageau M.A. (2013) Molecular mechanisms of multiple toxin-antitoxin systems are coordinated to govern the persister phenotype. Proc. Natl. Acad. Sci. USA. 110, E2528–2537.
Gelens L., Hill L., Vandervelde A., Danckaert J., Loris R. (2013) A general model for toxin-antitoxin module dynamics can explain persister cell formation in E. coli. PLoS Comput. Biol. 9, e1003190.
Unterholzner S.J., Poppenberger B., Rozhon W. (2013) Toxin-antitoxin systems: Biology, identification, and application. Mob. Genet. Elements. 3, e26219.
Gupta K., Tripathi A., Sahu A., Varadarajan R. (2017) Contribution of the chromosomal ccdAB operon to bacterial drug tolerance. J. Bacteriol. 199, e00397-17.
Gurnev P.A., Ortenberg R., Dörr T., Lewis K., Bezrukov S.M. (2012) Persister promoting bacterial toxin TisB produces anion-selective pores in planar lipid bilayers. FEBS Lett. 586, 2529–2534.
Kim Y., Wood T.K. (2010) Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli. Biochem. Biophys. Res. Commun. 391, 209–213.
Maisonneuve E., Castro-Camargo M., Gerdes K. (2013) (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell. 154, 1140–1150.
Wu Y., Vulić M., Keren I., Lewis K. (2012) Role of oxidative stress in persister tolerance. Antimicrob. Agents Chemother. 56, 4922–4926.
Molina-Quiroz R.C., Silva-Valenzuela C., Brewster J., Castro-Nallar E., Levy S.B., Camilli A. (2018) Cyclic AMP Regulates bacterial persistence through repression of the oxidative stress response and SOS-dependent DNA repair in uropathogenic Escherichia coli. MBio. 9, e02144-17.
Amato S.M., Brynildsen M.P. (2015) Persister heterogeneity arising from a single metabolic stress. Curr. Biol. 25, 2090–2098.
Amato S.M., Orman M.A., Brynildsen M.P. (2013) Metabolic control of persister formation in Escherichia coli. Mol. Cell. 50, 475–487.
Kotte O., Volkmer B., Radzikowski J.L., Heinemann M. (2014) Phenotypic bistability in Escherichia coli’s central carbon metabolism. Mol. Syst. Biol. 10, 736.
Namugenyi S.B., Aagesen A.M., Elliott S.R., Tisch-ler A.D. (2017) Mycobacterium tuberculosis PhoY proteins promote persister formation by mediating Pst/SenX3-RegX3 phosphate sensing. MBio. 8, e00494-17.
Shan Y., Brown Gandt A., Rowe S.E., Deisinger J.P., Conlon B.P., Lewis K. (2017) ATP-dependent persister formation in Escherichia coli. MBio. 8, e02267-16.
Cameron D.R., Shan Y., Zalis E.A., Isabella V., Lewis K. (2018) A genetic determinant of persister cell formation in bacterial pathogens. J. Bacteriol. 200, e00303-18.
Ghosh S., Sureka K., Ghosh B., Bose I., Basu J., Kundu M. (2011) Phenotypic heterogeneity in mycobacterial stringent response. BMC Syst. Biol. 5, 18.
Li Y., Zhang Y. (2007) PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob. Agents Chemother. 51, 2092–2099.
Shi W., Zhang Y. (2010) PhoY2 but not PhoY1 is the PhoU homologue involved in persisters in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 65, 1237–1242.
Ma C., Sim S., Shi W., Du L., Xing D., Zhang Y. (2010) Energy production genes sucB and ubiF are involved in persister survival and tolerance to multiple antibiotics and stresses in Escherichia coli. FEMS Microbiol. Lett. 303, 33–40.
Torrey H.L., Keren I., Via L.E., Lee J.S., Lewis K. (2016) High persister mutants in Mycobacterium tuberculosis. PLoS One. 11, e0155127.
Kim J.S., Cho D.H., Heo P., Jung S.C., Park M., Oh E.J., Sung J., Kim P.J., Lee S.C., Lee D.H., Lee S., Lee C.H., Shin D., Jin Y.S., Kweon D.H. (2016) Fumarate-mediated persistence of Escherichia coli against antibiotics. Antimicrob. Agents Chemother. 60, 2232–2240.
Cui P., Niu H., Shi W., Zhang S., Zhang W., Zhang Y. (2018) Identification of genes involved in bacteriostatic antibiotic-induced persister formation. Front. Microbiol. 9, 413.
Wang Y., Bojer M.S., George S.E., Wang Z., Jensen P.R., Wolz C., Ingmer H. (2018) Inactivation of TCA cycle enhances Staphylococcus aureus persister cell formation in stationary phase. Sci. Rep. 8, 10849.
Hansen S., Lewis K., Vulic M. (2008) Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob. Agents Chemother. 52, 2718–2726.
Hofsteenge N., van Nimwegen E., Silander O.K. (2013) Quantitative analysis of persister fractions suggests different mechanisms of formation among environmental isolates of E. coli. BMC Microbiol. 13, 25.
Kwan B.W., Valenta J.A., Benedik M.J., Wood T.K. (2013) Arrested protein synthesis increases persister-like cell formation. Antimicrob. Agents Chemother. 57, 1468–1473.
Likhoshvai V.A., Kogai V.V., Fadeev S.I., Khlebodarova T.M. (2016) Chaos and hyperchaos in a model of ribosome autocatalytic synthesis. Sci. Rep. 6, 38870.
Day T. (2016) Interpreting phenotypic antibiotic tolerance and persister cells as evolution via epigenetic inheritance. Mol. Ecol. 25, 1869–1882.
Лихошвай В.А., Хлебодарова Т.М. (2016) Фенотипическая множественность клеточного цикла бактерий: математическая модель. Матем. биол. биоинформ.11, 91–113.
Schaechter M., Maaloe O., Kjeldgaard N.O. (1958) Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J. Gen. Microbiol. 19, 592–606.
Хлебодарова Т.М., Лихошвай В.А. (2016) Фенотипическая множественность клеточного цикла – следствие универсальных свойств сопряженной системы транскрипции-трансляции. В: Матем. биол. и биоинформ. 6. Ред. Лахно В.Д. М.: МАКС Пресс, 98–99.
Khlebodarova T.M., Likhoshvai V.A. (2018) Persister cells – a plausible outcome of neutral coevolutionary drift. Sci. Rep. 8, 14309.
Kimura M. (1968) Evolutionary rate at the molecular level. Nature. 217, 624–626.
Kimura M. (1991) The neutral theory of molecular evolution: a review of recent evidence. Jpn. J. Genet. 66, 367–386.
King J.L., Jukes T.H. (1969) Non-Darwinian evolution. Science. 164, 788–798.
Ohta T. (2002) Near-neutrality in evolution of genes and gene regulation. Proc. Natl. Acad. Sci. USA. 99, 16134–16137.
Ohta T. (1973) Slightly deleterious mutant substitutions in evolution. Nature. 246, 96–98.
Koonin E.V. (2016) Splendor and misery of adaptation, or the importance of neutral null for understanding evolution. BMC Biol. 14, 114.
Волькенштейн М.В. (1985) Биополимеры и эволюция. Молекуляр. биология. 19, 55–66.
Волькенштейн М.В., Гольдштейн Б.Н. (1986) Ферментативные механизмы компенсации вредных мутаций. Молекуляр. биология. 20, 1645–1654.
Michaelis L., Menten M.L. (1913) Die Kinetik der Invertinwirkung. Biochem. Z. 49, 333–369.
Michaelis L., Menten M.M. (2013) The kinetics of invertin action. 1913. FEBS Lett. 587, 2712–2720.
Шмальгаузен И.И. (1968) Факторы эволюции: теория стабилизирующего отбора. Ред. Берг Р.Л., Махотин А.А., Яблоков А.В. М.: Наука.
Kimura M. (1985) The role of compensatory neutral mutations in molecular evolution. J. Genet. 64, 7–19.
Belinky F., Rogozin I.B., Koonin, E.V. (2017) Selection on start codons in prokaryotes and potential compensatory nucleotide substitutions. Sci. Rep. 7, 12422.
Frenkel-Morgenstern M., Tworowski D., Klipcan L., Safro M. (2009) Intra-protein compensatory mutations analysis highlights the tRNA recognition regions in aminoacyl-tRNA synthetases. J. Biomol. Struct. Dyn. 27, 115–126.
DiNardo S., Voelkel K.A., Sternglanz R., Reynolds A.E., Wright A. (1982) Escherichia coli DNA topoisomerase I mutants have compensatory mutations in DNA gyrase genes. Cell. 31, 43–51.
Pruss G.J., Manes S.H., Drlica K. (1982) Escherichia coli DNA topoisomerase I mutants: increased supercoiling is corrected by mutations near gyrase genes. Cell. 31, 35–42.
Raji A., Zabel D.J., Laufer C.S., Depew R.E. (1985) Genetic analysis of mutations that compensate for loss of Escherichia coli DNA topoisomerase I. J. Bacteriol. 162, 1173–1179.
Mao Y., Li Q., Zhang Y., Zhang J., Wei G., Tao S. (2013) Genome-wide analysis of selective constraints on high stability regions of mRNA reveals multiple compensatory mutations in Escherichia coli. PLoS One. 8, e73299.
Лихошвай В.А., Хлебодарова T.M. (2018) Один генотип → два фенотипа: “нейтрально-сопряженная коэволюция” и происхождение персистентных клеток. В: Матем. биол. биоинформ. 7. Ред. Лахно В.Д. Пущино: ИМПБ РАН, e67.
Likhoshvai V.A., Matushkin Yu.G. (2004) Sporadic emergence of latent phenotype during evolution. In: Bioinformatics of genome regulation and structure. Eds Kolchanov N., Hofestaedt R. Boston: Kluwer Acad. Publ., 231–243.
Germain E., Roghanian M., Gerdes K., Maisonneuve E. (2015) Stochastic induction of persister cells by HipA through (p)ppGpp-mediated activation of mRNA endonucleases. Proc. Natl. Acad. Sci. USA. 112, 5171–5176.
Klapper I., Gilbert P., Ayati B.P., Dockery J., Ste-wart P.S. (2007) Senescence can explain microbial persistence. Microbiology. 153, 3623–3630.
Shearwin K. (2009) Slow growth leads to a switch. Nat. Chem. Biol. 5, 784–785.
Haney P.J., Badger J.H., Buldak G.L., Reich C.I., Woese C.R., Olsen G.J. (1999) Thermal adaptation analyzed by comparison of protein sequences from mesophilic and extremely thermophilic Methanococcus species. Proc. Natl. Acad. Sci. USA. 96, 3578–3583.
Makarova K.S., Omelchenko M.V., Gaidamakova E.K., Matrosova V.Y, Vasilenko A., Zhai M., Lapidus A., Copeland A., Kim E., Land M., Mavrommatis K., Pitluck S., Richardson P.M., Detter C., Brettin T., Saunders E., Lai B., Ravel B., Kemner K.M., Wolf Y.I., Sorokin A., Gerasimova A.V., Gelfand M.S., Fredrickson J.K., Koonin E.V., Daly M.J. (2007) Deinococcus geothermalis: the pool of extreme radiation resistance genes shrinks. PLoS One. 2, e955.
Omelchenko M.V., Wolf Y.I., Gaidamakova E.K., Matrosova V.Y., Vasilenko A., Zhai M., Daly M.J., Koonin E.V., Makarova K.S. (2005) Comparative genomics of Thermus thermophilus and Deinococcus radiodurans: divergent routes of adaptation to thermophily and radiation resistance. BMC Evol. Biol. 5, 57.
Кунин Е.В. (2014) Логика случая. О природе и происхождении биологической эволюции. М.: Центрполиграф.
Bernander R., Poplawski A. (1997) Cell cycle characteristics of thermophilic archaea. J. Bacteriol. 179, 4963–4969.
Tan I.S., Ramamurthi K.S. (2014) Spore formation in Bacillus subtilis. Environ. Microbiol. Rep. 6, 212–225.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология