

Молекулярная биология, 2019, T. 53, № 5, стр. 725-740
“Свой среди чужих”: можно ли создать гипоиммуногенные линии плюрипотентных стволовых клеток?
М. Е. Богомякова a, b, А. В. Еремеев a, М. А. Лагарькова a, b, *
a Федеральный научно-клинический центр физико-химической медицины,
Федерального медико-биологического агентства
119435 Москва, Россия
b Кафедра иммунологии биологического факультета Московского государственного университета
им. М.В. Ломоносова
119234 Москва, Россия
* E-mail: lagar@rcpcm.org
Поступила в редакцию 24.02.2019
После доработки 01.04.2019
Принята к публикации 02.04.2019
Аннотация
Плюрипотентные стволовые клетки, к которым относятся эмбриональные стволовые клетки и индуцированные плюрипотентные клетки (ИПСК), способны к неограниченному делению и дифференцировке во все клетки организма. Эти клетки рассматривают как возможный источник различных типов клеток для трансплантации. Применение аутологичных ИПСК потенциально не связано с иммунным отторжением и не требует иммуносупрессии, необходимой при аллогенных трансплантациях. Тем не менее, высокая стоимость этой технологии и длительность получения ИПСК и дифференцированных клеток может ограничить использование аутологичных ИПСК в клинической практике. Кроме того, под сомнение неоднократно ставилась полная эквивалентность и иммунологическая совместимость даже аутологичных ИПСК и их производных. Одним из подходов к решению проблемы иммунологической совместимости аллогенных производных ИПСК может стать создание клеточных линий со сниженной иммуногенностью. Дифференцированные производные таких ИПСК могут быть пригодны для трансплантации любому пациенту. В представленном обзоре рассмотрены стратегии ускользания клеток от иммунного надзора в норме и при опухолевых процессах, которые можно использовать для создания линий стволовых клеток с пониженной иммуногенностью.
К плюрипотетным стволовым клеткам (ПСК) относятся эмбриональные стволовые клетки (ЭСК) и индуцированные плюрипотентные стволовые клетки (ИПСК). ПСК – это клеточные линии, которые существуют только in vitro. Главное свойство этих клеток – способность к неограниченному делению в условиях, способствующих самоподдержанию, и к дифференцировке в любые клетки, производные всех трех зародышевых листков, при изменении этих условий. ЭСК человека получают из внутренней клеточной массы бластоцист, невостребованных после процедуры экстракорпорального оплодотворения [1]. Генетическое репрограммирование соматических клеток (фибробластов) до плюрипотентного состояния в 2006 году осуществили Takahashi K. и Yamanaka S. [2] c помощью экзогенной экспрессии факторов транскрипции Oct4, Sox2, Klf4 и c-Myc. С момента получения первых линий ЭСК и ИПСК человека научное сообщество стало возлагать большие надежды на развитие заместительной клеточной терапии. Разработаны эффективные и воспроизводимые протоколы дифференцировки различных типов клеток из ПСК, включая кардиомиоциты, клетки пигментного эпителия сетчатки (ПЭС), нейроны и β-клетки поджелудочной железы [3]. Кроме того, ИПСК открыли широкие возможности для моделирования так называемой “болезни в чашке Петри” и скрининга лекарственных препаратов [4–6].
Технология получения аутологичных ИПСК делает возможной персонализированную клеточную терапию, которая снимает проблемы, связанные с иммунным отторжением. Однако высокая стоимость, длительность получения клинически сертифицированных линий ИПСК, их последующей дифференцировки в клетки, необходимые для трансплантации, а также проверка терапевтической безопасности клеточных продуктов являются факторами, которые замедляют внедрение технологии ИПСК в клиническую практику [3, 7]. Очень показательна работа, результаты которой свидетельствуют о существующих проблемах внедрения в производство клеточных продуктов из аутологичных ИПСК и их производных [8]. В ходе получения аутологичных ИПСК двух пациентов и их дифференцировки в клетки ПЭС для последующей трансплантации, авторы предложили показатели и методы оценки качества, проводя отбор клеточных линий на разных этапах культивирования и дифференцировки клонов ИПСК по ключевым характеристикам, которые потенциально могут влиять на безопасность: морфология, кариотип, ростовые свойства, генетическая стабильность, способность к направленной дифференцировке. Уже на этапе оценки морфологических характеристик были отбракованы 12 из 32 клонов ИПСК первого пациента и половина из 40 клонов ИПСК второго. В итоге получены два клона ИПСК первого пациента, и всего один клон второго, которые удовлетворяли всем критериям, связанным с безопасностью. Полагают, что для получения клинически сертифицированных линий аутологичных ИПСК требуется дорогостоящий и длительный анализ каждого полученного клона, который позволит снизить существующие риски, связанные с потенциальным мутагенезом и онкотрансформацией клеточных продуктов при трансплантации пациенту. Поэтому сейчас мировое научное сообщество придерживается мнения, что аллогенную трансплантацию тщательно охарактеризованных производных ИПСК и ЭСК, по крайней мере, в ближайшей перспективе можно рассматривать как более реалистичный подход к персонализированной клеточной терапии [7, 9].
Гистосовместимость – одна из основных проблем терапевтического применения аллогенных клеток и тканей, в том числе клеток – продуктов дифференцировки ПСК. Известно более 20 000 аллелей HLA (http://www.ebi.ac.uk/imgt/hla/). Такой полиморфизм создает проблемы подбора донора для трансплантации. В связи c неполным совпадением донора и реципиента по генам главного комплекса гистосовместимости (MHC) возникает необходимость в системном применении иммуносупрессивных препаратов для предотвращения иммунного ответа. Такая терапия имеет серьезные побочные эффекты, увеличивает риск возникновения инфекций и опухолей, часто бывает неэффективной [10].
Таким образом, оправдан высокий интерес к изучению механизмов иммуногенности и иммунологической толерантности в норме и при патологических процессах. Так, известно множество механизмов, с помощью которых опухолевые ткани уходят из-под контроля иммунной системы. Именно поэтому интересно выявить возможные пути и механизмы, участвующие в развитии толерантности, которые можно использовать для решения проблемы гистосовместимости аллогенных дифференцированных производных ПСК.
Одним из эффективных способов решения проблемы может быть создание универсально гистосовместимых линий ИПСК человека, подходящих для трансплантации любому реципиенту (рис. 1). Таким образом, персонализированный подход к будущим клеточным трансплантациям можно облегчить, используя более универсальный источник клеточного материала. Предполагается, что описанные в нашем обзоре генетические модификации ПСК могут применяться для разработки безопасной и эффективной стратегии, которая позволит уменьшить иммунный ответ потенциального реципиента на аллогенные клетки, дифференцированные из ПСК, без использования стандартных иммуносупрессивных препаратов.
Рис. 1.
Преимущества и недостатки потенциального использования аутологичных, гипоиммунногенных и аллогенных клеточных продуктов при трансплантации.
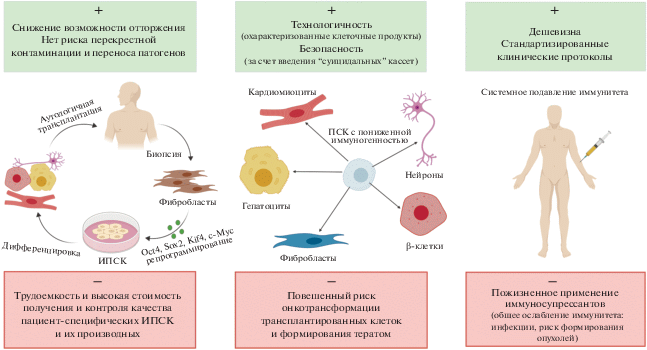
ИММУНОГЕННОСТЬ ПЛЮРИПОТЕТНЫХ СТВОЛОВЫХ КЛЕТОК И ИХ ПРОИЗВОДНЫХ
Изначально считалось, что ПСК могут уходить от иммунного надзора и не будут отторгаться при трансплантации из-за низкой экспрессии HLA классов I и II, костимуляторных молекул CD80 и CD86, а также из-за экспрессии иммуномодуляторных молекул, таких как серпин 6 (эндогенный ингибитор гранзима В) и TGFβ, ингибирующих пролиферацию Т-клеток [11]. Однако исследования на мышах с функциональной иммунной системой показали, что ПСК не обладают иммунологическими привилегиями и вызывают специфический иммунный ответ и отторжение, как и при трансплантации зрелых тканей и органов [12, 13].
В отличие от ЭСК мыши, на поверхности которых не детектируются молекулы MHC I и II [14], ЭСК человека экспрессируют HLA-I [15]. Как врожденная, так и адаптивная ветви иммунной системы вносят свой вклад в отторжение ЭСК. Аллогенные NK-клетки лизируют ПСК мыши и человека in vitro [16]. Считается, что ПСК не могут прямо активировать аллогенные Т-клетки через взаимодействие HLA и TCR in vitro или in vivo [17]. Однако ПСК экспрессируют иммуногенные антигены, в том числе белок Oct 3/4, которые могут активировать Т-клетки через антигенпредставляющие клетки (АПК) [18].
В одной из работ ЭСК человека и их дифференцированные производные трансплантировали под капсулу почки гуманизированной мыши [12]. Трансплантация кусочков кожи здоровых доноров этим мышам приводила к генерализованному отторжению трансплантата, а дифференцированные производные ЭСК вызывали только минимальные проявления воспаления в виде лейкоцитарной инфильтрации. Однако в дальнейшем эти данные не были подтверждены. Так, введение ЭСК мыши и их производных в сердечную мышцу аллогенных мышей-реципиентов приводило к повышению иммуногенности при увеличении степени дифференцировки трансплантируемых клеток [13]. Далее была получена коллекция линий ЭСК мыши, которые отличались друг от друга по определенным генетическим локусам с возрастающим уровнем иммунологического различия [19]. Степень иммунного ответа, вызываемого клетками, дифференцированными из ЭСК, оценивали, имплантируя эти клетки под капсулу почки без применения иммуносупрессии. Следует отметить, что дифференцированные производные ИПСК, гаплотип которых практически идентичен гаплотипу реципиентной линии мышей, элиминировались намного медленнее, чем дифференцированные из аллогенных ЭСК.
В связи с этим, важным остается вопрос о потенциальной возможности отторжения аутологичных производных плюрипотентных клеток. В 2011 году в экспериментах по аутологичной трансплантации ИПСК и ЭСК мыши подкожно ввели сингенным реципиентам [20]. Как и ожидалось, сингенные ЭСК формировали тератомы, однако ИПСК с тем же генетическим фоном вызывали образование существенных Т-клеточных инфильтратов, что вело к их отторжению. Полученные впоследствии сходные результаты также показали, что степень иммуногенности ПСК снижалась в ходе дальнейшей дифференцировки [21]. ПСК в недифференцированном состоянии не могут рассматриваться как материал для трансплантации. Таким материалом могут быть только клетки определенной тканевой специфичности, направленно дифференцированные из ПСК in vitro – нейроны и их клетки-предшественники, ПЭС, инсулинпродуцирующие клетки и так далее [3]. Неожиданное сообщение об иммуногенности аутологичных производных ИПСК в сингенных реципиентах вызвало пессимизм по поводу их терапевтического потенциала. Следует отметить, что результаты более поздних работ, наоборот, свидетельствуют об отсутствии иммуногенности и безопасности дифференцированных сингенных производных ИПСК [22, 23].
Из ЭСК и ИПСК получены эндотелиальные клетки, гепатоциты и нейрональные клетки [22]. Эти клетки экспрессировали MHC I и костимуляторные молекулы, т.е. теоретически они могли активировать Т-клеточный ответ. Однако сокультивирование с аллогенными T-лимфоцитами не выявило специфического Т-клеточного ответа на недифференцированные сингенные ИПСК или их производные. Трансплантация трех типов клеток сингенным мышам не приводила к инфильтрации трансплантата T-клетками, что согласуется с данными, полученными in vitro. Трансплантация аллогенных клеток немедленно вызывала генерализованный иммунный ответ in vivo и выраженную цитотоксичность in vitro, еще раз подтверждая, что ПСК и их производные не являются иммунологически инертными. Эти результаты говорят о том, что иммуногенные белки не продуцируются, по крайней мере, в эндотелиальных и нейрональных клетках, гепатоцитах, дифференцированных из ИПСК. К похожим выводам пришли и другие исследователи, показав, что тератомы из сингенных ЭСК и ИПСК не вызывают значимых иммунных реакций [23].
Высказано предположение, что иммуногенность полученных линий связана, скорее всего, с повышенной экспрессией опухолеассоциированных генов Hormad и Zg16 [24]. Тем не менее, не обнаружено различий в экспрессии этих генов в ЭСК и ИПСК [22, 23]. Причиной подобной неоднозначности результатов этих работ могут быть различия в способах получения ИПСК. Предполагается, что на иммуногенность клеточных линий может влиять вектор, выбранный для доставки факторов репрограммирования [25]. Так, для репрограммирования использовали ретровирусные конструкции и плазмиды [24], ИПСК получали с помощью плазмид [22], ЭСК и ИПСК – и плазмид, и ретровирусов [23]. Ретровирусные векторы встраиваются преимущественно в транскрипционно активные сайты, вызывая продолжительную активацию трансгенов или соседних генов около сайта интеграции, что может приводить к активации экспрессии потенциально иммуногенных белков [18]. Поэтому для клинического использования ИПСК следует получать с помощью методов репрограммирования соматических клеток, которые не вызывают мутагенез и не приводят к иммуногенности.
Иммуногенность производных ИПСК изучали также на приматах [26]. Обнаружено, что трансплантация аутологичных нейронов, дифференцированных из ИПСК, в мозг приматов вызывает минимальный иммунный ответ. Напротив, аллогенные нейроны вызывают активацию микроглии (IBA-1+/MHC class II+) и инфильтрацию трансплантата Т-лимфоцитами. В дальнейшем для исследования механизмов, возникающих при трансплантации клеток человека, использовали мышей с реконструированной иммунной системой человека [24]. При подкожном введении аутологичных ИПСК в образовавшихся тератомах формировались зоны явного воспаления и некроза, что означает иммунологическое отторжение, по крайней мере, некоторых типов клеток, дифференцированных из ИПСК. Кроме того, наблюдали также дифференциальную иммуногенность ИПСК-производных разного гистотипа: гладкомышечные клетки вызывали более сильный иммунный ответ, чем дифференцированные ПЭС даже при трансплантации не в область глаза. Следует отметить, что к настоящему времени единственное клиническое испытание аутологичных ИПСК-производных проведено на пациенте с возрастной макулярной дегенерацией, которое подтвердило отсутствие иммуногенности у аутологичных клеток ПЭС, дифференцированных из ИПСК [8]. В связи с противоречивыми данными об иммуногенности некоторых типов аутологичных производных ИПСК для дальнейшего развития протоколов клеточной терапии, может понадобиться оценка иммуногенности клеток, дифференцированных из ИПСК, на животных с гуманизированной иммунной системой. Поскольку клетки каждого типа содержат разные наборы белков, то, вероятно, перед клиническим применением необходимо будет проводить скрининг клеток каждого типа на иммуногенность [25].
Существует мнение, что ИПСК, в отличие от ЭСК, в значительной мере сохраняют “переходную” транскрипционную и эпигенетическую память о своем происхождении, так называемую “соматическую память”, особенно на ранних пассажах, однако молекулярные и функциональные различия утрачиваются в процессе продолжительного культивирования [27]. Это свойства могут быть специфичными для каждого конкретного клона ИПСК [28]. Эпигенетическими особенностями можно объяснить остаточную экспрессию иммуногенных белков, которые синтезируются при дифференцировке только в ИПСК, но не в ЭСК. Вклад эпигенетических механизмов в регуляцию экспрессии иммуногенных белков может быть тканеспецифичным, что наблюдали при дифференцировке ИПСК человека в ПЭС и гладкомышечные клетки [24]. Wang и соавт. показали, как феномен “соматической памяти” может влиять на дальнейшую иммуногенность полученных линий ИПСК [29]. Они репрограммировали клетки Сертоли семенников мыши, являющихся анатомически иммунопривилегированной областью. Полученные при этом клетки оказались менее иммуногенными при трансплантации в аллогенного реципиента, чем ИПСК из фибробластов кожи. Сходные данные получены и для ИПСК, репрограммированных из мезенхимальных клеток пуповины [30]. Дифференцированные нейрональные предшественники вызвали существенно меньшую пролиферацию и активацию цитотоксических лимфоцитов при сокультивировании in vitro.
Есть и другие причины, рассмотренные в связи с потенциальной иммуногенностью ИПСК. Во-первых, во многих линиях ИПСК выявлены соматические мутации, эти мутации могут создавать новые иммуногенные детерминатны, как и неоантигены, появляющиеся в опухолевых клетках [31]. Во-вторых, геномные транслокации, детектируемые в ИПСК, могут приводить к образованию слитых белков, которые могут дать новые иммуногенные детерминанты [31]. Причиной изменения функционирования генома может быть длительное культивирование и сам процесс репрограммирования, особенно на основе интегрирующих векторов, который сопровождается вставкой трансгена в случайное место. Поэтому в дополнение к иммуногенности клеток, дифференцированных из ИПСК, наиболее серьезное опасение вызывает возможная геномная нестабильность данных клеток, которая может увеличить риск канцерогенеза. Следует, тем не менее, отметить, что данные о генетической нестабильности ИПСК неоднозначны и противоречивы. Результаты сравнения генетической стабильности ЭСК и ИПСК свидетельствуют скорее об отсутствии разницы между ЭСК и ИПСК при полном репрограммировании последних [32].
СОЗДАНИЕ БАНКОВ ЛИНИЙ ИПСК, ГОМОЗИГОТНЫХ ПО ГЕНАМ HLA
Технология репрограмирования позволяет получать аутологичные пациент-специфичные ИПСК и их производные, что в большой степени снимает вопрос иммунного отторжения. Однако такая технология имеет ряд недостатков. Во-первых, для получения и характеристики линии ИПСК необходимо длительное время, исчисляемое месяцами [33]. Во-вторых, для дальнейшего производства и последующего клинического применения требуется разработка спецификации согласно требованиям законодательства и контроль качества репрограммированных клеток, что существенно увеличивает время подготовки клеточного продукта и стоимость персонализированной терапии [34]. Подобный подход может подойти лицам с хроническими заболеваниями, когда имеется время, необходимое для осуществления всех перечисленных процессов.
Альтернативой персонализированной терапии с помощью дифференцированных производных ИПСК может стать создание банка охарактеризованных линий ИПСК, гомозиготных по HLA, а также их производных [35]. Можно предположить, что такие гомозиготные линии будут совместимы с гетерозиготными реципиентами, c гетерозиготными реципиентами, у которых хотя бы один аллель совпадает с гаплотипом линии ИПСК. Так как несовместимость по системе АВО может стать причиной гиперострого отторжения в первично васкуляризированных тканях и органах, при создании гаплобанка необходимо подбирать здоровых доноров с группой крови I (0), что минимизирует риск отторжения [36]. Из-за сильного полиморфизма локуса HLA потребуется создать гаплобанк из сотен донорских линий, чтобы достичь совпадения для большинства реципиентов [35].
Некоторые эмпирические расчеты числа необходимых линий ПСК, гомозиготных по HLA, основаны на частоте встречаемости и этническом разнообразии выбранной популяции [36]. Так, например, банк ПСК, полученных из 150 типированных по HLA гомозигот, совпадет с 93% населения Великобритании с необходимостью минимальной иммуносупрессии [35]. Для потенциального совпадения с 90% населения Японии необходимо примерно 50 линий [37], однако для достижения такого уровня совпадения необходим скрининг более 60 000 волонтеров [38]. Формирование банка ИПСК в Японии ведется с 2012 года в Центре исследования и применения ИПСК в Киото (http://www.cira. kyoto-u.ac.jp/e/).
Для стран с этнически более разнообразным населением, в том числе США, Бразилии и России, таких линий потребуется больше. В 2012 году была предложена модель, согласно которой для создания гаплобанка из 20 доноров с наиболее часто встречаемыми гаплотипами HLA необходимо типировать 26 000 евроамериканцев и 110 000 афроамериканцев [39]. Однако потенциальное клиническое применение возможно только в условиях международного сотрудничества и типирования сотен тысяч добровольцев. Очевидно, что для массового создания специализированных банков клеток требуются огромные материальные вложения.
Несмотря на оптимистические прогнозы в отношении потенциального применения гаплобанков, следует помнить, что cовпадения по гаплотипу HLA может быть недостаточно для предотвращения аллогенного отторжения, и большинству пациентов может потребоваться иммуносупрессивная терапия. Способствовать аллореактивности также могут минорные mH-антигены, которые неизбежно будут отличаться у неродственного донорa, а также взаимодействие ингибирующих рецепторов KIR (killer cell immunoglobulin-like receptor) на NK-клетках с разными аллелями HLA-I. В процессе созревания NK-клетки приобретают толерантность к собственным клеткам, содержащим на поверхности определенный набор молекул HLA-I. Все молекулы HLA-C делятся на две группы ‒ С1 и С2 ‒ в зависимости от их способности связываться с KIR2DL3 и KIR2DL1 [40]. Поэтому всех реципиентов можно поделить на следующие группы: С1/С1, С1/С2 и С2/С2 согласно генотипу HLA-C. Предполагается, что при трансплантации ИПСК-производных, полученных от гомозиготного донора С1/С1, гетерозиготному реципиенту С1/С2 лицензированные NK-клетки реципиента будут отвечать на отсутствие KIR-лиганда (С2) и, в конечном счете, отторгнут аллографт из-за отсутствия ингибирующего сигнала KIR2DL1 в соответствии с традиционным механизмом распознавания “отсутствия своего”. Недавно обнаружено, что NK-клетки, выделенные из HLA-гетеро-C1/C2, атакуют in vitro Т-клетки и эндотелиальные клетки, дифференцированные из HLA-гомо-C1/C1 ИПСК [41]. С помощью эктопической экспрессии HLA-С2 на поверхности дифференцированных клеток удалось ингибировать ответ NK-клеток. Эти результаты подтверждают, что NK-клетки отвечают на несовпадение KIR-лиганда и могут убивать дифференцированные клетки за счет распознавания отсутствия экспрессии HLA-C, что необходимо учитывать при трансплантации производных ИПСК, гомозиготных по HLA.
В этой связи целесообразно рассмотреть возможные механизмы возникновения иммунотолерантности, реализуемые иммунной системой человека не только в норме, но и при патологиях, чтобы использовать эти стратегии для потенциального получения иммунопривилегированных линий ИПСК с минимальными рисками онкотрансформации.
СТРАТЕГИИ, КОТОРЫЕ ОПУХОЛЕВЫЕ КЛЕТКИ ИСПОЛЬЗУЮТ ДЛЯ ИЗБЕГАНИЯ ИММУННОГО НАДЗОРА
В попытках избежать уничтожения иммунными клетками организма опухоли могут становиться “невидимыми” для иммунной системы. Стратегии ускользания от иммунного надзора можно использовать для создания гипоиммуногенных линий ИПСК. Отторжение трансплантируемых органов и тканей происходит с помощью механизмов клеточного и гуморального ответа, которые преимущественно зависят от распознавания чужеродных антигенов HLA Т-клетками [42]. Чтобы “обмануть” иммунную систему, опухолевые клетки уменьшают экспрессию молекул, необходимых для представления антигена (HLA), костимуляции (CD80 и CD86) и адгезии (CD54), тем самым предотвращая их распознавание иммунными клетками, а также повышают экспрессию таких иммуносупрессирующих компонентов, как HLA-G, PD-L1 и CTLA-4 [43]. Эти механизмы играют большую роль в повышении активности регуляторных Т-клеток и последующей анергии цитотоксических Т- и NK-клеток.
CTLA-4 и PD-L1 ‒ важнейшие иммунологические чекпойнты в поддержании периферической толерантности T-клеток. Поэтому эти молекулы можно использовать для индукции иммунной толерантности к аллогенным трансплантатам [44]. CTLA-4 связывается с CD80 и CD86 на поверхности АПК, тем самым блокируя Т-клеточные костимуляторные пути, в то время как PD-L1 связывается с PD-1, который экспрессируется на активированных Т-клетках, для индукции ингибиторных сигнальных путей. Вместе они контролируют баланс костимуляторных и коингибиторных сигналов, что играет значительную роль в регуляции амплитуды и длительности Т-клеточного ответа. Экспрессия этих молекул повышена в клетках различных типов рака [45].
Далее мы более подробно рассмотрим основные стратегии, применяемые опухолевыми клетками для развития иммунотолерантности, которые можно использовать для решения проблемы гистосовместимости аллогенных дифференцированных производных плюрипотентных клеток.
HLA-I
Ускользание от иммунного надзора достаточно часто связано с потерей молекул HLA-I на поверхности опухолевых клеток. Молекулы HLA-I играют ключевую роль в представлении пептидов (включая опухолеассоциированные антигены) цитотоксическим Т-клеткам. Экспрессия этих антигенов и костимуляторных рецепторов, возможных сигналов процессов, происходящих в клетке, запускает иммунную активацию и способствует цитотоксическому разрушению опухолевых клеток [46]. Предполагается, что эффективность противоопухолевой терапии зависит от уровня экспрессии молекул HLA-I на поверхности опухолевых клеток [47]. Это, в первую очередь, зависит от молекулярных механизмов, лежащих в основе потери экспрессии HLA-I [48]. В так называемых “soft, мягких” новообразованиях изменения, ответственные за изменение уровня экспрессии HLA-I, нормализуются после стимуляции цитокинами или иммунотерапии. Возрастающий в таком случае специфический Т-клеточный ответ ведет к дальнейшему регрессу новообразования. В резистентных к терапии, или “hard, тяжелых” новообразованиях структурные дефекты генома необратимы, они приводят к возникновению различных мутаций, и тогда превалируют механизмы ускользания от иммунного надзора. Согласно этой идее, природа утраты экспрессии HLA-I в опухолевых клетках существенно влияет на успех противоопухолевой терапии [49]. Поэтому часто дефекты в экспрессии HLA-I опухолевыми клетками ведут к гипоиммуногенности, последующей эвазии и прогрессии метастазирования. Потеря экспрессии HLA-ABC обнаружена при целом ряде онкологических заболеваний, включая плоскоклеточный рак головы и шеи (примерно 70% случаев), рак молочной железы (96%), рак толстой кишки (87%) и меланому (63%) [50].
Белки HLA-I представляют собой гетеродимеры, состоящие из тяжелой α-цепи с высокой степенью полиморфизма и консервативной легкой цепи β-2-микроглобулина (b2m) – небольшоого белка с молекулярной массой около 12 кДа без трансмембранного домена, который входит в суперсемейство иммуноглобулинов. Связь b2m с α3-доменом тяжелой α-цепи HLA-I необходима для поддержания конформации гетеродимера и формирования функционального комплекса HLA-I на поверхности клетки, она также способствует усилению аффинности связывания пептидов [51].
Различные мутации, возникающие в гене b2m, могут препятствовать синтезу белка b2m и, соответственно, стабилизации функционально активной молекулы HLA-I. Такие мутации детектируются в различных клеточных линиях и опухолевых тканях, они могут быть представлены инсерциями и делециями нуклеотидов в мотивах с повторяющимися последовательностями, а также однобуквенными заменами в одном аллеле гена b2m в комбинации с потерей больших сегментов хромосомы 15q21, выключающей второй аллель гена b2m [52]. Эти мутации ингибируют экспрессию b2m, препятствуя транскрипции или, что случается более часто, приводят к сбою трансляции мРНК или синтезу нефункционального белка. Возникновение необратимых функциональных дефектов в гене b2m способствует селекции и прогрессии агрессивных клонов опухолевых клеток за счет отсутствия экспрессии HLA-I.
Еще одним механизмом эвазии, например, в случае гематологических опухолей, могут быть изменения экспрессии HLA-II, в том числе, вызванные мутациями в гене CIITA, трансактиваторе HLA-II. Такие мутации обнаружены у пациентов с классической лимфомой Ходжкина [53].
HLA-G – малоизученный член семейства HLA-I
HLA-G вместе с HLA-E, -F и -H принадлежит к “неклассическим” молекулам HLA-Ib. Молекулы HLA-Ib в отличие от “классических” молекул HLA-Ia с высоким уровнем полиморфизма сравнительно консервативны и представлены ограниченным числом аллелей. HLA-G не только выполняет основную функцию молекул HLA-I – представляет пептидные фрагменты специфическим субпопуляциям CD8+ Т-лимфоцитов, но и обладает иммуномодулирующей функцией [54]. В норме HLA-G не экспрессируется на поверхности здоровых клеток и детектируется только на клетках трофобласта, эпителиальных клетках тимуса, моноцитах, активированных цитокинами, зрелых миелоидных и плазмацитоидных дендритных клетках (ДК) и на воспаленных мышечных волокнах [55]. Основная физиологическая роль этой молекулы – формирование иммунной толерантности в гематоплацентарном барьере. Различные формы HLA-G экспрессируются клетками трофобласта. Они могут взаимодействовать с рецепторами на поверхности иммунных клеток, уменьшая материнский иммунный ответ против полуаллогенных тканей плода путем снижения цитотоксичности Т- и NK-клеток, пролиферации Т- и В-клеток и индукции апоптоза активированных CD8+ Т-клеток [56].
Молекулы HLA-G представлены не только на поверхности клетки, существуют также их растворимые изоформы. Известно семь изоформ HLA-G, которые образуются в процессе альтернативного сплайсинга одной мРНК ‒ мембраносвязанные mHLA-G1, -G2, -G3, -G4 и растворимые изоформы sHLA-G5, -G6 и -G7 [57]. Существует также растворимый sHLA-G1, идентичный sHLA-G5, который образуется путем вырезания трансмембранного домена металлопротеиназами [58]. Только HLA-G1 и -G5 могут представлять функциональную тяжелую α-цепь, ассоциированную с молекулой b2m и способную связывать малые пептиды.
Иммуномодулирующие свойства HLA-G опосредуются взаимодействием с иммуноглобулин-подобным транскриптом ILT2 (immunoglobulin-like transcript 2) на поверхности Т- и В-лимфоцитов, моноцитов/макрофагов, ДК и NK-клеток, а также с ILT4, который экспрессируется только на миелоидных клетках – ДК, моноцитах/макрофагах и нейтрофилах [54]. Кроме того, HLA-G взаимодействует с KIR2DL4 на поверхности NK-клеток [59] и CD160, который экспрессируется Т-лимфоцитами, NK-клетками и эндотелиальными клетками [60]. В покоящихся клетках эти рецепторы экспрессируются на низком уровне, но на активированных клетках в патологических состояниях, например при вирусной инфекции, экспрессия возрастает [61]. Взаимодействуя с этими рецепторами, HLA-G воздействует на функции различных клеточных популяций: препятствует активации эффекторных клеток, секреции цитокинов В-клетками, способствует апоптозу, ингибирует хемотаксис, уменьшая экспрессию некоторых поверхностных рецепторов хемокинов [54], а также уменьшает ангиогенез [60].
Повышенная экспрессия мембраносвязанного HLA-G и растворимого sHLA-G, обнаруженная в различных солидных и гематологических опухолях, коррелирует с повышенным риском прогрессии опухолей и метастазов и с общим плохим прогнозом [45].
Лиганды NKG2D
NKG2D (NK group 2 member D) – основной активирующий рецептор, который экспрессируется на поверхности NK-клеток. Он присутствует на поверхности цитотоксических, CD4+ и γδТ-клеток [62]. Поэтому рецептор NKG2D не только индуцирует цитотоксичность эффекторных клеток [63], но способствует продукции цитокинов и влияет на дифференцировку и пролиферацию Т-клеток [64]. Лигандами данного рецептора служат MICA, MICB (MIC – MHC class I-related chain) и семейство ULBP (UL-16 binding proteins), которые, в отличие от HLA-I, не связаны с b2m. Экспрессия этих так называемых “стресс-индуцированных” лигандов способствует иммунному надзору трансформированных, инфицированных или подвергшихся стрессу клеток. При увеличении экспрессии соответствующих лигандов NKG2D распознает повреждение ДНК, высокий уровень активных форм кислорода, повышенный уровень пролиферации и тепловой шок. Кроме того, пара лиганд-рецептор является одним из известных иммунологических чекпойнтов. Иммунные клетки, экспрессирующие NKG2D, атакуют трансформированные клетки до начала процесса изменения иммунологического фенотипа опухолевых клеток, служащего предпосылкой к иммунному ускользанию. Несмотря на это, ряд данных свидетельствует о противоречивой роли лигандов NKG2D в регуляции развития опухоли [65].
Раковые клетки способны управлять экспрессией лигандов NKG2D на посттранскрипционном и посттрансляционном уровнях. Регуляция экспрессии лигандов NKG2D недостаточно изучена. Однако недавно выявили множественные механизмы, которые раковые клетки используют для уменьшения индуцируемых стрессом лигандов, избегая таким образом иммунного распознавания [66]. Чаще всего встречается шеддинг, или слущивание молекул с поверхности клеточной мембраны. Металлопротеазы, в частности ADAM10, ADAM17 и MMP14, часто детектируемые в микроокружении опухоли, разрезают трансмембранный домен, убирая белки MICA, MICB и ULBP1-6 с поверхности злокачественных клеток [67‒69]. Кроме того, GPI-заякоренные лиганды (гликозилфосфатидилинозит – GPI-якорь), такие как ULBP1, ULBP3 и MICA∗008, очень часто высвобождаются в экзосомальных везикулах [70, 71], как и опухолеассоциированные антигены, молекулы HLA-I/II, лиганды “рецепторов смерти” и молекулы адгезии [72]. Растворимые и экзосомальные рецепторы связываются с соответствующими сайтами на NK- и Т-клетках с последующей интернализацией и деградацией [73]. В то время как различные сигналы клеточного стресса могут распознаваться функциональным рецептором, “слущивания” лиганда одного типа достаточно, чтобы сделать иммунные клетки “слепыми” по отношению ко всему семейству лигандов [66].
Уровень растворимых стресс-индуцированных лигандов в сыворотке онкологических больных считается значимым прогностическим фактором. Выявлена обратная корреляция между уровнем этих лигандов и активностью NK- и Т-клеток, корреляция со стадией рака, а также негативное влияние на выживание пациента [66].
CD47
CD47, или ассоциированный с интегрином белок (IAP) ‒ трансмембранный белок суперсемейства иммуноглобулинов. CD47, широко представленный в различных тканях взрослого организма, обладает множеством функций ‒ регулирует целый ряд процессов, включая апоптоз, пролиферацию, адгезию, миграцию, иммунные и ангиогенные реакции. Экспрессия CD47 существенно повышена в опухолевых клетках [74, 75].
С помощью блокирующих антител показано, что выключение CD47 подавляет миграцию и метастазирование клеток меланомы, рака предстательной железы и яичника [76]. У мышей с дефицитом CD47, используемых в качестве моделей множественной миеломы, число метастазов в кость было меньше, чем в контроле [77]. Аналогичные результаты получены в экспериментах на мышах с ксенографтом клеток неходжкинской лимфомы человека ‒ блокирование функции CD47 с помощью антител приводило к резкому снижению числа метастазов [78].
В 2012 году были опубликованы результаты, согласно которым CD47 препятствует фагоцитозу раковых клеток. Блокирование взаимодействий CD47 с его рецептором SIRPα (от Signal regulatory proteins) антителами к CD47 позволило замедлить прогрессию лейкосаркомы у мышей [79, 80]. Более того, оказалось, что фагоцитоз раковых клеток макрофагами, опосредованный блокирующими анти-CD47-антителами, может инициировать противоопухолевый иммунный ответ Т-клеток, в том числе при опухолях, которые ранее не поддавались иммунотерапии [81‒83]. Кроме того, связывание CD47 с SIRPα блокирует созревание незрелых ДК и ингибирует синтез цитокинов зрелыми ДК, снижая их функции по представлению антигенов. Взаимодействие между CD47 на клетках эндотелия и SIRP на лейкоцитах регулирует трансэндотелиальную миграцию Т-клеток. У мышей с нокаутом CD47 снижена доля Т-лимфоцитов, нейтрофилов и моноцитов в зонах воспаления [84]. CD47 может также уменьшать цитотоксичность NK-клеток в культурах опухолевых клеток in vitro [85].
Важная роль CD47 в регуляции фагоцитоза показана не только при опухолевых заболеваниях, но и в норме, в частности, взаимодействие CD47 с растворимым тромбоспондином 1 (THBS1) и SIRPα на поверхности макрофагов негативно регулирует фагоцитоз нормальных эритроцитов [86].
Таким образом, высокий уровень CD47 позволяет раковым клеткам избегать фагоцитоза макрофагами при взаимодействии с SIRPα, и подавляет распознавание цитотоксическими Т-лимфоцитами и, возможно, NK-клетками. В настоящее время CD47 рассматривается в качестве еще одной мишени для создания гипоиммуногенных линий ИПСК [87].
Основные молекулы, определяющие возникновение иммунологической толерантности в норме и патологии, представлены на рис. 2.
Рис. 2.
Обобщенные стратегии индукции иммунологической толерантности, используемые опухолевыми или нормальными клетками. Иммунологические чекпойнты CTLA-4 и PD-1L предотвращают активацию Т-лимфоцитов. Подавление экспрессии HLA-I, например за счет мутаций в гене b2m, затрудняет представление антигенов (включая опухолеассоциированные антигены) цитотоксическим Т-клеткам. Растворимые лиганды MICA, MICB и ULBP1-6 ингибируют NK-клеточный иммунный ответ. За счет взаимодействия с рецепторами ILT2 и KIR2DL4 HLA-G препятствует активации различных эффекторных клеток, включая Т-, В- и NK-клетки. Взаимодействие CD47 с SIRPα подавляет фагоцитоз макрофагов, а также снижает цитотоксичность NK-клеток. Тем не менее, механизм взаимодействия CD47 с рецепторами на NK-клетках не установлен.
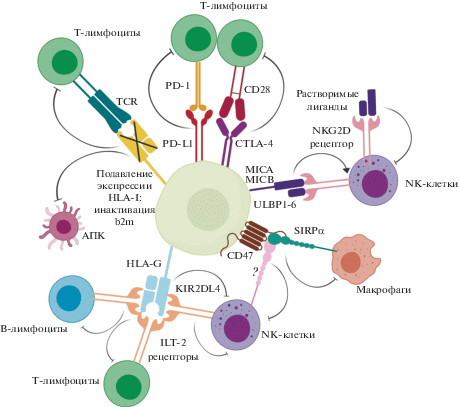
ПЕРСПЕКТИВЫ СОЗДАНИЯ ЛИНИЙ ПСК С ПОНИЖЕННОЙ ИММУНОГЕННОСТЬЮ
Геномное редактирование ПСК рассматривается как стратегия, позволяющая минимизировать иммуногенность стволовых клеток, что может способствовать внедрению клеточных технологий в регенеративную медицину, поскольку ткани, дифференцированные из стволовых клеток, содержащих измененные гены, также будут содержать данную модификацию. Вероятно, технология геномного редактирования может использоваться для создания “универсальной” линии стволовых клеток, подходящей для трансплантации любому пациенту. С этой целью подавляют или удаляют гены комплекса HLA или гены, необходимые для их экспрессии, а также индуцируют иммуносупрессирующие молекулы. Возможные стратегии предотвращения иммунного отторжения подробно перечислены в недавно опубликованном обзоре [88].
Подавить экспрессию HLA-I на поверхности ПСК можно путем ингибирования транскрипции или делеции гена тяжелой α-цепи или легкой цепи (b2m). Инактивация b2m, необходимого для поддержания конформации гетеродимера, приводит к нарушениям формирования функционального комплекса HLA-I. Клетки, на поверхности которых не экспрессируется HLA-I, становятся “невидимыми” для ДК и Т-клеток реципиента, поэтому они должны обладать пониженной иммуногенностью по отношению к аллогенным CD8+ Т-клеткам реципиента. Следует отметить, что клетки, не содержащие на поверхности молекулы HLA-I, могут стать мишенями для NK-клеток. Хотя CD8+ Т-клетки вносят более существенный вклад в отторжение трансплантата, чем NK-клетки, опосредованная NK клеточная смерть может стать проблемой для потенциального терапевтического применения линий ИПСК и их дифференцированных производных с делецией b2m.
Первая попытка создания линии плюрипотентных клеток (ЭСК человека) с пониженной иммуногенностью осуществлена в 2011 году. Эта попытка основана на нокдауне HLA-I с использованием малых интерферирующих РНК и внутриклеточных антител для подавления экспрессии гена на посттранскрипционном и посттрансляционном уровнях [89]. ЭСКKD (ЭСК человека с нокдауном HLA-I) характеризовались сниженной чувствительностью к цитотоксичности, опосредованной CD3+ фракцией мононуклеаров периферической крови in vitro. После трансплантации иммунокомпетентным мышам линии BALB/c, 4 из 10 тератом, сформированных ЭСКKD, выживали в течение 42 дней, в то время как контрольные ЭСК (шесть мышей) элиминировались в течение 10 дней. Несмотря на нокдаун HLA-I, ЭСКKD не инициировали значимую активность NK-клеток, что может быть связано c низкой экспрессией стимуляторных лигандов этих клеток. В 2013 году для нарушения экспрессии HLA-A в линии ЭСК WIBR3 использовали нуклеазы с мотивом цинковых пальцев (ZFN) [90]. Однако из-за полиморфизма тяжелой цепи HLА-I технически проще редактировать гены, необходимые для экспрессии комплекса HLA-I, например, консервативный b2m.
Для инактивации обеих копий гена b2m в ЭСК использовали также аденоассоциированные вирусные векторы (AAV) [91]. ЭСК b2m–/– получили с использованием нуклеаз TALEN [92] и гомологичной рекомбинации [93]. Полученные в этих работах клеточные линии не экспрессировали на своей поверхности b2m и HLA-I, обладали нормальным кариотипом, сохраняли характерную экспрессию маркеров плюрипотентности, были способны дифференцироваться в производные трех зародышевых листков in vitro и формировать тератомы in vivo. С помощью транскриптомного анализа показано, что экспрессия генов в клеточных линиях b2m–/– не отличалась от экспрессии в исходной материнской линии ЭСК [91]. Мононуклеарные клетки периферической крови проявляли сниженную реактивность на эмбриоидные тельца (ЭТ), полученные из b2m–/– ЭСК, в тесте MLR (mixed lymphocyte reaction). Кроме того, b2m–/– ЭТ, культивируемые с праймированными HLA-A*0201-CD8+ T-клетками, экспрессируют IFN-γ на более низком уровне, чем контрольная линия клеток. Тем не менее, инкубация b2m+/+ либо b2m–/– ЭТ с NK-клетками не вызывала повышенной экспрессии индикатора дегрануляции и цитотоксичности NK-клеток ‒ CD107a. Предполагается, что чтобы продемонстрировать феномен распознавания “отсутствия своего” в клетках, дифференцированных из ЭСК, необходима дальнейшая дифференцировка в гемопоэтическом направлении.
В работе китайских исследователей иммуногенность b2m–/– ЭСК проверена с помощью их сокультивирования in vitro с мононуклеарными клетками периферической крови [92]. Методом ELISPOT выявлено повышение секреции IFN-γ мононуклеарами на ЭСК дикого типа в сравнении с b2m–/– ЭСК. Также показана гипоиммуногенность in vivo: при инъекции b2m–/– ЭСК мышам линии BALB/c инфильтрация зоны имплантации лимфоцитами была значительно меньше, чем у мышей, инокулированных ЭСК дикого типа.
Проанализирована цитотоксическая активность аллогенных CD8+ Т-клеток в отношении ЭСК, B2M-null ЭСК и их производных при различных соотношениях эффекторных и таргетных клеток (E/T-effector/target) [93]. Как и ожидалось, B2M-null ЭСК и их производные оказались устойчивыми к цитотоксичности, опосредованной CD8+ Т-лимфоцитами in vitro. Известно, что продуцируемый в ходе иммунного ответа IFN-γ может регулировать экспрессию b2m, способствуя отторжению [15]. Оценка выживаемости после обработки провоспалительным цитокином показала, что контрольная линия ЭСК и ее производные стали более чувствительными к аллогенным CD8+ клеткам, при этом IFN-γ не влиял на резистентность B2M-null ЭСК к иммунному ответу. Также проверена восприимчивость B2M-null ЭСК к ответу на NK-клетки. Подтверждено, что линия ЭСК с нокаутом и ее производные, на поверхности которых отсутствуют HLA-I, узнаются и элиминируются NK-клетками. Чтобы проверить иммуногенность B2M-null ЭСК in vivo, клетки вводили внутримышечно в задние лапы мышей линии SCID. Тератомы формировались в течение 10 недель, после чего их изымали и анализировали. Размер опухолей, полученных из B2M-null ЭСК, был существенно меньше, нежели опухолей, полученных из контрольных ЭСК. Полагают, что это могло произойти из-за того, что мыши SCID обладают функциональными NK-клетками, которые и могли осуществлять лизис тератом, не экспрессирующих HLA-I. Далее изучали способность B2M-null ЭСК формировать тератомы в мышах линии SCID с редуцированным звеном NK-клеточного иммунитета. Как и ожидалось, размер таких опухолей был сравним с контрольными тератомами из ЭСК, выросшими в мышах SCID с функциональными NK-клетками.
В нашей лаборатории получены линии ИПСК с биаллельным нокаутом гена b2m. Фибробластоподобные CD105+ производные обладали повышенной резистентностью к аллогенным CD8+ Т‑лимфоцитам in vitro [94].
Клетки, негативные по HLA-I, становятся чувствительными к опосредованной NK-клеточной цитотоксичности, что является важным ограничением простого редактирования гена b2m [93]. Предполагается, что снизить NK-клеточный ответ можно путем ингибирования активирующего сигнала или экспрессии растворимого гомолога MHC – MICA [90, 93].
Кроме того, потенциальной защитой от NK-клеточного ответа может стать экспрессия неклассических молекул HLA-I ‒ HLA-E и HLA-G. Обнаружено снижение NK-клеточного лизиса трансгенной линии ЭСК, эктопически экспрессирующей мутантный mHLA-G, а также дифференцированных эпидермальных предшественников, в отличие от контрольных линий клеток [95]. Осуществленный недавно нокин HLA-E в локус b2m в линии ЭСК позволил получить стабильную экспрессию одиночной цепи HLA-E без экспрессии HLA-A, -B и -С [96]. HLA-E является лигандом ингибирующего рецептора CD94/NKG2A. Полученная линия ЭСК и ее дифференцированные производные не узнавались аллогенными CD8+ Т-клетками и были резистентными к NK-клеточному лизису. HLA-E также повышает выживаемость in vivo дифференцированных CD45+ клеток и тератом за счет подавления цитотоксичности, реализуемой линией NK-92, экспрессирующей CD94/NKG2A.
В 2016 году с помощью техники геномного редактирования TALEN из линии ЭСК были делетированы гены TAP1 (transporter associated with antigen presentation 1) и TAPBP (TAP-associated glycoprotein), участвующие в регуляции экспрессии HLA-I [97]. Экспрессия HLA-I на клеточной поверхности и иммуногенность линий с дефицитом TAP1 и TAPBP была ниже, чем в контрольных линиях клеток при сохранении нормального кариотипа и плюрипотентности.
Для обеспечения адекватной защиты от вирусов необходимо представление вирусных пептидов в комплексе молекул HLA-I, поэтому отсутствие HLA-I существенно повышает риск инфицирования аллогенных трансплантатов, полученных из ПСК. Этот потенциальный риск необходимо будет учитывать в будущем, особенно в случае заболеваний, ассоциированных с вирусной инфекцией. Димерные молекулы HLA-E могут обеспечить некоторую защиту в данных условиях, так как они способны представлять патогенспецифичные пептиды и, возможно, опухолевые пептиды для последующего распознавания цитотоксическими Т-лимфоцитами [96]. Стоит отметить, что NK-клетки являются важным элементом противовирусной и противоопухолевой иммунной защиты, поэтому длительная системная супрессия NK-клеток может существенно повышать риск оппортунистических инфекций и туморогенеза.
Гораздо меньше внимания привлекает репрессия или делеция молекул HLA-II для предотвращения реакции иммунного отторжения, так как экспрессия HLA-II ограничена профессиональными АПК. Тем не менее, аллогенные молекулы HLA-II могут вызывать ответ CD4+ Т-хелперных клеток и активировать, в первую очередь, В-клетки и макрофаги. Известно несколько транскрипционных факторов, которые регулируют экспрессию HLA-II, в том числе RFX, X2BP, NFY и CIITA. Используя технологию геномного редактирования TALEN, делетировали CIITA в ЭСК [98]. Линия ЭСК CIITA–/– сохранила характеристики плюрипотентной клеточной линии. Дифференцированные фибробласты и ДК не экспрессировали HLA-II. CIITA–/– Т-клетки и ДК могут использоваться в клеточной терапии, потому что они способны ускользать от CD4+ Т-клеточного ответа реципиента. Тем не менее, нокаута одного CIITA недостаточно для предупреждения иммунного отторжения. Для достижения максимальной защиты от иммунного ответа лучше использовать двойной нокаут – CIITA и b2m. В декабре 2018 года получены первые линии ИПСК с биалелльным нокаутом b2m и CIITA [99]. b2m/CIITA–/– кардиомиоциты, дифференцированные из ИПСК, не вызывают активацию Т-клеток in vitro, а также, в отличие от изогенного контроля дикого типа, обладают повышенной резистентностью к активности мононуклеарных клеток периферической крови, включая NK-клетки.
Проверена возможность использования CD47 в качестве иммуносупрессирующего агента [87]. С этой целью получены b2m–/– CIITA–/– линии ИПСК, экспрессирующие CD47. Показано формирование тератом при аллогенной трансплантации b2m–/– CIITA–/– Cd47tg+ ИПСК мыши. При этом выживаемость b2m–/– CIITA–/– ИПСК была выше по сравнению с соответствующим контролем. Далее проанализировали иммуногенность дифференцированных производных модифицированной линии. При аллогенной трансплантации мышам с функциональной иммунной системой b2m–/– CIITA–/– Cd47tg+ эндотелиальные клетки, гладкомышечные клетки и кардиомиоциты не вызывали ни иммунного ответа, ни образования антител, в то время как дифференцированные производные дикого типа вызывали острое отторжение трансплантатов. Аналогичный подход применили для получения b2m–/– CIITA–/– Cd47tg+ ИПСК человека c последующей проверкой реакции иммунной системы на гуманизированных мышах. Отмечено отсутствие клеточного иммунного ответа и образования антител. Сделан вывод, что одного CD47 достаточно для “ослепления” иммунной системы реципиента. Следует отметить, что в работе не обсуждается, каким образом CD47 взаимодействует с NK-клетками, так как рецепторы CD47 ‒ SIRPA (SIRPα, CD172a ) и СD61 на NK-клетках не экспрессируются [85, 100]. Продолжительность такой иммунотолерантности пока не установлена. Разовьется ли в отдаленный период (от нескольких месяцев) иммунный ответ на введение аллогенных b2m–/– CIITA–/– Cd47tg+, или же заявленного способа достаточно, и CD47 “выключает” NK-клетки посредством иного механизма? Для ответа на этот вопрос требуются дальнейшие исследования.
Pазработаны и другие стратегии обеспечения иммунной толерантности к аллогенным трансплантатам. Путем повышения экспрессии CTLA4-Ig и PD-L1 получена модифицированная линия СР-ЭСК [44]. Тератомы из СР-ЭСК, а также фибробласты и кардиомиоциты не отторгались после трансплантации гуманизированным мышам в отличие от контрольных линий клеток. В трансплантатах обнаружена существенно менее выраженная Т-клеточная инфильтрация, что позволяет сделать вывод о способности редактированных клеточных линий создавать локальную иммуносупрессивную нишу. Тем не менее, иммуногенность всех перечисленных линий еще предстоит проверить in vivo.
Уменьшение иммуногенности ПСК – это своего рода Дамоклов меч, так как гипоиммуногенность может не только снизить отторжение клеток – производных ПСК, но также увеличить риск образования опухолей. Например, из плюрипотентных клеток, которые могут остаться в трансплантате среди дифференцированных клеток, особенно у реципиентов, получающих иммуносупрессивные препараты, могут сформироваться тератомы. Элиминацию остаточных плюрипотентных клеток из культуры дифференцированных клеток перед трансплантацией можно провести с помощью сортировки клеток, центрифугирования в градиенте плотности или селекции на антибиотике в случае применения соответствующих векторов для создания ИПСК. Следует отметить, что из ПСК и их производных с генетическими аберрациями у модельных животных также могут формироваться злокачественные опухоли, хотя подобные сообщения крайне редки [88].
Возможным методом устранения злокачественных новообразований и тератом может стать введение “суицидальных” кассет в геном ПСК. Группой японских ученых получена линия ЭСК с трансгеном HSV-TK (herpes simplex virus-thymidine kinase) под промотором гена Oct4, экспрессия которого характерна только для плюрипотентных клеток [101]. Селективное удаление недифференцированных ЭСК, экспрессирующих HSV-TK, достигнуто при обработке клеток ганцикловиром. Еще одна стратегия преодоления проблемы туморогенности – использование суицидального гена индуцибельной каспазы-9 (iC9) [102]. Активация iC9 происходит при взаимодействии с химическим активатором димеризации, что инициирует каспазный каскад, который уничтожает iC9-ИПСК, их производные, а также iC9-тератомы in vivo. Стоит отметить, что 94‒99% клеток-мишеней были устранены в течение суток, это делает iC9 перспективным кандидатом на роль суицидального гена для возможного увеличения безопасности ПСК-терапии.
ЗАКЛЮЧЕНИЕ
Рассмотренные в этом обзоре пути и механизмы возникновения иммунотолерантности достаточно многообразны. Какой из них реализуется зачастую зависит от ткани, в которую проводится трансплантация или в которой возникает опухоль, и определяется локальным гомеостазом сигнальных молекул и клеток иммунной системы. Подавление или инактивация даже одного пути, отвечающего за иммунный надзор, может привести к существенному снижению распознавания T- или NK-клетками или к уменьшению выработки цитотоксических антител В-клеточной системой, которую инициируют АПК. При получении универсальной иммунотолерантной линии или линий ИПСК наиболее вероятным видится выключение всех основных звеньев, контролирующих толерантность. Вместе с тем, нельзя сбрасывать со счета и высокий риск туморогенеза или повышенной пролиферации гипоиммуногенных ИПСК и их производных. Подходы к снижению таких рисков только начинают разрабатываться в последние годы, и до применения в клинике таких “универсальных” линий еще предстоят многочисленные исследования их биобезопасности и эффективности на животных, в том числе с гуманизированной иммунной системой.
Работа поддержана Российским научным фондом (№ 17-75-10206).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. (1998) Embryonic stem cell lines derived from human blastocysts. Science. 282, 1145–1147.
Takahashi K., Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126, 663–676.
Sayed N., Liu C., Wu J.C. (2016) Translation of human iPSCs: from clinical trial in a dish to precision medicine. J. Am. Coll. Cardiol. 67, 2161–2176.
Богомазова А.Н., Васина Е.М., Киселев С.Л., Лагарькова М.А., Лебедева О.С., Некрасов Е.Д., Панова А.В., Филоненко Е.С., Хомякова Е.А., Цховребова Л.В., Честков И.В., Шутова М.В. (2015) Генетическое репрограммирование клеток: новая технология для фундаментальных исследований и практического использования. Генетика. 51, 466–478.
Лебедева О.С., Лагарькова М.А. (2018) Плюрипотентные стволовые клетки для моделирования и клеточной терапии болезни Паркинсона. Биохимия. 83, 1046–1056.
Харитонов А.Е., Сурдина А.В., Лебедева О.С., Богомазова А.Н., Лагарькова М.А. (2018) Возможности использования плюрипотетных стволовых клеток для восстановления поврежденного пигментного эпителия сетчатки глаза. Acta Naturae. 10, 30–39.
Neofytou E., O’Brien C.G., Couture L.A, Wu J.C. (2015) Hurdles to clinical translation of human induced pluripotent stem cells. J. Clin. Invest. 125, 2551–2557.
Mandai M., Watanabe A., Kurimoto Y., Hirami Y., Morinaga C., Daimon T., Fujihara M., Akimaru H., Sakai N., Shibata Y., Terada M., Nomiya Y., Tanishima S., Nakamura M., Kamao H., Sugita S., Onishi A., Ito T., Fujita K., Kawamata S., Go M.J., Shinohara C., Hata K.I., Sawada M., Yamamoto M., Ohta S., Ohara Y., Yoshida K., Kuwahara J., Kitano Y., Amano N., Umekage M., Kitaoka F., Tanaka A., Okada C., Takasu N., Ogawa S., Yamanaka S., Takahashi M. (2017) Autologous induced stem-cell–derived retinal cells for macular degeneration. N. Engl. J. Med. 376, 1038–1046.
Martin U. (2017) Therapeutic application of pluripotent stem cells: challenges and risks. Front. Med. 4, 229.
Bolton E.M., Bradley J.A. (2015) Avoiding immunological rejection in regenerative medicine. Regen. Med. 10, 287–304.
de Almeida P.E., Ransohoff J.D., Nahid M.A., Wu J.C. (2013) Immunogenicity of pluripotent stem cells and their derivatives. Circulation Res. 112, 549–561.
Drukker M., Katchman H., Katz G., Even-Tov Friedman S., Shezen E., Hornstein E., Mandelboim O., Reisner Y., Benvenisty N. (2006) Human embryonic stem cells and their differentiated derivatives are less susceptible to immune rejection than adult cells. Stem Cells. 24, 221–229.
Swijnenburg R.J., Schrepfer S., Cao F., Pearl J.I., Xie X., Connolly A.J., Robbins R.C., Wu J.C. (2008) In vivo imaging of embryonic stem cells reveals patterns of survival and immune rejection following transplantation. Stem Cells Dev. 17, 1023–1029.
Jaffe L., Robertson E.J., Bikoff E.K. (1991) Distinct patterns of expression of MHC class I and beta 2-microglobulin transcripts at early stages of mouse development. J. Immunol. 147, 2740–2749.
Drukker M., Katz G., Urbach A., Schuldiner M., Markel G., Itskovitz-Eldor J., Reubinoff B., Mandelboim O., Benvenisty N. (2002) Characterization of the expression of MHC proteins in human embryonic stem cells. Proc. Natl. Acad. Sci. USA. 99, 9864–9869.
Perez-Cunningham J., Ames E., Smith R.C., Pe-ter A.K., Naidu R., Nolta J.A., Murphy W.J. (2014) Natural killer cell subsets differentially reject embryonic stem cells based on licensing. Transplantation. 97, 992–998.
Li L., Baroja M.L., Majumdar A., Chadwick K., Rouleau A., Gallacher L., Ferber I., Lebkowski J., Martin T., Madrenas J., Bhatia M. (2004) Human embryonic stem cells possess immune-privileged properties. Stem Cells. 22, 448–456.
Dhodapkar K.M., Feldman D., Matthews P., Radfar S., Pickering R., Turkula S., Zebroski H., Dhodapkar M.V. (2010) Natural immunity to pluripotency antigen OCT4 in humans. Proc. Natl. Acad. Sci. USA. 107, 8718–8723.
Robertson N.J., Brook F.A., Gardner R.L., Cobbold S.P., Waldmann H., Fairchild P.J. (2007) Embryonic stem cell-derived tissues are immunogenic but their inherent immune privilege promotes the induction of tolerance. Proc. Natl. Acad. Sci. USA. 104, 20920–20925.
Zhao T., Zhang Z.N., Rong Z., Todorova D., Hu Z., Lin T., Rong Z., Kim J., He J., Wang M., Clegg D.O., Yang Y.G9. Zhang K., Friedlander M., Xu Y. (2011) Immunogenicity of induced pluripotent stem cells. Nature. 474, 212–215.
de Almeida P.E., Meyer E.H., Kooreman N.G., Diecke S., Dey D., Sanchez-Freire V., Hu S., Ebert A., Odegaard J., Mordwinkin N.M., Brouwer T.P., Lo D., Montoro D.T., Longaker M.T., Negrin R.S., Wu J.C. (2014) Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat. Commun. 5, 3903.
Guha P., Morgan J.W., Mostoslavsky G., Rodrigues N.P., Boyd A.S. (2013) Lack of immune response to differentiated cells derived from syngeneic induced pluripotent stem cells. Cell Stem Cell. 12, 407–412.
Araki R., Uda M., Hoki Y., Sunayama M., Nakamura M., Ando S., Sugiura M., Ideno H., Shimada A., Nifuji A., Abe M. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature. 494, 100–104.
Zhao T., Zhang Z.N., Westenskow P.D., Todorova D., Hu Z., Lin T., Rong Z., Kim J., He J., Wang M., Clegg D.O., Yang Y.G., Zhang K., Friedlander M., Xu Y. (2015) Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell. 17, 353–359.
Kaneko S., Yamanaka S. (2013) To be immunogenic, or not to be: that’s the iPSC question. Cell Stem Cell. 12, 385–386.
Morizane A., Doi D., Kikuchi T., Okita K., Hotta A., Kawasaki T., Hayashi T., Onoe H., Shiina T., Yamanaka S., Takahashi J. (2013) Direct comparison of autologous and allogeneic transplantation of iPSC-derived neural cells in the brain of a nonhuman primate. Stem Cell Rep. 1, 283–292.
Polo J.M., Liu S., Figueroa M.E., Kulalert W., Eminli S., Tan K.Y., Apostolou E., Stadtfeld M., Li Y., Shioda T., Natesan S., Wagers A.J., Melnick A., Evans T., Hochedlinger K. (2010) Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 28, 848–855.
Shutova M.V., Surdina A.V., Ischenko D.S., Naumov V.A., Bogomazova A.N., Vassina E.M., Alekseev D.G., Lagarkova M.A., Kiselev S.L. (2016) An integrative analysis of reprogramming in human isogenic system identified a clone selection criterion. Cell Cycle. 15, 986–997.
Wang X., Qin J., Zhao R.C., Zenke M. (2014) Reduced immunogenicity of induced pluripotent stem cells derived from Sertoli cells. PLoS One. 9, e106110.
Liu P., Chen S., Li X., Zenke M. (2013) Low immunogenicity of neural progenitor cells differentiated from induced pluripotent stem cells derived from less immunogenic somatic cells. PLoS One. 8, e69617.
Liu X., Li W., Fu X., Xu Y. (2017) The immunogenicity and immune tolerance of pluripotent stem cell derivatives. Front. Immunol. 8, 645.
Bhutani K., Nazor K.L., Williams R., Tran H., Dai H., Džakula Ž., Cho E.H., Pang A.W., Rao M., Cao H., Schork N.J., Loring J.F. (2016) Whole-genome mutational burden analysis of three pluripotency induction methods. Nat. Commun. 7, 10536.
Hanna J., Saha K., Jaenisch R. (2010) Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 143, 508–525.
Robinson D.A., Daley G.Q. (2012) The promise of induced pluripotent stem cells in research and therapy. Nature. 481, 295–305.
Taylor C.J., Peacock S., Chaudhry A.N., Bradley J.A., Bolton E.M. (2012) Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA Types. Cell Stem Cell. 11, 147–152.
Solomon S., Pitossi F., Rao M.S. (2015) Banking on iPSC- is it doable and is it worthwhile. Stem Cell Rev. 11, 1–10.
Nakatsuji N., Nakajima F., Tokunaga K. (2008) HLA-haplotype banking and iPS cells. Nat. Biotechnol. 26, 739–740.
Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K., Shibata T., Kunisada T., Takahashi M., Takahashi J., Saji H., Yamanaka S. (2011) A more efficient method to generate integration-free human iPS cells. Nat. Methods. 8, 409–412.
Gourraud P.-A., Gilson L., Girard M., Peschanski M. (2012) The role of human leukocyte antigen matching in the development of multiethnic “haplobank” of induced pluripotent stem cell lines. Stem Cells. 30, 180–186.
Moffett A., Colucci F. (2015) Co-evolution of NK receptors and HLA ligands in humans is driven by reproduction. Immunol. Rev. 267, 283–297.
Ichise H., Nagano S., Maeda T., Miyazaki M., Miyazaki Y., Kojima H., Yawata N., Yawata M., Tanaka H., Saji H., Masuda K., Kawamoto H. (2017) NK cell alloreactivity against KIR-ligand-mismatched HLA-haploidentical tissue derived from HLA haplotype-homozygous iPSCs. Stem Cell Rep. 9, 853–867.
Wood K.J., Goto R. (2012) Mechanisms of rejection: current perspectives. Transplantation. 93, 1–10.
de Charette M., Houot R. (2018) Hide or defend, the two strategies of lymphoma immune evasion: potential implications for immunotherapy. Haematologica. 103, 1256–1268.
Rong Z., Wang M., Hu Z., Stradner M., Zhu S., Kong H., Yi H., Goldrath A., Yang Y.G., Xu Y., Fu X. (2014) An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell. 14, 121–130.
Menter T., Tzankov A. (2018) Mechanisms of immune evasion and immune modulation by lymphoma cells. Front. Oncol. 8, 54.
Stewart T.J., Abrams S.I. (2008) How tumours escape mass destruction. Oncogene. 27, 5894–5903.
Garrido F., Aptsiauri N., Doorduijn E.M, Garcia Lora A.M., van Hall T. (2016) The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 39, 44–51.
Garrido F., Cabrera T., Aptsiauri N. (2010) “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer. 127, 249–256.
Garrido C., Romero I., Berruguilla E., Cancela B., Algarra I., Collado A., García-Lora A., Garrido F. (2011) Immunotherapy eradicates metastases with reversible defects in MHC class I expression. Cancer Immunol. Immunother. 60, 1257–1268.
Angell T.E., Lechner M.G., Jang J.K., LoPresti J.S., Epstein A.L. (2014) MHC class I loss is a frequent mechanism of immune escape in papillary thyroid cancer that is reversed by interferon and selumetinib treatment in vitro. Clin. Cancer Res. 20, 6034–6044.
Li L., Dong M., Wang X.G. (2016) The implication and significance of beta 2 microglobulin: a conservative multifunctional regulator. Chin. Med. J. 129, 448–455.
Bernal M., Ruiz-Cabello F., Concha A., Paschen A., Garrido F. (2012) Implication of the β2-microglobulin gene in the generation of tumor escape phenotypes. Cancer Immunol. Immunother. 61, 1359–1371
Steidl C., Shah S.P., Woolcock B.W., Rui L., Kawahara M., Farinha P., Johnson N.A., Zhao Y., Telenius A., Neriah S.B., McPherson A., Meissner B., Okoye U.C., Diepstra A., van den Berg A., Sun M., Leung G., Jones S.J., Connors J.M., Huntsman D.G., Savage K.J., Rimsza L.M., Horsman D.E., Staudt L.M., Steidl U., Marra M.A., Gascoyne R.D. (2011) MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 471, 377–381.
Morandi F., Rizzo R., Fainardi E., Rouas-Freiss N., Pistoia V. (2016) Recent advances in our understanding of HLA-G biology: lessons from a wide spectrum of human diseases. J. Immunol. Res. 2016, 4326495.
Rouas-Freiss N., Moreau P., LeMaoult J., Carosella E.D. (2014) The dual role of HLA-G in cancer. J. Immunol. Res. 2014, 359748.
Le Bouteiller P. (2015) HLA-G in human early pregnancy: control of uterine immune cell activation and likely vascular remodeling. Biomed. J. 38, 32–38.
González A., Rebmann V., LeMaoult J, Horn P.A., Carosella E.D., Alegre E. (2012) The immunosuppressive molecule HLA-G and its clinical implications. Crit. Rev. Clin. Lab. Sci. 49, 63–84.
Pistoia V., Morandi F., Wang X., Ferrone S. (2007) Soluble HLA-G: are they clinically relevant? Semin. Cancer Biol. 17, 469–479.
Le Page M.E., Goodridge J.P., John E., Christiansen F.T., Witt C.S. (2014) Killer Ig-like receptor 2DL4 does not mediate NK cell IFN-γ responses to soluble HLA-G preparations. J. Immunol. 192, 732–740.
Fons P., Chabot S., Cartwright J.E., Lenfant F., L’Faqihi F., Giustiniani J., Herault J.P., Gueguen G., Bono F., Savi P., Aguerre-Girr M., Fournel S., Malecaze F., Bensussan A., Plouët J., Le Bouteiller P. (2006) Soluble HLA-G1 inhibits angiogenesis through an apoptotic pathway and by direct binding to CD160 receptor expressed by endothelial cells. Blood. 108, 2608–2615.
Nakajima H., Asai A., Okada A., Ping L., Hamajima F., Sata T., Isobe K. (2003) Transcriptional regulation of ILT family receptors. J. Immunol. 171, 6611–6620.
Ogasawara K., Lanier L.L. (2005) NKG2D in NK and T cell-mediated immunity. J. Clin Immunol. 25, 534–540.
Wu J., Song Y., Bakker A.B., Bauer S., Spies T., La-nier L.L., Phillips J.H. (1999) An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 285, 730–732.
Verneris M.R., Karami M., Baker J., Jayaswal A., Negrin R.S. (2004) Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. 103, 3065–3072.
Zhang J., Basher F., Wu J.D. (2015) NKG2D ligands in tumor immunity: two sides of a coin. Front Immunol. 6, 97.
Schmiedel D., Mandelboim O. (2018) NKG2D ligands – critical targets for cancer immune escape and therapy. Front. Immunol. 9, 2040.
Salih H.R., Rammensee H.G., Steinle A. (2002) Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 169, 4098–4102.
Waldhauer I., Steinle A. (2006) Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. 66, 2520–2526.
Salih H.R., Goehlsdorf D., Steinle A. (2006) Release of MICB molecules by tumor cells: mechanism and soluble MICB in sera of cancer patients. Hum Immunol. 67, 188–195.
Fernandez-Messina L., Ashiru O., Boutet P., Agüera-González S., Skepper J.N., Reyburn H.T., Valés-Gómez M. (2010) Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J. Biol. Chem. 285, 8543–8551.
Ashiru O., Boutet P., Fernandez-Messina L., Agüera-González S., Skepper J.N., Valés-Gómez M., Reyburn H.T. (2010) Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. 70, 481–489.
Whiteside T.L. (2013) Immune modulation of t-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 41, 245–251.
Groh V., Wu J, Yee C, Spies T. (2002) Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 419, 734–738.
Sick E., Jeanne A., Schneider C., Dedieu S., Takeda K., Martiny L. (2012). CD47 update: a multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br. J. Pharmacol. 167, 1415–1430.
Chao M.P, Weissman I.L, Majeti R. (2012). The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 24, 225–232.
Shahan T.A, Fawzi A., Bellon G., Monboisse J.C, Kefalides N.A (2000). Regulation of tumor cell chemotaxis by type IV collagen is mediated by a Ca(2+)-dependent mechanism requiring CD47 and the integrin alpha(V)beta(3)”. J. Biol. Chem. 2757, 4796–4802.
Uluçkan O., Becker S.N., Deng H., Zou W., Prior J.L., Piwnica-Worms D., Frazier W.A., Weilbaecher K.N. (2009). CD47 regulates bone mass and tumor metastasis to bone. Cancer Res. 69, 3196–3204.
Chao M.P., Tang C., Pachynski R.K., Chin R., Majeti R., Weissman I.L. (2011) Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood. 118, 4890–4901.
Edris B., Weiskopf K., Volkmer A.K, Volkmer J.P., Willingham S.B., Contreras-Trujillo H., Liu J., Majeti R., West R.B., Fletcher J.A., Beck A.H., Weissman I.L., van de Rijn M. (2012) Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA. 109, 6656–6661.
Willingham S.B., Volkmer J.P., Gentles A.J., Sahoo D., Dalerba P., Mitra S.S., Wang J., Contreras-Trujillo H., Martin R., Cohen J.D., Lovelace P., Scheeren F.A., Chao M.P., Weiskopf K., Tang C., Volkmer A.K., Naik T.J., Storm T.A., Mosley A.R., Edris B., Schmid S.M., Sun C.K., Chua M.S., Murillo O., Rajendran P., Cha A.C., Chin R.K., Kim D., Adorno M., Raveh T., Tseng D., Jaiswal S., Enger P.Ø., Steinberg G.K., Li G., So S.K., Majeti R., Harsh G.R., van de Rijn M., Teng N.N., Sun-woo J.B., Alizadeh A.A., Clarke M.F., Weissman I.L. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA. 109, 6662–6667.
Tseng D., Volkmer J.P., Willingham S.B., Contreras-Trujillo H., Fathman J.W., Fernhoff N.B., Seita J., Inlay M.A., Weiskopf K., Miyanishi M., Weissman I.L. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA. 110, 11103–11108.
Unanue E. R. (2013) Perspectives on anti-CD47 antibody treatment for experimental cancer. Proc. Nat. Acad. Sci. USA. 110, 10886–10887.
Matlung H.L, Szilagyi K., Barclay N.A, van den Berg T.K. (2017) The CD47-SIRP alpha signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 276, 145–164.
Azcutia V., Stefanidakis M., Tsuboi N., Mayadas T., Croce K.J., Fukuda D., Aikawa M., Newton G., Luscinskas F.W. (2012) Endothelial CD47 promotes vascular endothelial-cadherin tyrosine phosphorylation and participates in T cell recruitment at sites of inflammation in vivo. J. Immunol. 189, 2553–2562.
Kim M.J., Lee J.C, Lee J.J, Kim S., Lee S.G., Park S.W., Sung M.W., Heo D.S. (2008) Association of CD47 with natural killer cell-mediated cytotoxicity of head-and-neck squamous cell carcinoma lines. Tumor Biol. 29, 28–34.
Soto-Pantoja D.R, Terabe M., Ghosh A., Ridnour L.A., DeGraff W.G., Wink D.A., Berzofsky J.A., Roberts D.D. (2014) CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res. 74, 6771–6783.
Deuse T., Hu X., Gravina A., Wang D., Tediashvili G., De C., Thayer W.O., Wahl A., Garcia J.V., Reichenspurner H., Davis M.M., Lanier L.L., Schrepfer S. (2019) Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 37(3), 252‒258.
Zheng D., Wang X., Xu R.-H. (2016) Concise review: one stone for multiple birds: generating universally compatible human embryonic stem cells. Stem Cells. 34, 2269–2275.
Deuse T., Seifert M., Phillips N., Tsao P.S., Hua X., Velden J., Eiermann T., Volk H.D., Reichenspurner H., Robbins R.C., Schrepfer S. (2011) Immunobiology of naïve and genetically modified HLA-class-I-knockdown human embryonic stem cells. J. Cell Sci. 124, 3029–3037.
Torikai H., Reik A., Soldner F., Warren E.H., Yuen C., Zhou Y., Crossland D.L., Huls H., Littman N., Zhang Z., Tykodi S.S., Kebriaei P., Lee D.A., Mil-ler J.C., Rebar E.J., Holmes M.C., Jaenisch R., Cham-plin R.E., Gregory P.D., Cooper L.J. (2013) Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 122, 1341–1349.
Riolobos L., Hirata R.K., Turtle C.J., Wang P.R., Gornalusse G.G., Zavajlevski M., Riddell S.R., Russell D.W. (2013) HLA engineering of human pluripotent stem cells. Mol. Ther. 21, 1232–1241.
Lu P., Chen J., He L., Ren J., Chen H., Rao L., Zhuang Q., Li H., Li L., Bao L., He J., Zhang W., Zhu F., Cui C., Xiao L. (2013) Generating hypoimmunogenic human embryonic stem cells by the disruption of beta 2-microglobulin. Stem Cell Rev. 9, 806–813.
Wang D., Quan Y., Yan Q., Morales J.E., Wetsel R.A. (2015) Targeted disruption of the β2-microglobulin gene minimizes the immunogenicity of human embryonic stem cells. Stem Cells Transl. Med. 4, 1234–1245.
Bogomiakova M.E., Bobrovsky P.A., Zhukova Y.N., Lazarev V.N., Lagarkova M.A. (2018) Derivation and characterization of induced pluripotent stem cells lines with inactivation of the beta-2-microglobulin gene by CRISPR/Cas9 genome editing. FEBS Open Bio. 8, 152–153.
Zhao L., Teklemariam T., Hantash B.M. (2014) Heterelogous expression of mutated HLA-G decreases immunogenicity of human embryonic stem cells and their epidermal derivatives. Stem Cell Res. 13, 342–354.
Gornalusse G.G., Hirata R.K., Funk S., Riolobos L., Lopes V.S., Manske G., Prunkard D., Colunga A.G., Hanafi L.A., Clegg D.O., Turtle C., Russell D.W. (2017) HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 35, 765–772.
Cui D., Wang J., Zeng Y., Rao L., Chen H., Li W., Li Y., Li H., Cui C., Xiao L. (2016) Generating hESCs with reduced immunogenicity by disrupting TAP1 or TAPBP. Biosci. Biotechnol. Biochem. 80, 1484–1491.
Chen H., Li Y., Lin X., Cui D., Cui C., Li H., Xiao L. (2015) Functional disruption of human leukocyte antigen II in human embryonic stem cell. Biol. Res. 48, 59.
Matapally S., Pawlik K.M, Fast V.G, Zumaquero E., Lund F.E., Randall T.D., Townes T.M., Zhang J. (2018) Human Leukocyte Antigen class I and II knockout human induced pluripotent stem cell-derived cells: universal donor for cell therapy. J. Am. Heart Assoc. 7, e010239.
Nath P.R., Gangaplara A., Pal-Nath D., Mandal A., Maric D., Sipes J.M., Cam M., Shevach E.M., Roberts D.D. (2018) CD47 expression in natural killer cells regulates homeostasis and modulates immune response to lymphocytic choriomeningitis virus. Front Immunol. 9, 2985.
Hara A., Aoki H., Taguchi A., Niwa M., Yamada Y., Kunisada T., Mori H. (2008) Neuron-like differentiation and selective ablation of undifferentiated embryonic stem cells containing suicide gene with Oct-4 promoter. Stem Cells Dev. 17, 619–627.
Yagyu S., Hoyos V., Del Bufalo F., Brenner M.K. (2015) An inducible caspase-9 suicide gene to improve the safety of therapy using human induced pluripotent stem cells. Mol. Ther. 23, 1475–1485.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология