

Молекулярная биология, 2019, T. 53, № 5, стр. 711-724
Как методы молекулярной биологии повлияли на понимание устройства кроветворной системы
А. Е. Бигильдеев a, *, Н. А. Петинати a, Н. И. Дризе a, b
a Национальный медицинский исследовательский центр гематологии Министерства здравоохранения
Российской Федерации
125167 Москва, Россия
b Кафедра иммунологии биологического факультета Московского государственного университета
им. М.В. Ломоносова
119234 Москва, Россия
* E-mail: bigildeev.ae@gmail.com
Поступила в редакцию 19.02.2019
После доработки 04.04.2019
Принята к публикации 07.04.2019
Аннотация
Кровь чрезвычайно важна для многоклеточного организма ‒ она связывает воедино все органы и ткани, снабжает их питательными веществами и кислородом, отводит углекислый газ и продукты обмена, поддерживает гомеостаз и обеспечивает защиту от инфекций. Вероятно, именно поэтому изучению крови всегда уделяли большое внимание. Древние считали, что душа находится в крови и временами “уходит в пятки”. Исходно кровь изучали морфологическими методами, к которым в ХХ веке добавились физиологические и клеточные. С их помощью получены важные знания о том, что зрелые клетки крови образуются из немногочисленной популяции стволовых кроветворных клеток, локализованных в костном мозге. Появление молекулярно-биологических методов и их сочетание с классическими физиологическими методами позволило совершить прорыв в понимании устройства кроветворной системы и изменить наши представления о природе стволовых клеток взрослого организма. В представленном обзоре рассмотрены методы молекулярной биологии, применяемые в экспериментальной гематологии, а также расширение знаний о кроветворении, получаемых с их помощью.
История изучения кроветворения началась в 1909 году, когда А.А. Максимов, наблюдая в световой микроскоп за клетками крови и костного мозга человека, расположил их в виде рядов морфологически распознаваемых предшественников той или иной линии дифференцировки (рис. 1). В каждом из таких рядов прослеживалось видоизменение (созревание) исходных морфологически слабо различимых клеток в терминально дифференцированные клетки крови [1, 2]. В настоящее время аналогичные дифференцировочные ряды составляют отдел морфологически узнаваемых унипотентных клеток-предшественниц.
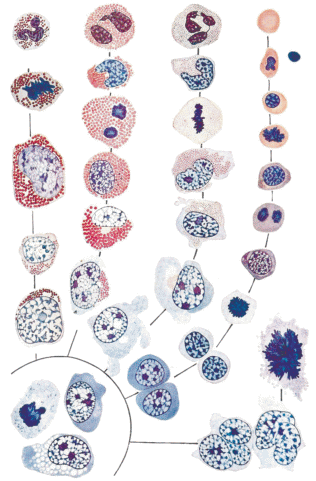
На основании сделанных наблюдений А.А. Максимов выдвинул гипотезу образования всех клеток крови из единой стволовой кроветворной клетки (СКК). Эта унитарная гипотеза подтвердилась почти через 50 лет после перехода от изучения морфологии костного мозга к экспериментам, связанным с физиологией. Толчком послужили ядерные взрывы в 1945 году, после которых стало ясно, что радиация убивает клетки крови. В 1950-х годах две группы исследователей обнаружили, что внутривенное введение клеток аллогенного костного мозга летально облученным мышам и морским свинкам приводило к значительному увеличению выживаемости реципиентов [4, 5]. Это показало, что в костном мозге содержатся клетки, способные восстанавливать кроветворение. Доказательство существования единой СКК получены Till J.E. и McCulloch E.A. [6], которые показали, что если облученным мышам трансплантировать клетки костного мозга от сингенных животных, то в селезенке образуются кроветворные колонии. Количество таких колоний линейно зависит от количества введенных клеток. По типу доминирующих клеток различают эритроидные, гранулоцитарные, мегакариоцитарные и недифференцированные колонии. Введение летально облученным мышам клеток костного мозга, несущих радиационные маркеры, показало, что все клетки одной колонии содержат один и тот же маркер, а клетки разных колоний отличаются своими маркерами. Таким образом было доказано, что каждая колония образована одной клеткой-предшественницей – колониеобразующей единицей селезеночной (КОЕс). Если ввести клетки одной колонии сингенному облученному реципиенту, то в его селезенке образуются колонии всех четырех типов, т.е. отдельные кроветворные клетки-предшественницы могут давать многолинейное потомство [7]. Долгое время считалось, что КОЕс – это настоящие СКК. Затем обнаружили, что эти полипотентные клетки могут только непродолжительное время заселять кроветворную систему и являются потомками истинных СКК, способных в течение длительного времени поддерживать кроветворение у мышей [8–12]. Во всех описанных работах исследуемую популяцию клеток трансплантировали сингенным облученным реципиентам.
Параллельно появились методы, позволившие визуализировать развитие клеток крови во времени [13], c помощью которых была подтверждена созданная Максимовым схема развития клеток крови. Эти методы включали получение химерных зародышей мышей [14], приживление клеток одного вида к другому – создание ксеногенных химер, таких как перепел-курица [15], и введение красителей в одну материнскую клетку мыши [16].
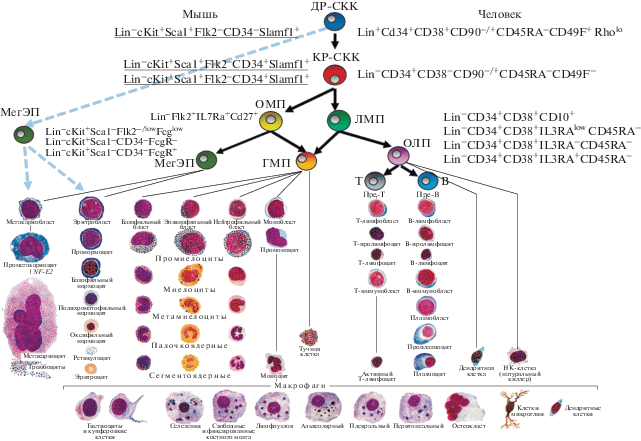
Следующий этап изучения кроветворения начался с появлением возможности идентификации молекул, представленных на поверхностной клеточной мембране, с помощью иммунофенотипирования. В 1980-х годах с помощью моноклональных антител [17] и клеточного сортера [18] впервые были очищены СКК мыши. Оказалось, что они способны производить все типы клеток крови после трансплантации [19]. Иммунофенотипический анализ в сочетании с физиологическим тестом – трансплантацией выделенных клеток, долгое время считался золотым стандартом определения дифференцировочного потенциала клеток. Такой подход лежал в основе изучения СКК мыши [20]. Он позволил уточнить классическую иерархию кроветворных предшественников и определить их поверхностные маркеры [21–23]. В 2000-х годах иерархия кроветворения была пересмотрена в связи с идентификацией новых типов клеток [24–26] (рис. 2). Что касается стволовых клеток человека, то указанные на рис. 2 иммунофенотипы тестировали или в системе in vitro, или на ксеногенной системе, когда клетки человека трансплантировали мышам с иммунодефицитом [27, 28]. Таким образом, данные о клетках человека получены не прямым, а косвенным образом.
Рис. 2.
Иерархическая модель устройства отдела стволовых кроветворных клеток. Самообновляющиеся СКК находятся на вершине иерархии, далее появляется ряд мультипотентных предшественников. Мультипотентные предшественники дают начало олигопотентным предшественникам, включая общий лимфоидный предшественник (ОЛП), образующий зрелые B-лимфоциты, T-лимфоциты и естественные киллерные клетки. Общий миелоидный предшественник (ОMП) дает начало гранулоцитарно-макрофагальным предшественникам, которые дифференцируются в моноциты/макрофаги и гранулоциты, и предшественникам мегакариоцитов/эритроцитов, которые дифференцируются в мегакариоциты/тромбоциты и эритроциты. Предполагалось, что и ОМП, и ОЛП дают начало дендритным клеткам. Фенотип клеточной поверхности многих из этих типов клеток определен у мыши и человека. Некоторые маркеры, такие как Thy1.1 и Sca1, информативны не во всех линиях мышей. Развитие от олигопотентных предшественников до зрелых клеток крови происходит через ряд промежуточных морфологически распознаваемых предшественников. Недавно показано, что линии мегакариоцитов и эритроцитов могут происходить из мультипотентного предшественника и не проходить через промежуточный миелоидный предшественник (выделено пунктирными линиями) [29].
Методами иммунофенотипирования и сортировки клеток в сочетании с трансплантацией выявлена и подтверждена гетерогенность популяции СКК у стареющих мышей. Показано, что соотношение гранулоцитов и лимфоцитов в периферической крови с возрастом изменяется [30]. Такое изменение связано с тем, что судьба СКК предопределена, и они образуют потомство с преобладанием лимфоидного или миелоидного дифференцировочного потенциала [31]. Продолжительность жизни СКК, предрасположенных к лимфоидной дифференцировке, оказалась меньше, чем у СКК, предрасположенных к миелоидной дифференцировке. Более тонкое исследование позволило выявить СКК со сбалансированной дифференцировкой [32].
Однородность популяции отсортированных по поверхностному иммунофенотипу СКК всегда вызывала некоторые сомнения. В описанных подходах отбирали клетки, на поверхности которых не было молекул, ассоциированных с линейной дифференцировкой (Lin–), но присутствовали пять–шесть поверхностных маркеров, ассоциированных со “стволовостью”. Что происходит при этом в самих клетках не ясно. Например, у мышей клетки такой популяции могут находиться в разных фазах клеточного цикла [33], тогда популяция теряет однородность [34], поскольку свойства СКК зависят от ее пролиферативной предыстории и фазы клеточного цикла, в которой клетка находится в данный момент.
Следующим методом, продвинувшим представление о кроветворении далеко вперед, стало маркирование клеток. Первыми маркерами были вызванные облучением хромосомные перестройки, которые использовали в работах на мышах [35, 36]. Однако они позволяли проследить судьбу лишь небольшого числа клеток, поскольку после облучения не все клетки приобретали устойчивый радиационный маркер.
Более подробные сведения получены при генетическом маркировании СКК с помощью переноса чужеродного гена. Этот подход позволил доказать, что одна стволовая клетка способна дифференцироваться во все клеточные линии кроветворной ткани [37, 38]. Используя перенос маркерных генов с помощью ретровирусных векторов, удалось проследить судьбу маркированных клонов СКК из костного мозга и эмбриональной печени в течение всей жизни мыши [39, 40]. Установлено, что кроветворение у мышей поликлонально и поддерживается в основном большим количеством короткоживущих клонов, сменяющих друг друга [39, 41]. Более того, оказалось, что кроветворение поликлонально не только на уровне ранних полипотентных предшественников – КОЕс, но и зрелых терминально дифференцированных плазматических клеток [42]. Однако этот метод имеет свои недостатки. Маркирование СКК с помощью ретровирусов, способных встраиваться только в делящиеся клетки, не позволяло выявить клоны СКК, не делившиеся в момент маркирования. К тому же методам выявления маркерных клонов не хватало чувствительности [43]. Кроме того, вставка ретровируса может в значительной степени изменить клональный вклад в кроветворение как у мышей, так и у человека ‒ от небольших изменений в участии маркированного клона до явного клонального доминирования и даже лейкоза [44–47]. Последний недостаток частично устраняется, если использовать векторы на основе лентивирусов, таких как ВИЧ [48]. Развитие методов маркирования СКК с помощью лентивирусов, способных заражать не только клетки, находящиеся в процессе митоза, но и неделящиеся клетки, позволило выявить гораздо больше функциональных клонов и еще раз убедительно показало возможность клональной сукцессии, т.е. постоянной смены активных кроветворных клонов [46].
Следующий прорыв в изучении кроветворения произошел при использовании не одного маркерного гена, а библиотеки маркерных последовательностей – “штрих-кодов”. Такая библиотека состоит из большого количества вирусных частиц, отличающихся друг от друга только короткой последовательностью (несколько десятков нуклеотидов) [49]. Олигонуклеотиды штрих-кодов получают с помощью химического синтеза и клонируют в одну из плазмид, используемых для трансфекции клеточной линии-продуцента вирусных частиц. Полученные вирусные супернатанты используют для маркирования кроветворных клеток. Подбор оптимального соотношения разнообразия штрих-кодов и количества заражаемых клеток позволяет добиться того, что каждая трансдуцированная клетка будет маркирована штрих-кодом, не встречающимся в остальных клетках. В идеальном варианте после интеграции провируса из такой библиотеки каждая клетка популяции приобретает уникальный, стабильный и наследуемый маркер, позволяющий проследить судьбу потомства маркированной клетки и отличать его от потомства других меченых стволовых клеток. После экспериментов с маркированной клеточной популяцией необходимо определить состав и относительную представленность штрих-кодов в тестируемых клеточных популяциях (клетки периферической крови или костного мозга) с помощью современных методов высокопроизводительного параллельного секвенирования (NGS) [50]. Сочетание методов генетического штрих-кодирования и NGS позволило определить наиболее полный клональный состав популяции кроветворных клеток и его динамику в кроветворной системе мыши и человека [48, 49, 51–54]. Еще раз подтверждено, что кроветворение поддерживается множеством сменяющих друг друга клонов кроветворных клеток. Применение столь чувствительного метода маркирования популяции СКК, тщательно выделенной с помощью флуоресцентного сортера, позволило показать, что большинство трансплантированных клонов непрерывно в течение длительного времени вносит вклад в кроветворение, хотя клональный состав гранулоцитов, Т- и В-клеток существенно различается. Вклад отдельных клонов в кроветворение постоянно изменяется [54]. В этой работе выявлено множество клонов (примерно 70 у одной мыши), функционирующих одновременно. Удалось доказать гетерогенность популяции СКК и обнаружить изменения в ней, связанные с возрастом. Набор клонов в периферической крови и костном мозге не всегда совпадал. Представительство клона менялось с течением времени, многие клоны наблюдались более 12 недель. Полученные результаты показывают интересную особенность: лимфоциты и гранулоциты часто имеют различные маркеры. Это указывает на то, что трансплантированные популяции СКК, при их кажущейся однородности, неравноценны и состоят из клеток, предрасположенных к лимфоидной или миелоидной дифференцировке.
Однако известно, что свойства СКК зависят от их локализации в различных местах кроветворной территории. В длинных костях мест для СКК меньше, чем в губчатой кости, и качество СКК в этих участках различно и у мышей, и у человека [55, 56]. Проведено маркирование достаточно однородной популяции СКК, основанное на поверхностном иммунофенотипе. Анализ функционирования этих клеток в облученном организме в течение длительного отрезка времени выявил отличия между индивидуальными клонами, что связывают с попаданием клеток в разные участки кроветворной ткани.
Маркирование генетическими штрих-кодами не только выявило неоднородность популяции СКК, но и показало, что СКК после их внутривенного введения мышам при ретрансплантации распределяются неравномерно по различным территориям костного мозга и после этого не мигрируют активно [57]. Однако введение в периферическую кровь одной мобилизирующей СКК дозы гранулоцитарного колониестимулирующего фактора приводит к перераспределению СКК. В результате исходная неравномерность распределения исчезает, так как СКК открепляются от стромальных клеток костного мозга и выходят в кровяное русло.
С помощью аналогичного подхода обнаружены долгоживущие клоны генетически маркированных in vivo Т-клеток человека [58], и на модели ксенотрансплантации СКК изучен процесс восстановления Т-клеточного звена кроветворения [59].
Сравнение клонального состава СКК у приматов и мышей выявило как их сходство, так и существенные отличия. Отслеживание тысяч маркированных СКК и клеток-предшественниц у макаков-резус в течение 12 лет показало, что примерно половина проанализированных клонов выявлялась несколько раз на протяжении длительного времени (более 3–10 лет). Вероятно, клетки-предшественницы таких маркированных клонов представляют собой самообновляющиеся СКК [60]. Остальные клоны исчезали в основном в течение первого года после трансплантации. Обнаружена чрезвычайно низкая средняя частота (<0.0002) большого количества клонов (43–71%), составляющая <7% от общего представительства клонов в кроветворной системе в течение всего времени наблюдения. На 5% наиболее часто обнаруживаемых клонов приходится в среднем 49–72% от общего количества маркированных клонов, в зависимости от животного и типа клеток. У одного животного обнаруживали примерно 330 клонов. Выявлено приблизительно равное количество длительно и коротко функционирующих клонов. Клональный состав СКК у всех животных становился более стабильным только через 1–2 года после трансплантации.
Проведен анализ клонального состава СКК у больных с тяжелой формой комбинированного иммунодефицита, вызванного отсутствием функционального гена аденозиндезаминазы (ADA-SCID), и с синдромом Вискотта-Олдрича (WAS-синдром), которым СКК трансплантировали после проведения генотерапии. Введение нормального гена ADA создало уникальные условия, которые позволяли идентифицировать каждый маркированный вектором предшественник и его потомство в крови по сайтам интеграции вектора. Проведен in vivo анализ клонального состава кроветворных клеток и СКК в динамике. Определены временные рамки краткосрочного, среднего и длительного функционирования СКК. Показано, что множество клонов CD34+ клеток, активных через 3–6 месяцев после трансплантации, не обнаруживались на более поздних сроках [51, 58]. Повторный анализ трансдуцированных ADA СКК у пациентов, находящихся в стабильном состоянии, выявил примерно по 200 клонов у каждого из них. Эти клоны стабильно вносили вклад в репертуар предшественников в течение 3 лет после трансплантации CD34+ клеток с исправленными генами. Длительность сохранения активности трансплантированных СКК оценивали, используя результаты определения сайтов интеграции в 4845 клонах пациентов с ADA-SCID, выполненных в течение 6 лет после проведения генной терапии. Идентичные сайты интеграции постоянно обнаруживались в нескольких линиях кроветворных клеток через несколько лет после трансплантации. Поразительно, что с помощью полуколичественной ПЦР можно детектировать изменения относительной представленности клонов СКК с исправленным геном в общей популяции кроветворных клеток. Обнаружен последовательный выход клонов СКК из состояния покоя в фазу активного деления и дифференцировки в течение 5 лет без возвращения в фазу покоя. Поскольку гаммаретровирусный вектор, использованный для введения исправленного гена ADA, способен встраиваться в геном только активно делящихся клеток, было не ясно сколь долго смогут функционировать трансдуцированные СКК. В этой работе получено первое доказательство того, что активированные in vitro СКК, которые не находились в фазе покоя в момент трансдукции, могут долго сохраняться в организме после инфузии, обеспечивая в течение длительного времени “исправленное” кроветворение [58]. Таким образом, у людей и приматов обнаружены сотни клонов СКК, потомство которых представлено в разных кроветворных линиях дифференцировки. Многие клоны и клональная нестабильность наблюдались в течение первого года после трансплантации, а затем система стабилизировалась.
Показано, что только 10% клеток из популяции СКК мышей способны вернуться в состояние покоя после деления и могут функционировать на протяжении всей жизни животного [24]. Совокупность полученных данных указывает на возможность функционирования СКК в течение длительного срока и на их способность вернуться в состояние покоя.
Клональный состав СКК изменяется с возрастом, и поликлональное кроветворение постепенно становится олигоклональным. С помощью глубокого полноэкзомного секвенирования ДНК клеток периферической крови 12 380 [61] и 17 182 лиц [62] идентифицированы редкие соматические мутации, позволившие выявить отдельные клоны. В первом исследовании доминирование отдельного клона с соматическими мутациями обнаружено у 10% лиц старше 65 лет и у 9.5% лиц старше 70 лет во втором. Эти данные доказывают, что с возрастом поликлональное кроветворение заменяется олигоклональным. В результате глубокого полногеномного секвенирования в некодирующих AT-богатых регионах генома нормальных кроветворных клеток 115-летней женщины обнаружено приблизительно 450 соматических мутаций [63]. Распределение частот вариантов аллелей этих мутаций позволило предположить, что большинство периферических лейкоцитов были потомками двух родственных клонов СКК, и теломеры в них были значительно короче, чем в других клетках. Эти результаты говорят о том, что ограниченная продолжительность жизни СКК, а не эффекты соматических мутаций могут приводить к клональной эволюции в экстремальном возрасте. Вероятнее всего, количество СКК, заложенных в онтогенезе, ограничено и его может не хватить для поддержания кроветворения у долгожителей. Показано, что у лиц старше 90 лет наблюдается олигоклональное кроветворение [61–63].
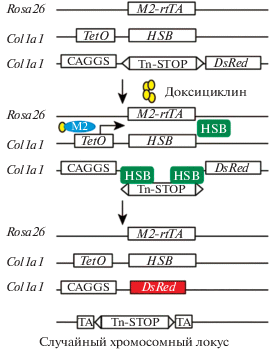
Большинство описанных методов исследования клонального состава опирается на выделение популяции СКК, манипулирование с ней ex vivo и трансплантацию СКК в организм реципиента. Такой методический подход может вносить существенные возмущения в картину кроветворения, наблюдаемую у реципиента. Во-первых, свойства СКК могут изменяться при их разобщении с костномозговой нишей и вследствие культивирования in vitro. Во-вторых, облученный или кондиционированный организм отличается от интактного: после облучения или применения цитостатиков могут изменяться уровни цитокинов, сигнальных молекул, активироваться воспаление и происходить другие изменения. В силу этого исследователи стремились придумать способы наблюдения за кроветворением в условиях, приближенных к гомеостатическим. Разработан новый подход к индивидуальному маркированию клеток с использованием системы транспозонов, клонированных в мышах [64]. Суть метода заключается в том, что в результате активации транспозазы “Sleeping Beauty” тетрациклином транспозон перемещается в случайное место генома (рис. 3). Эта транспозиция уникальна для каждой клетки и наследуется ее потомками. Клетки, происходящие от общего предшественника, могут быть идентифицированы по одинаковой локализации транспозона. Такая система слежения дала возможность изучать кроветворение без использования трансплантации [65]. Анализ локализации транспозонов позволил получить результаты, поразительно отличающиеся от результатов предыдущих исследований. Оказалось, что в каждой временной точке исследования кроветворение поддерживают совершенно разные клоны. Согласно [65], у мыши одновременно функционируют тысячи клонов. Таким образом, результаты данной работы свидетельствуют о том, что длительное кроветворение поддерживается последовательной сменой большого количества клонов.
Рис. 3.
Создание метода индуцируемого мечения транспозонов. Трансгенные аллели и стратегия, используемая для индуцируемого генетического мечения. M2-rtTA ‒ тетрациклин-чувствительный активатор транскрипции; HSB ‒ гиперактивная транспозаза “Sleeping Beauty”; Tn ‒ HSB-транспозон; STOP ‒ сигнал полиаденилирования; CAGGS ‒ промотор гена β-актина курицы; TetO ‒ тетрациклин-зависимый элемент. Рисунок адаптирован из работы [64]. После воздействия доксициклина начинает экспрессироваться белок М2, который связывается с тетрациклин-чувствительным транскрипционным активатором транспозазы “Sleeping Beauty”. Экспрессия этого фермента приводит к перемещению транспозона в случайное место генома и одновременно к активации экспрессии красного флуоресцентного белка, что позволяет визуализировать клетки, в которых транспозон переместился в случайный хромосомный локус.
Хотя наблюдаемая клональная динамика согласуется с моделью клональной сукцессии, предполагаемое клональное разнообразие намного выше, чем показано методами трансплантации. В дальнейшем костный мозг мышей с перемещенными транспозонами ретрансплантировали облученным реципиентам. При сравнении клонального состава кроветворных клеток доноров и реципиентов не обнаружили ни одного совпадающего клона. Далее сравнили клональный состав СКК, промежуточных предшественников и зрелых клеток у каждой мыши. Оказалось, что менее 5% клонов СКК представлены в популяциях зрелых клеток, тогда как основные участники продукции зрелых клеток – промежуточные мультипотентные (MPP) и миелоидные (MyP) предшественники. По результатам этих двух экспериментов был сделан вывод, что клетки, которые обеспечивают стабильное кроветворение в нормальных условиях, не способны к трансплантации и поэтому не обнаруживаются в общем пуле СКК. Полученные результаты указывают на то, что большое количество клеток-предшественниц, включая MPP и MyP, могут быть основным источником поддержания нормального кроветворения в обычных условиях. Однако эти результаты не исключают возможности участия СКК в стабильном кроветворении.
Вслед за этим были созданы мыши Tie2MCM/+RosaYFP с индуцируемым генетическим маркированием наиболее примитивных Tie2+ СКК в костном мозге. Под действием тамоксифена в клетках Tie2MCM/+ активировалась экспрессия Cre-рекомбиназы, которая удаляла стоп-сигнал транскрипции гена желтого флуоресцентного белка (YFP). Через 20 дней YFP выявлялся только в примитивных СКК. Проведена количественная оценка распределения маркера с помощью метода лимитирующих разведений при восстановлении кроветворения у мышей с иммунодефицитом. Выявлено более 5000 клонов СКК, одномоментно участвующих в кроветворении [66]. Стало понятно, что существуют принципиальные различия между поддержанием нормальной кроветворной системы и восстановлением кроветворения после трансплантации СКК.
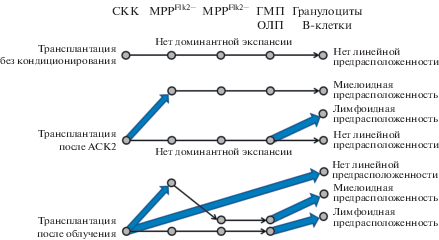
Изучение активности СКК при нативном (не трансплантационном) кроветворении улучшилось благодаря усовершенствованной технологии, включая разработку инструментов клеточного штрих-кодирования и отслеживания клонов в мышах [67, 68]. В соответствии с тем, что трансплантируемые СКК находятся в костном мозге, как правило, в состоянии покоя, обнаружено, что при стабильном кроветворении СКК минимально участвуют в образовании дифференцированных клеток крови. В основном, эту функцию выполняют популяции фенотипически различающихся клеток-предшественниц (особенно MPP). Использование молекулярной системы штрих-кодирования показало, что источником мегакариоцитов при нативном кроветворении могут быть непосредственно СКК, которые минуют стадию ОМП [67, 68]. Более того, в случае дифференцировки индивидуальных СКК в условиях гомеостаза все клоны СКК равномерно дифференцируются во все линии кроветворных клеток. Напротив, после возмущения кроветворной системы облучением или антагонистическими антителами против сKit дифференцируется только небольшая часть донорских клонов СКК [69] (рис. 4).
Рис. 4.
Модель путей дифференцировки СКК in vivo на клональном уровне. Более жирные (синие) восходящие стрелки показывают доминантную дифференцировку клонов СКК. После трансплантации СКК мышам, предварительно кондиционированным в различных режимах, преобладают различные линии дифференцировки. ГМП – гранулоцитарно-макрофагальный предшественник Lin−(CD3, CD4, CD8, B220, Gr1, Mac1, Ter119)−/IL7Rα−/ckit+/Sca1−/CD34+/FcγR+), MPPFlk2− – Flk2− мультипотентный предшественник (CD3, CD4, CD8, B220, Gr1, Mac1, Ter119)−/IL7Rα−/ckit+/ Sca1+/Flk2−/CD34+), MPPFlk2− – Flk2+ мультипотентный предшественник (CD3, CD4, CD8, B220, Gr1, Mac1, Ter119)−/IL7Rα−/ckit+/Sca1+/Flk2+) ОЛП – общий лимфоидный предшественник (CD3, CD4, CD8, B220, Gr1, Mac1, Ter119)−/IL7Rα+/Flk2+). Рисунок адаптирован из статьи [69] с модификациями.
Новые данные меняют наше представление об иерархическом устройстве кроветворной системы (рис. 2).
Изменение наших представлений об устройстве кроветворной системы также связано с достижениями в технологии изучения транскриптома индивидуальных клеток [70]. Этот метод позволяет изучить транскриптомы тысяч отдельных клеток, идентифицировать тип клеток и проанализировать их дифференцировку [71–75]. Такой подход решает обратную задачу. Если раньше исследователи выделяли популяцию клеток по внешним признакам (морфология/поверхностный иммунофенотип/локализация в организме) и изучали ее свойства, в том числе экспрессию генов, то теперь стало возможно, проанализировав транскриптомы большого количества несортированных клеток, определить их линию и степень дифференцировки.
Новые экспериментальные технологии основаны на сочетании секвенирования транскриптома большого числа индивидуальных клеток и их генетического маркирования. Этот подход позволяет проследить за введенным или предварительно созданным внутренним маркером в клеточных поколениях и одновременно получить информацию о прохождении клетками этапов созревания и их транскрипционном профиле [76–80] (рис. 5). Большинство методов анализа транскриптома опирается на предположение, что клетки со схожим профилем экспрессии генов принадлежат одной линии дифференцировки, и такие клетки могут быть близкородственными. Анализ транскриптома индивидуальных клеток дает возможность описывать разнообразие клеточных типов в гетерогенных клеточных популяциях, что очень важно для кроветворной системы. Первая работа, в которой представлены результаты секвенирования транскриптома одной клетки, опубликованы в 2009 году [81]. Сегодня тысячи и десятки тысяч клеток можно анализировать подобным образом с помощью микрофлюидики или робот-ассоциированных технологий [74, 75]. Сложность применения анализа транскриптома индивидуальных клеток состоит в том, что этому методу присущ высокий уровень шума и выпадения данных (гены с низким уровнем экспрессии сложно детектировать из-за технических ограничений).
Рис. 5.
Комбинация генетического маркирования индивидуальных клеток с отслеживанием маркера в клеточном потомстве и анализа транскриптома индивидуальных клеток.
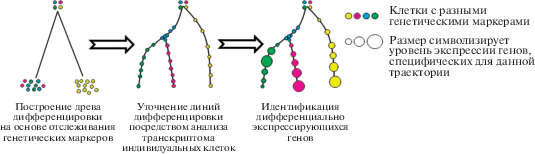
При изучении клеточной дифференцировки с использованием сочетания методов индивидуального клеточного маркирования и анализа транскриптомов отдельных клеток применяют, как правило, следующую последовательность действий (рис. 5). На основе иммунофенотипических и генетических маркеров строится филогенетическое дерево отдельных клеток. Это дерево может быть уточнено с помощью алгоритмов реконструкции пути дифференцировки на основе транскриптомного анализа. Затем уровни экспрессии генов могут быть наложены на филогенетическое дерево, чтобы идентифицировать динамику экспрессии в системе.
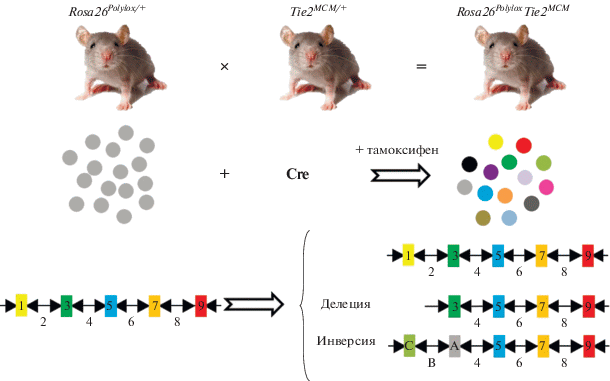
В последние годы линии дифференцировки анализируют в основном с помощью генетических маркеров, вводимых экспериментально или присущих системе. Такое исследование называют проспективным, или ретроспективным отслеживанием соответственно. Проспективное отслеживание основано на введении репортерных трансгенов, таких как гены β-галактозидазы или зеленого флуоресцентного белка (GFP) [82, 83]. В этом случае все потомки маркированной клетки наследуют трансген. Поскольку трансген кодирует белок, легко обнаруживаемый с помощью микроскопии, можно увидеть клон и проследить за потомками клетки-родоначальницы. В настоящее время вирусные стратегии расширены и включают специфичные для каждой клетки штрих-коды ДНК [48]. Создание системы Cre-LoxP позволяет анализировать дифференцировку в любой момент времени и в широком диапазоне тканей (ввиду того, что существует много линий трансгенных животных, в которых Cre-рекомбиназа контролируется различными тканеспецифическими промоторами). Активация этой системы снабжает материнскую клетку одним или несколькими маркерами. Недавно опубликовали методику, в которой используемые ранее флуоресцентные белки заменены ДНК-штрих-кодами, что совместило гибкость индукции системы Cre-Lox с резким увеличением максимального количества клонов, которые можно отслеживать одновременно [84]. Создана мышиная модель, содержащая массив штрих-кодов, перемежающихся сайтами LoxP (кассета Polylox) [84]. Скрещивание таких животных с мышами, несущими ген Cre-рекомбиназы, приводит к делеции или инверсии некоторых штрих-кодов, образуя специфические для клетки генетические метки (рис. 6)
Рис. 6.
Индуцируемая Cre-loxP мышиная модель маркирования СКК. Генетические штрих-коды перемежаются сайтами loxP (черные треугольники). При активации Cre-рекомбиназы происходят инверсии и делеции участков, ограниченных одинаково направленными и ориентированными навстречу друг другу последовательностями loxP соответственно. Это приводит к формированию уникального маркера в каждой клетке.
Теоретическое разнообразие штрих-кодов, достигаемое с помощью этой рекомбинации, превышает 106. Поскольку животные этой линии легко скрещиваются со всеми видами тканеспецифичных и индуцируемых моделей Cre-мышей, исследователи могут маркировать тысячи уникальных клонов в разных тканях и в разные временные точки.
Другой недавно разработанный метод проспективного анализа дифференцировки использует CRISPR-Cas9-направленное редактирование генома. Действие нуклеазы Cas9, затрагивающее конкретный локус, часто влечет введение небольших вставок или делеций (indels) [85, 86]. Этот процесс называют “нанесение шрамов”. Все потомки маркированной таким образом материнской клетки наследуют эти мутации, которые служат маркерными элементами.
Все перечисленные методы генетического отслеживания происхождения/дифференцировки предоставили бесценную новую информацию о развитии кроветворной ткани в норме и патологии. Исследования как транскриптомов индивидуальных клеток [73, 87], так и анализ путей дифференцировки отдельных клеток [64, 67] показали, что классическое дерево иерархии, где стволовые клетки имеют неограниченный пролиферативный и дифференцировочный потенциал, а каждая из множества популяций клеток-предшественниц имеет предопределенную судьбу, может быть неточным. Считалось, что клетка не может изменить направление своей дифференцировки, и только при патологии может возникать “линейная неверность”, когда на поверхности опухолевых клеток экспрессируется набор антигенов, не свойственный нормальным кроветворным клеткам той или иной линии дифференцировки. Новые данные делают все более очевидной возможность разнолинейной дифференцировки не только стволовых клеток, но и более поздних клеток-предшественниц с одинаковым фенотипом. Наше понимание дифференцировки как предопределенных последовательных шагов (что мы и видим при изучении морфологических изменений в кроветворных предшественниках, рис. 1) преобразуется в непрерывный процесс изменения эпигенетического состояния транскриптома и протеома клетки, в котором возможны модификации сценария дифференцировки. В ходе этого процесса потенциал СКК и клеток-предшественниц может регулироваться внешними факторами и потребностями организма [88].
Анализ транскриптома отдельных клеток [73, 87] и изучение путей дифференцировки индивидуальных клеток [64, 67] дает веские основания полагать, что дифференцировка кроветворных клеток при гомеостазе отличается от дифференцировки при трансплантации [69, 89].
При использовании проспективных методов маркирования и слежения за потомством клеток остается вероятность влияния введенного клонального маркера на основные свойства СКК. Разработаны методы ретроспективного слежения за потомством отдельных клеток, основанные на определении и отслеживании спонтанных мутаций, однонуклеотидных полиморфизмов или микросателлитных последовательностей в ДНК отдельных клеток [70]. Эти подходы ограничены необходимостью массивного секвенирования ДНК огромного количества отдельных клеток. Однако можно ожидать, что с совершенствованием технологии секвенирования эти подходы станут более доступными, а их применение станет более эффективным.
Внедрение методов молекулярной биологии позволило значительно продвинуться в понимании процесса кроветворения и функционирования СКК. Эти знания позволили улучшить диагностику и лечение лейкозов, лимфом, наследственных патологий и других тяжелых заболеваний. За разработку метода трансплантации костного мозга Edward Donnall Thomas в 1990 году получил Нобелевскую премию по физиологии и медицине. В настоящее время трансплантация костного мозга стала единственным методом излечения острых лейкозов. По статистике ВОЗ ежегодно выполняется более 50 000 трансплантаций. Генотерапия, основанная на методах редактирования генома и трансплантации СКК, успешно применяется при ранее некурабельных заболеваниях, связанных с кроветворением [90, 91]. Накопление фундаментальных знаний, получаемых с помощью современных методов молекулярной биологии, позволяет добиться улучшения результатов лечения широкого спектра гематологических заболеваний [58, 92], а ряд ранее неизлечимых заболеваний поддается терапии.
Написание настоящего обзора не потребовало специального финансирования.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Maximow A. (1909) Der Lymphozyt als gemeinsame Stammzelle der verschiedenen Blutelemente in der embryonalen Entwicklung und im postfetalen Leben der Säugetiere. Folia Haematol. 125–134.
Maximow A.A. (1927) Development of non-granular leucocytes (lymphocytes and monocytes) into polyblasts (macrophages) and fibroblasts in vitro. Proc. Soc. Exp. Biol. Med. 24, 570–572.
Maximow A.A., Bloom W. (1952) A Textbook of Histology. 6th edition. Philadelphia and London: W.B. Saunders Comp.
Lorenz E., Uphoff D., Reid T.R., Shelton E. (1951) Modification of irradiation injury in mice and guinea pigs by bone marrow injections. J. Natl. Cancer. Inst. 12, 197–201.
Jacobson L.O., Simmons E.L., Marks E.K., Eldredge J.H. (1951) Recovery from radiation injury. Science. 113, 510–511.
Till J.E., McCulloch E.A. (1961) A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res. 14, 213–222.
Becker A.J., McCulloch E.A., Till J.E. (1963) Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature. 197, 452–454.
Abramson S., Miller R.G., Phillips R.A. (1977) The identification in adult bone marrow of pluripotent and restricted stem cells of the myeloid and lymphoid systems. J. Exp. Med. 145, 1567–1579.
Lemischka I.R., Raulet D.H., Mulligan R.C. (1986) Developmental potential and dynamic behavior of hematopoietic stem cells. Cell. 45, 917–927.
Morrison S.J., Uchida N., Weissman I.L. (1995) The biology of hematopoietic stem cells. Annu. Rev. Cell Dev. Biol. 11, 35–71.
Weissman I.L. (2000) Stem cells: Units of development, units of regeneration, and units in evolution. Cell. 100, 157–168.
Morrison S.J., Weissman I.L. (1994) The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1, 661–673.
Deppe U., Schierenberg E., Cole T., Krieg C., Sch-mitt D., Yoder B., von Ehrenstein G. (1978) Cell lineages of the embryo of the nematode Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 75, 376–380.
Mintz B. (1967) Gene control of mammalian pigmentary differentiation. I. Clonal origin of melanocytes. Proc. Natl. Acad. Sci. USA. 58, 344–351.
Le Douarin N.M., Teillet M.A. (1973) The migration of neural crest cells to the wall of the digestive tract in avian embryo. J. Embryol. Exp. Morphol. 30, 31–48.
Lawson K.A., Meneses J.J., Pedersen R.A. (1986) Cell fate and cell lineage in the endoderm of the presomite mouse embryo, studied with an intracellular tracer. Dev. Biol. 115, 325–339.
Köhler G., Milstein C. (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 256, 495–497.
Spangrude G.J., Heimfeld S., Weissman I.L. (1988) Purification and characterization of mouse hematopoietic stem cells. Science. 241, 58–62.
Muller-Sieburg C.E., Whitlock C.A., Weissman I.L. (1986) Isolation of two early B lymphocyte progenitors from mouse marrow: A committed pre-pre-B cell and a clonogenic Thy-1-lo hematopoietic stem cell. Cell. 44, 653–662.
Osawa M., Hanada K., Hamada H., Nakauchi H. (1996) Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 273, 242–245.
Akashi K., Traver D., Miyamoto T., Weissman I.L. (2000) A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 404, 193–197.
Spangrude G.J. (1994) Biological and clinical aspects of hematopoietic stem cells. Annu. Rev. Med. 45, 93–104.
Kondo M., Weissman I.L., Akashi K. (1997) Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 91, 661–672.
Wilson A., Laurenti E., Oser G., van der Wath R.C., Blanco-Bose W., Jaworski M., Offner S., Dunant C.F., Eshkind L., Bockamp E., Lió P., Macdonald H.R., Trumpp A. (2008) Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 135, 1118–1129.
Adolfsson J., Månsson R., Buza-Vidas N., Hultquist A., Liuba K., Jensen C.T., Bryder D., Yang L., Borge O.J., Thoren L.A., Anderson K., Sitnicka E., Sasaki Y., Sigvardsson M., Jacobsen S.E. (2005) Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 121, 295–306.
Воробьев А.А., Дризе Н.И., Чертков И.Л. (1995) Схема кроветворения. Проблемы гематологии. 1, 7–14.
Doulatov S., Notta F., Laurenti E., Dick J.E. (2012) Hematopoiesis: A human perspective. Cell Stem Cell. 10, 120–136.
Cheung A.M., Leung D., Rostamirad S., Dhillon K., Miller P.H., Droumeva R., Brinkman R.R., Hogge D., Roy D.C., Eaves C.J. (2012) Distinct but phenotypically heterogeneous human cell populations produce rapid recovery of platelets and neutrophils post-transplant. Blood. 3431–3439.
Yamamoto R., Wilkinson A.C., Nakauchi H. (2018) Changing concepts in hematopoietic stem cells. Science. 362, 895–896.
Cho R.H., Sieburg H.B., Muller-Sieburg C.E. (2008) A new mechanism for the aging of hematopoietic stem cells: Aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood. 111, 5553–5561.
Muller-Sieburg C., Sieburg H.B., Bernitz J.M., Cattarossi G. (2012) Stem cell heterogeneity: Implications for aging and regenerative medicine. Blood. 119, 3900–3907.
Ema H., Morita Y., Suda T. (2014) Heterogeneity and hierarchy of hematopoietic stem cells. Exp. Hematol. 42, 74–82.e2.
Quesenberry P.J., Goldberg L.R., Dooner M.S. (2015) Concise reviews: A stem cell apostasy: a tale of four H words. Stem Cells. 33, 15–20.
Quesenberry P.J., Goldberg L., Aliotta J., Dooner M. (2014) Marrow hematopoietic stem cells revisited: They exist in a continuum and are not defined by standard purification approaches; then there are the microvesicles. Front Oncol. 4. 56.
Siminovitch L., Mcculloch E. A., Till J.E. (1963) The distribution of colony-forming cells among spleen colonies. J. Cell. Physiol. 62, 327–336.
Wu A.M., Till J.E., Siminovitch L., McCulloch E.A. (1968) Cytological evidence for a relationship between normal hemotopoietic colony-forming cells and cells of the lymphoid system. J. Exp. Med. 127, 455–464.
Magli M.C., Dick J.E., Huszar D., Bernstein A., Phillips R.A. (1987) Modulation of gene expression in multiple hematopoietic cell lineages following retroviral vector gene transfer. Proc. Natl. Acad. Sci. USA. 84, 789–793.
Dick J.E., Magli M.C., Huszar D., Phillips R.A., Bernstein A. (1985) Introduction of a selectable gene into primitive stem cells capable of long-term reconstitution of the hemopoietic system of W/Wv mice. Cell. 42, 71–79.
Drize N.J., Keller J.R., Chertkov J.L. (1996) Local clonal analysis of the hematopoietic system shows that multiple small short-living clones maintain life-long hematopoiesis in reconstituted mice. Blood. 88, 2927–2938.
Drize N.I., Chertkov I.L. (2000) Clone-forming activity of embryonal stem hemopoietic cells after transplantation to newborn or adult sublethally irradiated mice. Bull. Exp. Biol Med. 130, 709–711.
Drize N., Chertkov J., Sadovnikova E., Tiessen S., Zander A. (1997) Long-term maintenance of hematopoiesis in irradiated mice by retrovirally transduced peripheral blood stem cells. Blood. 89, 1811–1817.
Olovnikova N.I., Drize N.J., Ershler M.A., Nifontova I.N., Belkina E. V, Nikolaeva T.N., Proskurina N.V., Chertkov J.L. (2003) Developmental fate of hematopoietic stem cells: The study of individual hematopoietic clones at the level of antigen-responsive B lymphocytes. Hematol. J. 4, 146–150.
Bystrykh L.V., Verovskaya E., Zwart E., Broekhuis M., de Haan G. (2012) Counting stem cells: Methodological constraints. Nat. Methods. 9, 567–574.
Deichmann A., Hacein-Bey-Abina S., Schmidt M., Garrigue A., Brugman M.H., Hu J., Glimm H., Gyapay G., Prum B., Fraser C.C., Fischer N., Schwarzwaelder K., Siegler M.L., de Ridder D., Pike-Overzet K., Howe S.J., Thrasher A.J., Wagemaker G., Abel U., Staal F.J., Delabesse E., Villeval J.L., Aronow B., Hue C., Prinz C., Wissler M., Klanke C., Weissenbach J., Alexander I., Fischer A., von Kalle C., Cavazzana-Calvo M. (2007) Vector integration is nonrandom and clustered and influences the fate of lymphopoiesis in SCID-X1 gene therapy. J. Clin. Invest. 117, 2225–2232.
Bushman F.D., Torrey N., Rd P., Jolla L. (2003) Targeting survival : Minireview by retroviruses and LTR-retrotransposons. 115, 135–138.
Kustikova O., Fehse B., Modlich U., Yang M., Düllmann J., Kamino K., von Neuhoff N., Schlegelberger B., Li Z., Baum C. (2005) Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science. 308, 1171–1174.
Stein S., Ott M.G., Schultze-Strasser S., Jauch A., Burwinkel B., Kinner A., Schmidt M., Krämer A., Schwäble J., Glimm H., Koehl U., Preiss C., Ball C., Martin H., Göhring G., Schwarzwaelder K., Hof-mann W.K., Karakaya K., Tchatchou S., Yang R., Reinecke P., Kühlcke K., Schlegelberger B., Thra-sher A.J., Hoelzer D., Seger R., von Kalle C., Grez M. (2010) Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 16, 198–204.
Glimm H., Ball C.R., von Kalle C. (2011) You can count on this: Barcoded hematopoietic stem cells. Cell Stem Cell. 9, 390–392.
Gerrits A., Dykstra B., Kalmykowa O.J., Klauke K., Verovskaya E., Broekhuis M.J.C., de Haan G., Bystrykh L.V. (2010) Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood. 115, 2610–2618.
Ребриков Д.В., Коростин Д.О., Шубина Е.С., Ильинский В.В. (2015) NGS: высокопроизводительное секвенирование. Москва: Бином. Лаборатория знаний.
Biasco L., Pellin D., Scala S., Dionisio F., Basso-Ricci L., Leonardelli L., Scaramuzza S., Baricordi C., Ferrua F., Cicalese M.P., Giannelli S., Neduva V., Dow D.J., Schmidt M., Von Kalle C., Roncarolo M.G., et al. (2016) In vivo tracking of human hematopoiesis reveals patterns of clonal dynamics during early and steady-state reconstitution phases. Cell Stem Cell. 19, 107–119.
Lu R., Neff N.F., Quake S.R., Weissman I.L. (2011) Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat. Biotechnol. 29, 928–933.
Maetzig T., Brugman M.H., Bartels S., Heinz N., Kustikova O.S., Modlich U., Li Z., Galla M., Schie-dlmeier B., Schambach A., Baum C. (2011) Polyclonal fluctuation of lentiviral vector-transduced and expanded murine hematopoietic stem cells. Blood. 117, 3053–3064.
Verovskaya E., Broekhuis M.J.C., Zwart E., Ritsema M., van Os R., de Haan G., Bystrykh L.V. (2013) Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood. 122, 523–532.
Xie Y., Yin T., Wiegraebe W., He X.C., Miller D., Stark D., Perko K., Alexander R., Schwartz J., Grindley J.C., Park J., Haug J.S., Wunderlich J.P., Li H., Zhang S., Johnson T., Feldman R.A., Li L. (2009) Detection of functional haematopoietic stem cell niche using real-time imaging. Nature. 457, 97–101.
Guezguez B., Campbell C.J. V, Boyd A.L., Karanu F., Casado F.L., Di Cresce C., Collins T.J., Shapovalova Z., Xenocostas A., Bhatia M. (2013) Regional localization within the bone marrow influences the functional capacity of human HSCs. Cell Stem Cell. 13, 175–189.
Bystrykh L.V., de Haan G., Verovskaya E. (2014) Barcoded vector libraries and retroviral or lentiviral barcoding of hematopoietic stem cells. Methods Mol. Biol. 1185, 345–360.
Biasco L., Scala S., Basso Ricci L., Dionisio F., Baricordi C., Calabria A., Giannelli S., Cieri N., Barzaghi F., Pajno R., Al-Mousa H., Scarselli A., Cancrini C., Bordignon C., Roncarolo M.G., Montini E., Bonini C., Aiuti A. (2015) In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci. Transl. Med. 7, 273ra13–273ra13.
Brugman M.H., Wiekmeijer A.-S., van Eggermond M., Wolvers-Tettero I., Langerak A.W., de Haas E.F.E., Bystrykh L.V., van Rood J.J., de Haan G., Fibbe W.E., Staal F.J. (2015) Development of a diverse human T-cell repertoire despite stringent restriction of hematopoietic clonality in the thymus. Proc. Natl. Acad. Sci. USA. 112, E6020–6027.
Kim S., Kim N., Presson A.P., Metzger M.E., Bonifacino A.C., Sehl M., Chow S.A., Crooks G.M., Dunbar C.E., An D.S., Donahue R.E., Chen I.S. (2014) Dynamics of HSPC repopulation in nonhuman primates revealed by a decade-long clonal-tracking study. Cell Stem Cell. 14, 473–485.
Genovese G., Kähler A.K., Handsaker R.E., Lindberg J., Rose S.A., Bakhoum S.F., Chambert K., Mick E., Neale B.M., Fromer M., Purcell S.M., Svantesson O., Landén M., Höglund M., Lehmann S., Gabriel S.B., Moran J.L., Lander E.S., Sullivan P.F., Sklar P., Grönberg H., Hultman C.M., McCarroll S.A. (2014) Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487.
Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P.V., Mar B.G., Lindsley R.C., Mermel C.H., Burtt N., Chavez A., Higgins J.M., Moltchanov V., Kuo F.C., Kluk M.J., Henderson B., Kinnunen L., Koistinen H.A., Ladenvall C., Getz G., Correa A., Banahan B.F., Gabriel S., Kathiresan S., String-ham H.M., McCarthy M.I., Boehnke M., Tuomilehto J., Haiman C., Groop L., Atzmon G., Wilson J.G., Neuberg D., Altshuler D., Ebert B.L. (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498.
Holstege H., Pfeiffer W., Sie D., Hulsman M., Nicholas T.J., Lee C.C., Ross T., Lin J., Miller M.A., Ylstra B., Meijers-Heijboer H., Brugman M.H., Staal F.J., Holstege G., Reinders M.J., Harkins T.T., Levy S., Sistermans E.A. (2014) Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res. 24, 733–742.
Sun J., Ramos A., Chapman B., Johnnidis J.B., Le L., Ho Y.-J., Klein A., Hofmann O., Camargo F.D. (2014) Clonal dynamics of native haematopoiesis. Nature. 514, 322–327.
Lu R. (2014) Sleeping beauty wakes up the clonal succession model for homeostatic hematopoiesis. Cell Stem Cell. 15, 677–678.
Busch K., Klapproth K., Barile M., Flossdorf M., Holland-Letz T., Schlenner S.M., Reth M., Höfer T., Rodewald H.R. (2015) Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 518, 542–546.
Rodriguez-Fraticelli A.E., Wolock S.L., Weinreb C.S., Panero R., Patel S.H., Jankovic M., Sun J., Calogero R.A., Klein A.M., Camargo F.D. (2018) Clonal analysis of lineage fate in native haematopoiesis. Nature. 553, 212–216.
Yamamoto R., Wilkinson A.C., Ooehara J., Lan X., Lai C.-Y., Nakauchi Y., Pritchard J.K., Nakauchi H. (2018) Large-scale clonal analysis resolves aging of the mouse hematopoietic stem cell compartment. Cell Stem Cell. 22, 600–607.e4.
Lu R., Czechowicz A., Seita J., Jiang D., Weissman I.L. (2019) Clonal-level lineage commitment pathways of hematopoietic stem cells in vivo. Proc. Natl. Acad. Sci. USA.116, 1447–1456.
Kester L., van Oudenaarden A. (2018) Single-cell transcriptomics meets lineage tracing. Cell Stem Cell. 23, 166–179.
Grün D., van Oudenaarden A. (2015) Design and analysis of single-cell sequencing experiments. Cell. 163, 799–810.
Haber A.L., Biton M., Rogel N., Herbst R.H., Shekhar K., Smillie C., Burgin G., Delorey T.M., Howitt M.R., Katz Y., Tirosh I., Beyaz S., Dionne D., Zhang M., Raychowdhury R., Garrett W.S., Rozenblatt-Rosen O., Shi H.N., Yilmaz O., Xavier R.J., Regev A. (2017) A single-cell survey of the small intestinal epithelium. Nature. 551, 333–339.
Jaitin D.A., Kenigsberg E., Keren-Shaul H., Elefant N., Paul F., Zaretsky I., Mildner A., Cohen N., Jung S., Tanay A., Amit I. (2014) Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 343, 776–779.
Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W. (2015) Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 161, 1187–1201.
Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., Trombetta J.J., Weitz D.A., Sanes J.R., Shalek A.K., Regev A., McCarroll S.A. (2015) Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161, 1202–1214.
Alemany A., Florescu M., Baron C.S., Peterson-Maduro J., van Oudenaarden A. (2018) Whole-organism clone tracing using single-cell sequencing. Nature. 556, 108–112.
Frieda K.L., Linton J.M., Hormoz S., Choi J., Chow K.-H.K., Singer Z.S., Budde M.W., Elo-witz M.B., Cai L. (2017) Synthetic recording and in situ readout of lineage information in single cells. Nature. 541, 107–111.
Raj B., Wagner D.E., McKenna A., Pandey S., Klein A.M., Shendure J., Gagnon J.A., Schier A.F. (2018) Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 36, 442–450.
Spanjaard B., Hu B., Mitic N., Olivares-Chauvet P., Janjuha S., Ninov N., Junker J.P. (2018) Simultaneous lineage tracing and cell-type identification using CRISPR-Cas9-induced genetic scars. Nat. Biotechnol. 36, 469–473.
Yao Z., Mich J.K., Ku S., Menon V., Krostag A.-R., Martinez R.A., Furchtgott L., Mulholland H., Bort S., Fuqua M.A., Gregor B.W., Hodge R.D., Jayabalu A., May R.C., Melton S., Nelson A.M., Ngo N.K., Shapovalova N.V., Shehata S.I., Smith M.W., Tait L.J., Thompson C.L., Thomsen E.R., Ye C., Glass I.A., Kaykas A., Yao S., Phillips J.W., Grimley J.S., Levi B.P., Wang Y., Ramanathan S. (2017) A single-cell roadmap of lineage bifurcation in human ESC models of embryonic brain development. Cell Stem Cell. 20, 120–134.
Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B.B., Siddiqui A., Lao K., Surani M.A. (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods. 6, 377–382.
Frank E., Sanes J.R. (1991) Lineage of neurons and glia in chick dorsal root ganglia: Analysis in vivo with a recombinant retrovirus. Development. 111, 895–908.
Turner D.L., Cepko C.L. (1987) A common progenitor for neurons and glia persists in rat retina late in development. Nature. 328, 131–136.
Pei W., Feyerabend T.B., Rössler J., Wang X., Pos-trach D., Busch K., Rode I., Klapproth K., Dietlein N., Quedenau C., Chen W., Sauer S., Wolf S., Höfer T., Rodewald H.R. (2017) Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature. 548, 456–460.
Jao L.-E., Wente S.R., Chen W. (2013) Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA. 110, 13904–13909.
Varshney G.K., Pei W., LaFave M.C., Idol J., Xu L., Gallardo V., Carrington B., Bishop K., Jones M., Li M., Harper U., Huang S.C., Prakash A., Chen W., Sood R., Ledin J., Burgess S.M. (2015) High-throughput gene targeting and phenotyping in zebrafish using CRISPR/ Cas9. Genome Res. 25, 1030–1042.
Paul F., Arkin Y., Giladi A., Jaitin D.A., Kenigsberg E., Keren-Shaul H., Winter D., Lara-Astiaso D., Gury M., Weiner A., David E., Cohen N., Lauridsen F.K., Haas S., Schlitzer A., Mildner A., Ginhoux F., Jung S., Trumpp A., Porse B.T., Tanay A., Amit I. (2015) Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. 163, 1663–1677.
Laurenti E., Göttgens B. (2018) From haematopoietic stem cells to complex differentiation landscapes. Nature. 553, 418–426.
Carrelha J., Meng Y., Kettyle L.M., Luis T.C., Norfo R., Alcolea V., Boukarabila H., Grasso F., Gambardella A., Grover A., Högstrand K., Lord A.M., Sanjuan-Pla A., Woll P.S., Nerlov C., Jacobsen S.E.W. (2018) Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature. 554, 106–111.
Blaese R.M., Culver K.W., Miller A.D., Carter C.S., Fleisher T., Clerici M., Shearer G., Chang L., Chiang Y., Tolstoshev P., Greenblatt J.J., Rosenberg S.A., Klein H., Berger M., Mullen C.A., Ramsey W.J., Muul L., Morgan R.A., Anderson W.F. (1995) T lymphocyte-directed gene therapy for ADA- SCID: Initial trial results after 4 years. Science. 270, 475–480.
Bordignon C., Notarangelo L.D., Nobili N., Ferrari G., Casorati G., Panina P., Mazzolari E., Maggioni D., Rossi C., Servida P., Ugazio A.G., Mavilio F. (1995) Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science. 270, 470–475.
Hilgendorf I., Greinix H., Halter J.P., Lawitschka A., Bertz H., Wolff D. (2015) Long-term follow-up after allogeneic stem cell transplantation. Dtsch Aerzteblatt Online. 112, 51–58.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология