

Молекулярная биология, 2019, T. 53, № 6, стр. 911-923
3D геномика
С. В. Разин a, b, *, С. В. Ульянов a, b, А. А. Гаврилов a
a Институт биологии гена Российской академии наук
119334 Москва, Россия
b Биологический факультет Московского государственного университета им. М.В. Ломоносова
119991 Москва, Россия
* E-mail: sergey.v.razin@usa.net
Поступила в редакцию 17.04.2019
После доработки 01.06.2019
Принята к публикации 03.06.2019
Аннотация
Развитие новых методов исследования существенно изменило наши взгляды на роль 3D организации генома в реализации его функциональной активности. Показано, что на уровне пространственной организации геном разделяется на структурно-функциональные блоки, ограничивающие сферу работы энхансеров. Стало ясно, что пространственная реконфигурация протяженного сегмента генома может быть механизмом, обеспечивающим активацию либо репрессию расположенных в нем генов. Соответственно, изменения 3D организации генома часто приводят к возникновению различных заболеваний, в том числе онкологических. Все эти наблюдения способствовали возникновению нового научного направления – 3D геномики. В настоящем обзоре мы суммируем наиболее важные открытия в области 3D геномики и обсуждаем дальнейшие пути развития этого научного направления.
Хорошо известно, что ДНК является линейной молекулой. Соответственно, расстояния между различными геномными элементами часто измеряют в парах нуклеотидов, указывая длину фрагмента ДНК, разделяющего эти элементы. Все существующие геномные браузеры устроены таким образом, что показывают позиции тех или иных регионов генома на линейной ДНК. Между тем геномные элементы, находящиеся далеко друг от друга на молекуле ДНК, могут оказаться рядом благодаря определенному способу укладки ДНК внутри клеточного ядра. С использованием техники флуоресцентной гибридизации in situ (FISH) показано, что гены, находящиеся на разных хромосомах, могут привлекаться к общим транскрипционным фабрикам [1, 2]. Показано также, что энхансеры и контролируемые ими гены располагаются рядом в объеме клеточного ядра даже в том случае, когда разделяющий их сегмент ДНК составляет сотни тысяч пар нуклеотидов [3]. Развитие методов исследования, основанных на процедуре лигирования близкорасположенных фрагментов ДНК (так называемых С-методов) [4], позволило изучать пространственные контакты регуляторных элементов в рамках всего генома [5, 6]. В результате использования наиболее распространенного высокопроизводительного Hi-C-метода выявлены многочисленные функционально-значимые пространственные контакты между удаленными элементами генома [7, 8]. Стало ясно, что простая реконфигурация протяженной области генома, обеспечивающая сближение промоторов с энхансерами и cуперэнхансерами, может быть причиной активации транскрипции [9, 10]. Также установлено, что в геноме существуют структурно-функциональные домены, ограничивающие сферу действия энхансеров [11]. Нарушение границ этих доменов, приводящее к их слиянию, оказалось причиной возникновения различных заболеваний [12‒15]. В то же время трехмерная организация генома может служить мишенью для противоопухолевых агентов. Так, перспективные противоопухолевые агенты кураксины подавляют работу онкогенов, нарушая пространственные контакты между промоторами и энхансерами [16].
3D геномика возникла в начале XXI века, когда стало ясно, что на уровне макроукладки генома в ядре работают эпигенетические регуляторные механизмы. С учетом этого удалось разрешить ряд накопившихся противоречий, например, объяснить, каким образом функционально связанные эпигенетические домены (сегменты генома, содержащие определенные модификации гистонов и обогащенные теми или иными негистоновыми белками) оказываются разнесенными на молекуле ДНК [17]. Так, анализ профиля ацетилирования гистонов на линейном геноме позволяет констатировать, что высокий уровень ацетилирования характерен для работающих генов и контролирующих их работу энхансеров, тогда как разделяющие их сегменты генома характеризуются существенно более низким уровнем ацетилирования гистонов. Если бы ацетилирование гистонов распространялось процессивно вдоль хроматиновой фибриллы посредством привлечения гистонацетилаз к уже ацетилированным гистонам, то формирующийся домен, скорее всего, был непрерывным. Однако в компактной хроматиновой глобуле ацетилирование будет распространяться на все участки фибриллы, находящиеся в непосредственной близости, даже в том случае, если они разделены протяженным выпетленным участком. Те эпигенетические домены, которые на линейном геноме выглядят, как разорванные, в действительности составляют единый трехмерный домен [17, 18]. 3D геномика ‒ одно из наиболее активно развивающихся направлений современной молекулярной биологии. В фокусе исследований находятся механизмы формирования и поддержания структурно-функциональных доменов генома, механизмы установления промотор-энхансерных взаимодействий, способы влияния на пространственную организацию генома с помощью различных низкомолекулярных агентов. Трендом времени является переход от клеточных популяций к индивидуальным клеткам, в том числе к оценке динамичности и пластичности 3D организации генома. Это представляется особенно важным для выявления причин возникновения таких редких событий, как нарушение нормальной работы регуляторных механизмов в отдельных клетках, приводящее к возникновению клонов опухолевых клеток. В этом обзоре мы попытаемся проанализировать наиболее важные результаты и концепции 3D геномики.
ОБЩИЕ ПРИНЦИПЫ УПАКОВКИ ХРОМАТИНОВОЙ ФИБРИЛЛЫ В КЛЕТОЧНОМ ЯДРЕ
В течение нескольких десятилетий общепринятыми были представления об иерархической упаковке ДНК в хроматиновые структуры различных уровней компактизации, включая 10 и 30 нм нуклеосомные фибриллы и ряд иерархических соленоидов, либо петли 30 нм фибриллы [19]. Результаты работ последних лет поставили эту модель под сомнение. С использованием электронной микроскопии и компьютерной томографии в клеточном ядре не были обнаружены никакие регулярные структуры, кроме 10 нм фибриллы [20‒22]. Эти 10 нм фибриллы образуют компактные массы, стабилизированные электростатическими взаимодействиями между нуклеосомами соседних фибрилл (так называемые “клатчи” [23]). Компактность этих масс негативно коррелирует с транскрипционным статусом и может регулироваться посредством модификаций гистонов и/или включения в нуклеосомы вариантных гистонов [24‒27]. Микроскопические исследования с более низким разрешением выявляют в интерфазном ядре относительно обособленные хромосомные территории, построенные из хроматиновых доменов размером 1 млн.п.н. и кластеров таких доменов [28‒31]. Полногеномный анализ профилей пространственных контактов различных участков хроматиновой фибриллы также свидетельствует о том, что геномная ДНК организована в относительно обособленные контактные домены, размер которых составляет ~1 млн.п.н. у млекопитающих и ~100 т.п.н. у дрозофилы [32‒34]. Эти структуры обычно называют топологически-ассоциированными доменами, или ТАДами [35]. Результаты Hi-C-анализа принято представлять в виде тепловых карт пространственных взаимодействий. На таких картах ТАДы имеют вид треугольников (рис. 1а), внутри которых часто можно видеть вложенные треугольники, отражающие существование нескольких иерархических уровней контактных доменов (рис. 1б). При проведении анализа с более высоким разрешением удается различить два типа контактных доменов. Одна группа представляет собой петли хроматина, о чем можно судить на основании предпочтительного взаимодействия между концами домена. На тепловых картах это взаимодействие выглядит как более яркий сигнал на вершине треугольника (pис. 1в). Другая группа контактных доменов представлена доменами, которые не организованы в петли. Их предложено называть “обычными доменами” (ordinary domains) [6]. С использованием флуоресцентной гибридизации in situ с ТАД-специфичными пробами показано, что ТАДы представляют собой относительно компактные хроматиновые глобулы, которые частично разворачиваются в том случае, когда ТАД содержит активные гены [36]. Компьютерное моделирование, выполненное на основании результатов Hi-C-анализа индивидуальных клеток, показывает, что форма ТАДа достаточно пластична и может изменяться в широких пределах [37].
Рис. 1.
Варианты паттернов Hi-C-карт (сверху) и их интерпретация (снизу). а ‒ Обособленные треугольники на Hi-C-карте обычно интерпретируют как хроматиновые глобулы. б ‒ Объединение мелких треугольников в более крупные может соответствовать иерархической организации глобул (мелкие глобулы, сливающиеся в более крупные). в ‒ Яркий сигнал в вершине треугольника свидетельствует о взаимодействии границ ТАДа с формированием хроматиновой петли, внутренние участки которой могут также взаимодействовать между собой с формированием глобулы. г ‒ Рисунок типа “шахматная доска” свидетельствует о формировании хроматиновых компартментов.
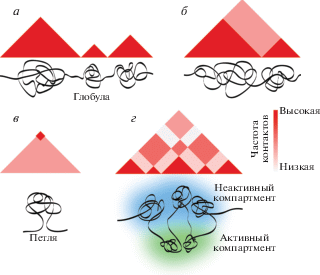
Хотя основным свойством контактных доменов считается существенно большая частота контактов внутри домена, чем между доменами, однако последние все же возможны. При этом ТАДы, содержащие активный хроматин, предпочтительно контактируют друг с другом, но не с ТАДами с неактивным хроматином. Обратное тоже справедливо. ТАДы с неактивным хроматином, чаще контактируют друг с другом, чем с ТАДами, содержащими активный хроматин. Это позволяет говорить о том, что в клеточном ядре существует пространственная сегрегация активного и неактивного хроматиновых компартментов. На контактных тепловых картах взаимодействие между компартментами напоминает шахматную доску (рис. 1г).
ТОПОЛОГИЧЕСКИ-АССОЦИИРОВАННЫЕ ДОМЕНЫ ЯВЛЯЮТСЯ СТРУКТУРНО-ФУНКЦИОНАЛЬНЫМИ БЛОКАМИ ГЕНОМА
Идея о том, что геном подразделяется на относительно независимые структурно-функциональные блоки, была высказана около 30 лет назад в контексте осмысления результатов экспериментов по характеристике предпочтительной чувствительности протяженных участков генома к нуклеазам [38, 39]. Модель стимулировала поиск регуляторных элементов, контролирующих транскрипционный статус протяженных участков генома (доменов) в целом и ограничивающих домены. Такие регуляторные элементы (области контроля локуса и инсуляторы соответственно) действительно обнаружены [40‒44]. В то же время, в дальнейшем показали, что тканеспецифичные гены часто располагаются в тех же сегментах генома, где и постоянно экспрессирующиеся гены “домашнего хозяйства”. Такую ситуацию трудно объяснить в рамках доменной модели организации генома. Действительно, эта модель постулирует, что домен в целом может быть либо активен, либо неактивен. Однако оказалось, что комплекс регуляторных элементов в кластерах эритроидспецифичных альфа-глобиновых генов расположен внутри гена домашнего хозяйства, находящегося рядом с кластером [45, 46]. Такие ситуации, которые сначала рассматривали как исключительные, оказались, напротив, весьма типичными [47, 48]. Доменная модель организации генома была в этой связи забыта и стала вновь востребованной только после появления 3D геномики.
В геноме высших эукариот транскрипция контролируется промоторами, расположенными в начале транскрипционных единиц, и энхансерами, которые могут находиться на значительном расстоянии от промоторов. Число энхансеров в разы превосходит число известных генов [49, 50]. В этой связи есть основания полагать, что в разных ситуациях работа одного и того же гена может контролироваться разными комбинациями энхансеров. Механизм действия энхансеров до сих пор остается предметом дискуссий [51]. Согласно наиболее популярной в настоящее время модели, связанные с энхансером белки, в том числе факторы ремоделирования хроматина, транскрипционные факторы, РНК-полимераза и медиатор, способствуют формированию “жидкого” активаторного компартмента, возникающего в силу разделения жидких фаз. Последнее происходит, скорее всего, благодаря взаимодействию неструктурированных доменов связанных с энхансером белков [52‒55]. Жидкие компартменты до известной степени похожи на капли жира, суспендированные в водной среде. Эти компартменты собираются вокруг определенной “платформы”, сродством к которой обладают все или значительная часть макромолекул, составляющих жидкий компартмент. В случае энхансерного компартмента роль платформы могут выполнять связанные с энхансером транскрипционные факторы, медиатор и некоторые другие компоненты транскрипционной машины, а также энхансерная РНК. Чтобы энхансер мог стимулировать транскрипцию контролируемого им гена, этот ген должен оказаться внутри созданного вокруг энхансера активаторного компартмента. Понятно, что в тех случаях, когда ген не находится рядом с энхансером на молекуле ДНК, это возможно лишь посредством выпетливания разделяющего энхансер и промотор сегмента хроматиновой фибриллы (рис. 2а, б). Эта организация допускает участие в сборке активаторного компартмента одновременно нескольких энхансеров, равно как и одновременную активацию энхансером нескольких генов (рис. 2в, г). Такие ситуации хорошо известны. Можно напомнить хотя бы о том, что начало 3D геномике положили работы по изучению активаторного блока домена бета-глобиновых генов мыши, к которому привлекаются несколько генов, работающих на определенной стадии развития [56, 57]. Сходные наблюдения сделаны и при изучении пространственной организации как отдельных геномных областей, так и генома в целом [8, 58‒63].
Рис. 2.
Модель жидкостного активаторного компартмента, формируемого вокруг энхансера за счет разделения фаз. а ‒ Если энхансер расположен близко от гена в линейной последовательности ДНК, то попадание промотора гена в активаторный компартмент возможно без выпетливания участка ДНК между промотором и энхансером. б ‒ Если энхансер расположен на значительном расстоянии от гена, то попадание промотора гена в активаторный компартмент требует выпетливания участка ДНК между промотором и энхансером. в ‒ Активация гена двумя энхансерами. г ‒ Активация двух генов одним энхансером.
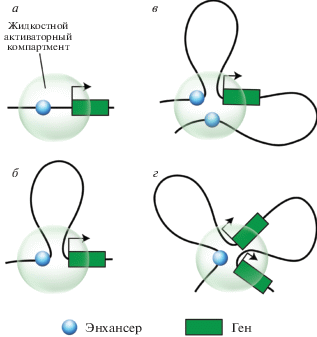
Если для активации гена действительно необходим контакт промотора с энхансером, то границы ТАДов, препятствующие установлению пространственных контактов, должны ограничивать сферу действия энхансеров. Существующие экспериментальные наблюдения подтверждают эту точку зрения. С использованием различных подходов показано, что в геноме существуют функциональные домены, в границах которых энхансер может активировать различные, в том числе и интегрированные в этот домен, репортерные гены. Такие функциональные домены получили названия “регуляторные домены” (regulatory domains) [64], “регуляторные архипелаги” (regulatory archipelagoes) [65, 66], “инсулированные окрестности” (insulated neigbourhoods) [67‒69]. Существенно, что границы этих функциональных доменов колокализуются с границами ТАДов и хроматиновых петель [64, 69, 70]. Более того, в ряде случаев прямо показано, что нарушение границ ТАДов изменяет профиль энхансер-промоторных контактов и профиль транскрипции [12‒14, 68]. Таким образом, можно утверждать, что ТАДы это структурно-функциональные домены генома. Об этом косвенно свидетельствует и тот факт, что ТАДы достаточно стабильны в эволюции. Сравнение геномов различных позвоночных животных показало, что сохранившиеся в эволюции хромосомные перестройки происходили, как правило, по границам ТАДов. Соответственно, ТАДы “перетасовывались”, но не перемешивались в перестроенных геномах [71].
МЕХАНИЗМЫ ФОРМИРОВАНИЯ И ИНСУЛЯЦИИ КОНТАКТНЫХ ДОМЕНОВ ХРОМАТИНА
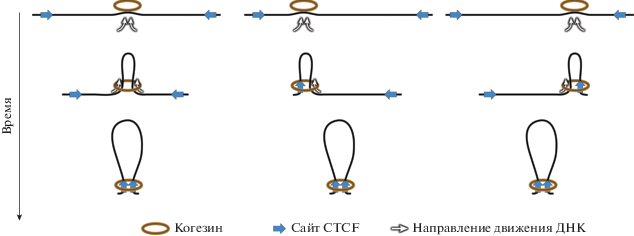
Учитывая роль контактных доменов хроматина (ТАДов) в определении сферы действия энхансеров, чрезвычайно важно понять, как достигается их инсуляция. Сопоставление профилей ТАДов в клетках млекопитающих с профилями связывания различных негистоновых белков показало, что границы ТАДов существенно обогащены сайтами связывания инсуляторного белка CTCF, когезина и топоизомеразы 2β [33, 72‒75]. Также оказалось, что индуцированная деградация CTCF приводит к утрате петлевых доменов хроматина и нарушению инсуляции ТАДов [76]. На уровне индивидуальных доменов генома аналогичные эффекты наблюдали при удалении сайтов связывания CTCF [77, 78]. СТСF ‒ мультифункциональный белок, впервые обнаруженный как транскрипционный фактор, контролирующий экспрессию гена c-myc [79]. Однако в дальнейшем выявили важную роль этого белка в работе энхансер-блокирующих элементов [80‒82] и в поддержании 3D организации генома, в том числе в формировании хроматиновых петель [72, 83‒85]. В этой связи логично было предположить, что взаимодействия молекул CTCF, связанных с границами ТАДа, могут организовать ТАД в петлю. Само возникновение хроматиновой петли не объясняет, однако, каким образом формируется ТАД. Действительно, на тепловых картах пространственных контактов удаленные участки хроматиновой фибриллы, расположенные в основании петли, должны дать локальный сигнал на вершине треугольника, но для заполнения всего треугольника нужны множественные контакты внутри петли (см. рис. 1в). Объяснения требовало еще одно наблюдение. Оказалось, что ТАДы ограничиваются конвергентными сайтами связывания CTCF [6]. Изменение направления сайта (сайтов) связывания CTCF на границе ТАДа приводит к утрате этой границы [86, 87]. С учетом упомянутых наблюдений для объяснения механизма формирования ТАДов предложена модель экструзии хроматиновых петель (рис. 3) [88, 89]. Основной постулат этой модели состоит в том, что в клеточном ядре существуют молекулярные моторы, которые постоянно формируют петли хроматиновой фибриллы. Место начала выпетливания является, скорее всего, случайным и различается в разных клетках. При этом область выпетливания ограничивается конвергентными сайтами связывания CTCF. По мнению авторов модели роль экструзионного мотора может выполнять когезин. Прямо показана способность близкородственного белка конденсина выпетливать ДНК [90]. ТАД рассматривается авторами как популяционный феномен. Согласно модели, контактные домены на популяционных Hi-C-картах возникают благодаря наложению сигналов от взаимодействия оснований разнообразных петель, существующих в индивидуальных клетках в момент их фиксации формальдегидом, предшествующей проведению процедуры Hi-C. Модель подтверждается результатами компьютерных симуляций [89], а также целым рядом косвенных данных. Так, показано, что процесс формирования петлевых контактных доменов является энергозатратным (не идет при истощении пула АТР), но не зависит от осуществления транскрипции [91]. Петлевые контактные домены утрачиваются при индуцированной деградации когезина [92]. Средний размер экспериментально выявляемых петлевых контактных доменов увеличивается при истощении фактора WAPL, способствующего снятию когезина с ДНК (cohesion unloading factor WAPL) [73].
Рис. 3.
Гипотетическая модель работы “экструзионной машины”, формирующей петли ДНК. Формирование петли начинается в произвольных участках хроматиновой фибриллы (показаны три участка) путем затягивания фибриллы в когезиновое кольцо. В качестве стопоров выпетливания выступают сайты связывания белка CTCF в определенной ориентации.
Обычные контактные домены [6] в экструзионной модели рассматриваются как хроматиновые компартменты [93, 94]. При анализе Hi-C-карт низкого разрешения компартменты представлялись достаточно протяженными [5]. Однако с появлением карт высокого разрешения стало ясно, что компартменты могут быть сопоставимы по длине с петлевыми доменами [95, 96]. Компартменты возникают в силу тенденции к ассоцииции хроматиновых доменов одного типа (активных доменов, нективных доменов, неактивных доменов Polycomb). Механизмы их формирования не вполне ясны. У дрозофилы петлевых контактных доменов вообще не обнаружено, и границы контактных доменов не обогащены CTCF [96, 97]. Результаты нашей работы позволяют полагать, что ключевую роль в формировании ТАДов у этого организма играют электростатические межнуклеосомные взаимодействия, которые подавляются посредством ацетилирования гистонов [97]. Соответственно, транскрипционно активные участки, для которых характерен высокий уровень ацетилирования гистонов, располагаются преимущественно между ТАДами, обеспечивая их инсуляцию [97‒99]. Сила границ контактных доменов у дрозофилы прямо зависит от протяженности разделяющего их участка и степени обогащения этого участка активным хроматином [100]. Механизмы сегрегации разных типов хроматиновых доменов (компартментов) еще предстоит понять. Предполагается, что определенную роль в этом процессе может играть разделение жидких фаз [94, 101]. Так или иначе, складывается впечатление, что обычные домены (ordinary, или compartmental domains) эволюционно более древние. В том или ином виде они существуют у самых разных организмов, тогда как петлевые домены, появляются только у позвоночных [96]. У этих животных архитектура интерфазных хромосом складывается посредством наложения, а возможно, и конкуренции двух типов доменов. Удаление петлевых доменов посредством деградации когезина или деплеции загрузчика когезина позволяет лучше видеть структуру компартментов [93].
Учитывая иерархичность контактных доменов хроматина (т.е. внутри больших и относительно рыхлых контактных доменов можно выделить несколько уровней более плотных вложенных доменов) [97, 100, 102, 103], закономерным представляется вопрос о том, контактные домены какого уровня следует считать ТАДами. Однозначного ответа на этот вопрос не существует. Некоторые считают, что для ответа на этот вопрос следует исходить из того, какие контактные домены соответствуют функциональным единицам генома [104]. В этой связи стоит обратить внимание на то, что у позвоночных животных ограничение действия энхансеров обеспечивают главным образом границы петлевых доменов. По этой причине предлагается считать ТАДами именно петлевые домены [95]. Проблема, однако, состоит в том, что и петлевые домены могут включать вложенные петлевые домены. Так что основным критерием остается все же соответствие контактных доменов того или иного уровня функциональным доменам генома.
TAДы В ИНДИВИДУАЛЬНЫХ КЛЕТКАХ
За последние 2 года опубликовано несколько работ по Hi-C-анализу индивидуальных клеток. Стратегии получения Hi-C-карт индивидуальных клеток в этих работах существенно различались [105]. В настоящее время для построения Hi-C-карт индивидуальных клеток используются два принципиально разных подхода. Первый из них состоит в непосредственном анализе Hi-C-библиотек из индивидуальных клеток [37, 106‒108]. Второй состоит во внесении специфичных для индивидуальных клеток баркодов на одном из этапов приготовления Hi-C-библиотек с последующим вычленением информации об индивидуальных клетках из популяционной Hi-C-библиотеки на этапе биоинформатического анализа [109]. Главная проблема, возникающая при анализе Hi-C-карт индивидуальных клеток, – относительная разряженность контактных матриц, что не позволяет построить карты высокого разрешения. На текущий момент удалось показать, что в индивидуальных клетках присутствуют контактные домены хроматина, границы которых не всегда совпадают с границами ТАДов, выявляемыми при анализе популяционных карт [108]. В то же время при сложении карт нескольких индивидуальных клеток можно воспроизвести профили популяционных ТАДов [108]. На основании Hi-C-карт индивидуальных клеток построены модели индивидуальных ТАДов, которые показали, что форма ТАДов существенно варьирует от клетки к клетке [37]. В равной мере вариабельной оказалась и форма индивидуальных хромосомных территорий (способ макроукладки ДНК внутри хромосомной территории) [106]. Вопрос о том, организованы ли контактные домены в индивидуальных клетках иерархически, остается пока открытым. Существующие данные не исключают возможности того, что видимая на популяционных картах иерархия контактных доменов возникает в результате наложения вариабельных конфигураций контактных доменов, существующих в индивидуальных клетках. Аналогичная неопределенность сохраняется и в вопросе о том, существует ли в индивидуальных клетках пространственная сегрегация активного и неактивного хроматиновых компартментов.
ПРОСТРАНСТВЕННАЯ ОРГАНИЗАЦИЯ ГЕНОМА В КОНТЕКСТЕ ФУНКЦИОНАЛЬНОЙ КОМПАРТМЕНТАЛИЗАЦИИ КЛЕТОЧНОГО ЯДРА
Упакованный геном занимает большую часть клеточного ядра и служит платформой для функциональной компартментализации нуклеоплазмы. Совокупность хромосомных территорий составляет хроматиновый домен, пронизанный интерхроматиновыми каналами, которые образуют интерхроматиновый домен. Последние служат местом сборки различных ядерных телец (функциональных компартментов) ‒ реакционных центров, обеспечивающих выполнение тех или иных функциональных процессов [28, 29, 110, 111]. Ядерные тельца возникают в силу разделения жидких фаз и представляют собой динамичные образования. Они могут собираться и разбираться в зависимости от ряда условий. Дезинтеграцию ядерных телец можно, например, стимулировать, искусственно понизив уровень макромолекулярного скопления в клеточном ядре [112]. Некоторые из этих функциональных компартментов формируются непосредственно вокруг определенных локусов генома. К числу этих реакционных центров относятся, в частности, ядрышко, локус гистоновых генов и транскрипционные фабрики [113]. Другие функциональные компартменты собираются в интерхроматиновом домене без прямого участия генома. Однако по ходу осуществления тех или иных процессов различные гены могут привлекаться к этим функциональным центрам. В качестве примера можно привести привлечение генов, содержащих интроны, к спеклам (ядерным тельцам, в которых сосредоточены факторы сплайсинга) [114] и генов U РНК к тельцам Кахаля [115]. Важный структурный компонент ядра ‒ ядерная ламина. В геноме существуют протяженные участки, которые предпочтительно локализуются на ядерной ламине (ламина-ассоциированные домены, ЛАДы) [116‒118] ‒ это в основном неактивные домены хроматина [119, 120]. Более того, индуцированное перемещение гена к ядерной ламине способствует его инактивации [121]. Другая репрессивная область клеточного ядра в клетках млекопитающих ‒ околоядрышковое пространство [122, 123]. Возможно, это связано с тем, что на границах ядрышка расположены неактивные копии рибосомных генов, которые инициируют формирование здесь кластера неактивного хроматина [124]. Примечательно, что в индивидуальной клетке после митоза наблюдается случайное перемещение неактивных доменов хроматина между ламиной и околоядрышковым пространством; регионы, до митоза расположенные в приламинарном пространстве, могут оказаться около ядрышка и наоборот [125].
Стандартные экспериментальные протоколы, основанные на процедуре лигирования близкорасположенных фрагментов ДНК и использующие секвенирование с длиной рида 50‒150 н. [4], позволяют анализировать только попарные взаимодействия. Многокомпонентные комплексы нельзя однозначно обнаружить с помощью этой процедуры. Нельзя детектировать и приближение тех или иных участков генома к функциональным компартментам ядра в силу того, что расстояния между контактирующими с таким компартментом фрагментами ДНК могут быть больше тех, которые позволяют осуществить кросс-лигирование. В силу этого взаимосвязь между 3D организацией генома и компартментализацией клеточного ядра в течение многих лет изучали с использованием исключительно цитологических (микроскопических) методов. В настоящее время разработано несколько экспериментальных процедур, которые свободны от ограничений, накладываемых процедурой лигирования близкорасположенных фрагментов ДНК. Первым стоит упомянуть метод, основанный на массированном секвенировании фрагментов ДНК, экстрагированных из серийных срезов замороженных клеток [126]. Понятно, что два фрагмента ДНК будут тем чаще присутствовать в одном и том же срезе, чем ближе друг к другу они находятся внутри клеточного ядра. Процедура позволяет строить тепловые карты пространственных взаимодействий геномных элементов, аналогичные Hi-C-картам. Однако она также позволяет анализировать сближенность нескольких фрагментов ДНК. Существует возможность включить в анализ молекулы РНК после конвертации их в кДНК. Другая процедура основана на мягкой фрагментации фиксированного хроматина, позволяющей сохранить достаточно большие блоки, с последующим лигированием специфичных для каждого блока наборов баркодов. Это достигается посредством нескольких циклов распределения суспензии блоков хроматина по ячейкам, лигирования баркодов, объединения материала, повторного распределения по ячейкам, лигирования баркодов следующего типа и т.д. В результате вся совокупность фрагментов, присутствующих в составе каждого хроматинового блока, оказывается меченной индивидуальным набором баркодов. Последующий анализ позволяет установить, какие наборы фрагментов ДНК часто оказываются в одном и том же блоке хроматина [127]. Третья заслуживающая упоминания процедура заключается во внесении биотиновой метки в ДНК, прилежащей к представляющему интерес ядерному компартменту [128]. Для биотинилирования используется хорошо известный цитологам протокол тирамидной амплификации [129]. Синтез тирамид-биотиновых радикалов инициируется пероксидазой хрена, конъюгированной с антителами. Место(а) внутри ядра, вокруг которого(ых) происходит биотинилирование, определяется специфичностью используемых антител. Это позволяет иницировать биотинилирование рядом с любым представляющим интерес ядерным компартментом, к компонентам которого имеются антитела [128]. С использованием перечисленных протоколов сделан ряд важных наблюдений. Прежде всего, убедительно показано, что многокомпонентные хроматиновые блоки, в том числе и блоки, включающие фрагменты разных хромосом, встречаются достаточно часто. Особенно типична кластеризация суперэнхансеров и активно транскрибирующихся участков генома [126]. Что касается привлечения различных участков генома к тем или иным ядерным компартментам и функциональным зонам, то обнаружена четкая пространственная сегрегация активного и неактивного хроматина. Активный хроматин кластеризуется вокруг ядерных спеклов (компартментов, содержащих факторы сплайсинга) [127, 128], тогда как неактивный хроматин присутствует преимущественно в околоядрышковом [127] и приламинарном слое [125]. Роль ядерной ламины в создании неактивного хроматинового компартмента определена и в целом ряде исследований [118, 120]. То же можно сказать и о прилегающей к ядрышку области клеточного ядра [122, 123]. Однако масштабная пространственная кластеризация активных генов рядом со спеклами обнаружена впервые.
С учетом всего сказанного можно утверждать, что функциональная компартментализация ядра и 3D организация генома взаимосвязаны. Те или иные манипуляции с клеточным ядром прямо влияют и на пространственную организацию генома. Так, активаторный блок домена бета-глобиновых генов декомпактизуется в условиях гипотонического шока, когда объем ядра значительно увеличивается [130]. Также показано, что заякоренный на ядерной ламине и ядрышке хроматин находится в напряженном состоянии [131 ] . Разрушение ламины, наблюдаемое при истощении ламина, индуцированного посредством РНК-интерференции, приводит с сокращению объема, занимаемого хроматином. Одновременно существенно снижается уровень сегрегации активного и неактивного хроматинового компартментов [131 ] . Таким образом, клеточное ядро представляет собой интегральную систему, свойства которой зависят от взаимодействия всех присутствующих в нем компонентов. При частичном разрушении этой системы изменяются ее свойства в целом, а не только той части, где локализуются повреждения.
Работа выполнена при поддержке Российского научного фонда (проект 19-14-00016).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Osborne C.S., Chakalova L., Brown K.E., Carter D., Horton A., Debrand E., Goyenechea B., Mitchell J.A., Lopes S., Reik W., Fraser P. (2004) Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 36(10), 1065‒1071.
Schoenfelder S., Sexton T., Chakalova L., Cope N.F., Horton A., Andrews S., Kurukuti S., Mitchell J.A., Umlauf D., Dimitrova D.S., Eskiw C.H., Luo Y., Wei C.L., Ruan Y., Bieker J.J., Fraser P. (2010) Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 42(1), 53‒61.
Williamson I., Lettice L.A., Hill R.E., Bickmore W.A. (2016) Shh and ZRS enhancer colocalization is specific to the zone of polarizing activity. Development. 143(16), 2994‒3001.
Dekker J., Rippe K., Dekker M., Kleckner N. (2002) Capturing chromosome conformation. Science. 295(5558), 1306‒1311.
Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., Sandstrom R., Bernstein B., Bender M.A., Groudine M., Gnirke A., Stamatoyannopoulos J., Mirny L.A., Lander E.S., Dekker J. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326(5950), 289‒293.
Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S., Aiden E.L. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159(7), 1665‒1680.
Dekker J., Misteli T. (2015) Long-range chromatin interactions. Cold Spring Harb. Perspect. Biol. 7(10), a019356.
Sanyal A., Lajoie B.R., Jain G., Dekker J. (2012) The long-range interaction landscape of gene promoters. Nature. 489(7414), 109‒113.
Deng W., Lee J., Wang H., Miller J., Reik A., Gre-gory P.D., Dean A., Blobel G.A. (2012) Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 149(6), 1233‒1244.
Breda L., Motta I., Lourenco S., Gemmo C., Deng W., Rupon J.W., Abdulmalik O.Y., Manwani D., Blobel G.A., Rivella S. (2016) Forced chromatin looping raises fetal hemoglobin in adult sickle cells to higher levels than pharmacologic inducers. Blood. 128(8), 1139‒1143.
Nora E.P., Dekker J., Heard E. (2013) Segmental folding of chromosomes: a basis for structural and regulatory chromosomal neighborhoods? BioEssays. 35(9), 818‒828.
Valton A.L., Dekker J. (2016) TAD disruption as oncogenic driver. Curr. Opin. Genet. Dev. 36, 34‒40.
Lupianez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R, Santos-Simarro F., Gilbert-Dussardier B., Wittler L., Borschiwer M., Haas S.A., Osterwalder M., Franke M., Timmermann B., Hecht J., Spielmann M., Visel A., Mundlos S. (2015) Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 161(5), 1012‒1025.
Lupianez D.G., Spielmann M., Mundlos S. (2016) Breaking TADs: How alterations of chromatin domains result in disease. Trends Genet. 32(4), 225‒237.
Krijger P.H., de Laat W. (2016) Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell. Biol. 17(12), 771‒782.
Kantidze O.L., Luzhin A.V., Nizovtseva E.V., Safina A., Valieva M.E., Golov A.K., Velichko A.K., Lyubitelev A.V., Feofanov A.V., Gurova K.V., Studitsky V.M., Razin S.V. (2019) The anti-cancer drugs curaxins target spatial genome organization. Nat. Comm. 10(1), 1441.
Razin S.V., Vassetzky Y.S. (2017) 3D genomics imposes evolution of the domain model of eukaryotic genome organization. Chromosoma. 126, 59‒69.
Razin S.V., Gavrilov A.A. (2014) Chromatin without the 30-nm fiber: constrained disorder instead of hierarchical folding. Epigenetics. 9(5), 653‒657.
Getzenberg R.H., Pienta K.J., Ward W.S., Coffey D.S. (1991) Nuclear structure and the three-dimensional organization of DNA. J. Cell. Biochem. 47(4), 289‒299.
Ou H.D., Phan S., Deerinck T.J., Thor A., Ellisman M.H., O’Shea C.C. (2017) ChromEMT: visua-lizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 357(6349), eaag0025.
Fussner E., Strauss M., Djuric U., Li R., Ahmed K., Hart M., Ellis J., Bazett-Jones D.P. (2012) Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Rep. 13(11), 992‒996.
Eltsov M., Maclellan K.M., Maeshima K., Frangakis A.S., Dubochet J. (2008) Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc. Natl. Acad. Sci. USA. 105(50), 19732‒19737.
Ricci M.A., Manzo C., Garcia-Parajo M.F., Lakadamyali M., Cosma M.P. (2015) Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 160(6), 1145‒1158.
Maeshima K., Imai R., Hikima T., Joti Y. (2014) Chromatin structure revealed by X-ray scattering analysis and computational modeling. Methods. 70(2‒3), 154‒161.
Maeshima K., Imai R., Tamura S., Nozaki T. (2014) Chromatin as dynamic 10-nm fibers. Chromosoma. 123(3), 225‒237.
Maeshima K., Rogge R., Tamura S., Joti Y., Hikima T., Szerlong H., Krause C., Herman J., Seidel E., DeLuca J., Ishikawa T., Hansen J.C. (2016) Nucleosomal arrays self-assemble into supramolecular globular structures lacking 30-nm fibers. EMBO J. 35(10), 1115 ‒1132. https://doi.org/10.15252/embj.201592660
Hansen J.C., Connolly M., McDonald C.J., Pan A., Pryamkova A ., Ray K., Seidel E., Tamura S., Rogge R., Maeshima K. (2018) The 10-nm chromatin fiber and its relationship to interphase chromosome organization. Biochem. Soc. Trans. 46(1), 67‒76.
Cremer T., Cremer M. (2010) Chromosome territories. Cold Spring Harb. Perspect. Biol. 2(3), a003889.
Cremer T., Cremer M., Dietzel S., Muller S., Solovei I., Fakan S. (2006) Chromosome territories‒a functional nuclear landscape. Curr. Opin. Cell. Biol. 18(3), 307‒316.
Kolbl A.C., Weigl D., Mulaw M., Thormeyer T., Bohlander S.K., Cremer T., Dietzel S. (2012) The radial nuclear positioning of genes correlates with features of megabase-sized chromatin domains. Chromosome Res. 20(6), 735‒752.
Smeets D., Markaki Y., Schmid V.J., Kraus F., Tattermusch A., Cerase A., Sterr M., Fiedler S., Demmerle J., Popken J., Leonhardt H., Brockdorff N., Cremer T., Schermelleh L., Cremer M. (2014) Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenetics Chromatin. 7, 8.
Dixon J.R., Gorkin D.U., Ren B. (2016) Chromatin domains: the unit of chromosome organization. Mol. Cell. 62(5), 668‒680.
Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485(7398), 376‒380.
Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G. (2012) Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 148(3), 458‒472.
Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J., Gribnau J., Barillot E., Bluthgen N., Dekker J., Heard E. (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 485(7398), 381‒385.
Szabo Q., Jost D., Chang J.M., Cattoni D.I., Papadopoulos G.L., Bonev B., Sexton T., Gurgo J., Jacquier C., Nollmann M., Bantignies F., Cavalli G. (2018) TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv. 4(2), eaar8082.
Stevens T.J., Lando D., Basu S., Atkinson L.P., Cao Y., Lee S.F., Leeb M., Wohlfahrt K.J., Boucher W., O’Shaughnessy-Kirwan A., Cramard J., Faure A.J., Ralser M., Blanco E., Morey L., Sanso M., Palayret M.G.S., Lehner B., Di Croce L., Wutz A., Hendrich B., Klenerman D., Laue E.D. (2017) 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature. 544(7648), 59‒64.
Bodnar J.W. (1988) A domain model for eukaryotic DNA organization: a molecular basis for cell differentiation and chromosome evolution. J. Theor. Biol. 132(4), 479‒507.
Goldman M.A. (1988) The chromatin domain as a unit of gene regulation. BioEssays. 9, 50‒55.
Forrester W.C., Epner E., Driscoll M.C., Enver T., Brice M., Papayannopoulou T., Groudine M. (1990) A deletion of the human β-globin locus activation region causes a major alteration in chromatin structure and replication across the entire β-globin locus. Gene Dev. 4, 1637‒1649.
Grosveld F., van Assandelt G.B., Greaves D.R., Kollias B. (1987) Position-independent, high-level expression of the human β-globin gene in transgenic mice. Cell. 51, 975‒985.
Kellum R., Schedl P. (1991) A position-effect assay for boundaries of higher-order chromosomal domains. Cell. 64, 941‒950.
Kellum R., Schedl P. (1992) A group of scs elements function as boundaries in enhancer-blocking assay. Mol. Cell. Biol. 12, 2424‒2431.
Chung J.H., Whiteley M., Felsenfeld G. (1993) A 5' element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 74(3), 505‒514.
Jarman A.P., Wood W.G., Sharpe J.A., Gourdon G., Ayyub H., Higgs D.R. (1991) Characterization of the major regulatory element upstream of the human α‑globin gene cluster. Mol. Cell. Biol. 11, 4679‒4689.
Recillas-Targa F., Razin S.V. (2001) Chromatin domains and regulation of gene expression: familiar and enigmatic clusters of chicken globin genes. Crit. Rev. Eukaryot. Gene Expr. 11, 227‒242.
Razin S.V., Farrell C.M., Recillas-Targa F. (2003) Genomic domains and regulatory elements operating at the domain level. Int. Rev. Cytol. 226, 63‒25.
Dillon N., Sabbatini P. (2000) Functional gene expression domains: defining the functional units of eukaryotic gene regulation. BioEssays. 22, 657‒665.
Arnold C.D., Gerlach D., Stelzer C., Boryn L.M., Rath M., Stark A. (2013) Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 339(6123), 1074‒1077.
Consortium E.P., Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature. 489(7414), 57‒74.
Furlong E.E.M., Levine M. (2018) Developmental enhancers and chromosome topology. Science. 361(6409), 1341‒1345.
Hnisz D., Shrinivas K., Young R.A., Chakraborty A.K., Sharp P.A. (2017) A phase separation model for transcriptional control. Cell. 169(1), 13‒23.
Sabari B.R., Dall’Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C., Li C.H., Guo Y.E., Day D.S., Schuijers J., Vasile E., Malik S., Hnisz D., Lee T.I., Cisse II, Roeder R.G., Sharp P.A., Chak-raborty A.K., Young R.A. (2018) Coactivator condensation at super-enhancers links phase separation and gene control. Science. 361(6400), eaar3958.
Gurumurthy A., Shen Y., Gunn E.M., Bungert J. (2019) Phase separation and transcription regulation: are super-enhancers and locus control regions primary sites of transcription complex assembly? BioEssays. 41(1), e1800164.
Hahn S. (2018) Phase separation, protein disorder, and enhancer function. Cell. 175(7), 1723‒1725.
Tolhuis B., Palstra R.J., Splinter E., Grosveld F., de Laat W. (2002) Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell. 10(6), 1453‒1465.
Palstra R.J., Tolhuis B., Splinter E., Nijmeijer R., Grosveld F., de Laat W. (2003) The beta-globin nuclear compartment in development and erythroid differentiation. Nat. Genet. 35(2), 190‒194.
Philonenko E.S., Klochkov D.B., Borunova V.V., Gavrilov A.A., Razin S.V., Iarovaia O.V. (2009) TMEM8 ‒ a non-globin gene entrapped in the globin web. Nucl. Acids Res. 37(22), 7394‒7406.
Vernimmen D., Marques-Kranc F., Sharpe J.A., Sloane-Stanley J.A., Wood W.G., Wallace H.A., Smith A.J., Higgs D.R. (2009) Chromosome looping at the human alpha-globin locus is mediated via the major upstream regulatory element (HS-40). Blood. 114(19), 4253‒4260.
Novo C.L., Javierre B.M., Cairns J., Segonds-Pichon A., Wingett S.W., Freire-Pritchett P., Furlan-Magaril M., Schoenfelder S., Fraser P., Rugg-Gunn P.J. (2018) Long-range enhancer interactions are prevalent in mouse embryonic stem cells and are reorganized upon pluripotent state transition. Cell Rept. 22(10), 2615‒2627.
Schoenfelder S., Javierre B.M., Furlan-Magaril M., Wingett S.W., Fraser P. (2018) Promoter capture Hi-C: high-resolution, genome-wide profiling of promoter interactions. J. Vis. Exp. 136, e57320. https://doi.org/10.3791/57320
Li T., Jia L., Cao Y., Chen Q., Li C. (2018) OCEAN-C: mapping hubs of open chromatin interactions across the genome reveals gene regulatory networks. Genome Biol. 19(1), 54.
Zheng M., Tian S.Z., Capurso D., Kim M., Maurya R., Lee B., Piecuch E., Gong L., Zhu J.J., Li Z., Wong C.H., Ngan C.Y., Wang P., Ruan X., Wei C.L., Ruan Y. (2019) Multiplex chromatin interactions with single-molecule precision. Nature. 566(7745), 558‒562.
Symmons O., Uslu V.V., Tsujimura T., Ruf S., Nassari S., Schwarzer W., Ettwiller L., Spitz F. (2014) Functional and topological characteristics of mammalian regulatory domains. Genome Res. 24(3), 390‒400.
Montavon T., Soshnikova N., Mascrez B., Joye E., Thevenet L., Splinter E., de Laat W., Spitz F., Duboule D. (2011) A regulatory archipelago controls Hox genes transcription in digits. Cell. 147(5), 1132‒1145.
Montavon T., Duboule D. (2012) Landscapes and archipelagos: spatial organization of gene regulation in vertebrates. Trends Cell Biol. 22(7), 347‒354.
Hnisz D., Day D.S., Young R.A. (2016) Insulated neighborhoods: structural and functional units of mammalian gene control. Cell. 167(5), 1188‒1200.
Hnisz D., Weintraub A.S., Day D.S., Valton A.L., Bak R.O., Li C.H., Goldmann J., Lajoie B.R., Fan Z.P., Sigova A.A., Reddy J., Borges-Rivera D., Lee T.I., Jaenisch R., Porteus M.H., Dekker J., Young R.A. (2016) Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 351(6280), 1454‒1458.
Sun F., Chronis C., Kronenberg M., Chen X.F., Su T., Lay F.D., Plath K., Kurdistani S.K., Carey M.F. (2019) Promoter-enhancer communication occurs primarily within insulated neighborhoods. Mol. Cell. 73(2), 250‒263. e255.
Smith E.M., Lajoie B.R., Jain G., Dekker J. (2016) Invariant TAD boundaries constrain cell-type-specific looping interactions between promoters and distal elements around the CFTR locus. Am. J. Hum. Genet. 98(1), 185‒201.
Krefting J., Andrade-Navarro M.A., Ibn-Salem J. (2018) Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biol. 16(1), 87.
Ong C.T., Corces V.G. (2014) CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet. 15(4), 234‒246.
Wutz G., Varnai C., Nagasaka K., Cisneros D.A., Stocsits R.R., Tang W., Schoenfelder S., Jessberger G., Muhar M., Hossain M.J., Walther N., Koch B., Kueblbeck M., Ellenberg J., Zuber J., Fraser P., Peters J.M. (2017) Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 36(24), 3573‒3599.
Uuskula-Reimand L., Hou H., Samavarchi-Tehrani P., Rudan M.V., Liang M., Medina-Rivera A., Mohammed H., Schmidt D., Schwalie P., Young E.J., Reimand J., Hadjur S., Gingras A.C., Wilson M.D. (2016) Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 17(1), 182.
Van Bortle K., Nichols M.H., Li L., Ong C.T., Takenaka N., Qin Z.S., Corces V.G. (2014) Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol. 15(5), R82.
Nora E.P., Goloborodko A., Valton A.L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G. (2017) Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 169(5), 930‒944 e922.
Narendra V., Bulajic M., Dekker J., Mazzoni E.O., Reinberg D. (2016) CTCF-mediated topological boundaries during development foster appropriate gene regulation. Genes Dev. 30(24), 2657‒2662.
Narendra V., Rocha P.P., An D., Raviram R., Skok J.A., Mazzoni E.O., Reinberg D. (2015) CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science. 347(6225), 1017‒1021.
Klenova E.M., Nicolas R.H., Paterson H.F., Carne A.F., Heath C.M., Goodwin G.H., Neiman P.E., Lobanenkov V.V. (1993) CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol. Cell. Biol. 13(12), 7612‒7624.
Bell A.C., West A.G., Felsenfeld G. (1999) The protein CTCF is required for the enhancer-blocking activity of vertebrate insulators. Cell. 98, 387‒396.
Farrell C.M., West A.G., Felsenfeld G. (2002) Conserved CTCF insulator elements flank the mouse and human β-globin loci. Mol. Cell. Biol. 22, 3820‒3831.
Hark A.T., Schoenherr C.J., Katz D.J., Ingram R.S., Levorse J.M., Tilghman S.M. (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 405, 486‒489.
Holwerda S.J., de Laat W. (2013) CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 368(1620), 20120369.
Hou C., Zhao H., Tanimoto K., Dean A. (2008) CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc. Natl. Acad. Sci. USA. 105(51), 20398‒20403.
Phillips J.E., Corces V.G. (2009) CTCF: master weaver of the genome. Cell. 137(7), 1194‒1211.
de Wit E., Vos E.S., Holwerda S.J., Valdes-Quezada C., Verstegen M.J., Teunissen H., Splinter E., Wijchers P.J., Krijger P.H., de Laat W. (2015) CTCF binding polarity determines chromatin looping. Mol. Cell. 60(4), 676‒684.
Guo Y., Xu Q., Canzio D., Shou J., Li J., Gorkin D.U., Jung I., Wu H., Zhai Y., Tang Y., Lu Y., Wu Y., Jia Z., Li W., Zhang M.Q., Ren B., Krainer A.R., Maniatis T., Wu Q. (2015) CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 162(4), 900‒910.
Sanborn A.L., Rao S.S., Huang S.C., Durand N.C., Huntley M.H., Jewett A.I., Bochkov I.D., Chinnappan D., Cutkosky A., Li J., Geeting K.P., Gnirke A., Melnikov A., McKenna D., Stamenova E.K., Lan-der E.S., Aiden E.L. (2015) Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA. 112(47), E6456‒E6465.
Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L.A. (2016) Formation of chromosomal domains by loop extrusion. Cell Rept. 15(9), 2038‒2049.
Ganji M., Shaltiel I.A., Bisht S., Kim E., Kalichava A., Haering C.H., Dekker C. (2018) Real-time imaging of DNA loop extrusion by condensin. Science. 360(6384), 102‒105.
Vian L., Pekowska A., Rao S.S.P., Kieffer-Kwon K.R., Jung S., Baranello L., Huang S.C., El Khattabi L., Dose M., Pruett N., Sanborn A.L., Canela A., Maman Y., Oksanen A., Resch W., Li X., Lee B., Kovalchuk A.L., Tang Z., Nelson S., Di Pierro M., Cheng R.R., Machol I., St Hilaire B.G., Durand N.C., Shamim M.S., Stamenova E.K., Onuchic J.N., Ruan Y., Nussenzweig A., Levens D., Aiden E.L., Casellas R. (2018) The energetics and physiological impact of cohesin extrusion. Cell. 173(5), 1165‒1178. e1120
Rao S.S.P., Huang S.C., Glenn St Hilaire B., Engreitz J.M., Perez E.M., Kieffer-Kwon K.R., Sanborn A.L., Johnstone S.E., Bascom G.D., Bochkov I.D., Huang X., Shamim M.S., Shin J., Tur-ner D., Ye Z., Omer A.D., Robinson J.T., Schlick T., Bernstein B.E., Casellas R., Lander E.S., Aiden E.L. (2017) Cohesin loss eliminates all loop domains. Cell. 171(2), 305‒320. e324
Schwarzer W., Abdennur N., Goloborodko A., Pekowska A., Fudenberg G., Loe-Mie Y., Fonseca N.A., Huber W., C H.H., Mirny L., Spitz F. (2017) Two independent modes of chromatin organization revealed by cohesin removal. Nature. 551(7678), 51‒56.
Nuebler J., Fudenberg G., Imakaev M., Abdennur N., Mirny L.A. (2018) Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA. 115(29), E6697–E6706.
Rowley M.J., Corces V.G. (2018) Organizational principles of 3D genome architecture. Nat. Rev. Genet. 19(12), 789‒800.
Rowley M.J., Nichols M.H., Lyu X., Ando-Kuri M., Rivera I.S.M., Hermetz K., Wang P., Ruan Y., Corces V.G. (2017) Evolutionarily conserved principles predict 3D cromatin organization. Mol. Cell. 67(5), 837‒852. e7
Ulianov S.V., Khrameeva E.E., Gavrilov A.A., Flyamer I.M., Kos P., Mikhaleva E.A., Penin A.A., Logacheva M.D., Imakaev M.V., Chertovich A., Gelfand M.S., Shevelyov Y.Y., Razin S.V. (2016) Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res. 26(1), 70‒84.
Gavrilov A.A., Shevelyov Y.Y., Ulianov S.V., Khrameeva E.E., Kos P., Chertovich A., Razin S.V. (2016) Unraveling the mechanisms of chromatin fibril packaging. Nucleus. 7(3), 319‒324.
Razin S.V., Gavrilov A.A., Vassetzky Y.S., Ulianov S.V. (2016) Topologically-associating domains: gene warehouses adapted to serve transcriptional regulation. Transcription. 7(3), 84‒90.
Luzhin A.V., Flyamer I.M., Khrameeva E.E., Ulia-nov S.V., Razin S.V., Gavrilov A.A. (2019) Quantitative differences in TAD border strength underly the TAD hierarchy in Drosophila chromosomes. J. Cell. Biochem. 120(3), 4494‒4503.
Rada-Iglesias A., Grosveld F.G., Papantonis A. (2018) Forces driving the three-dimensional folding of eukaryotic genomes. Mol. Systems Biol. 14(6), e8214.
Weinreb C., Raphael B.J. (2016) Identification of hierarchical chromatin domains. Bioinformatics. 32(11), 1601‒1609.
Wang X.T., Cui W., Peng C. (2017) HiTAD: detecting the structural and functional hierarchies of topologically associating domains from chromatin interactions. Nucl. Acids Res. 45(19), e163.
Zhan Y., Mariani L., Barozzi I., Schulz E.G., Bluthgen N., Stadler M., Tiana G., Giorgetti L. (2017) Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res. 27(3), 479‒490.
Ulianov S.V., Tachibana-Konwalski K., Razin S.V. (2017) Single-cell Hi-C bridges microscopy and genome-wide sequencing approaches to study 3D chromatin organization. BioEssays. 39(10), 1700104.
Nagano T., Lubling Y., Stevens T.J., Schoenfelder S., Yaffe E., Dean W., Laue E.D., Tanay A., Fraser P. (2013) Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 502(7469), 59‒64.
Nagano T., Lubling Y., Varnai C., Dudley C., Leung W., Baran Y., Mendelson Cohen N., Wingett S., Fraser P., Tanay A. (2017) Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 547(7661), 61‒67.
Flyamer I.M., Gassler J., Imakaev M., Brandao H.B., Ulianov S.V., Abdennur N., Razin S.V., Mirny L.A., Tachibana-Konwalski K. (2017) Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature. 544(7648), 110‒114.
Ramani V., Deng X., Qiu R., Gunderson K.L., Steemers F.J., Disteche C.M., Noble W.S., Duan Z., Shendure J. (2017) Massively multiplex single-cell Hi-C. Nat. Meth. 14(3), 263‒266.
Cremer T., Cremer C. (2001) Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2(4), 292‒301.
Cremer T., Cremer M., Cremer C. (2018) 4D-нуклеом: компартментализация генома в контексте эволюции. Биохимия. 83(4), 452‒466.
Hancock R. (2004) A role for macromolecular crowding effects in the assembly and function of compartments in the nucleus. J. Struct. Biol. 146(3), 281‒290.
Ulianov S.V., Gavrilov A.A., Razin S.V. (2015) Nuclear compartments, genome folding, and enhancer-promoter communication. Internat. Rev. Cell Mol. Biol. 315, 183‒244.
Brown J.M., Green J., das Neves R.P., Wallace H.A., Smith A.J., Hughes J., Gray N., Taylor S., Wood W.G., Higgs D.R., Iborra F.J., Buckle V.J. (2008) Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J. Cell. Biol. 182(6), 1083‒1097.
Wang Q., Sawyer I.A., Sung M.H., Sturgill D., Shevtsov S.P., Pegoraro G., Hakim O., Baek S., Hager G.L., Dundr M. (2016) Cajal bodies are linked to genome conformation. Nat. Commun. 7, 10966. https://doi.org/10.1038/ncomms10966
Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W., van Steensel B. (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 453(7197), 948‒951.
Pickersgill H., Kalverda B., de Wit E., Talhout W., Fornerod M., van Steensel B. (2006) Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet. 38(9), 1005‒1014.
van Steensel B., Belmont A.S. (2017) Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell. 169(5), 780‒791.
Kind J., van Steensel B. (2010) Genome-nuclear lamina interactions and gene regulation. Curr. Opin. Cell. Biol. 22(3), 320‒325.
Shevelyov Y.Y., Nurminsky D.I. (2012) The nuclear lamina as a gene-silencing hub. Curr. Issues Mol. Biol. 14(1), 27‒38.
Reddy K.L., Zullo J.M., Bertolino E., Singh H. (2008) Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 452(7184), 243‒247.
Fedoriw A.M., Starmer J., Yee D., Magnuson T. (2012) Nucleolar association and transcriptional inhibition through 5S rDNA in mammals. PLoS Genet. 8(1), e1002468.
Matheson T.D., Kaufman P.D. (2016) Grabbing the genome by the NADs. Chromosoma. 125(3), 361‒371.
Politz J.C., Scalzo D., Groudine M. (2013) Something silent this way forms: the functional organization of the repressive nuclear compartment. Annu. Rev. Cell Dev. Biol. 29, 241‒270.
Kind J., Pagie L., de Vries S.S., Nahidiazar L., Dey S.S., Bienko M., Zhan Y., Lajoie B., de Graaf C.A., Amendola M., Fudenberg G., Imakaev M., Mirny L.A., Jalink K., Dekker J., van Oudenaarden A., van Steensel B. (2015) Genome-wide maps of nuclear lamina interactions in single human cells. Cell. 163(1), 134‒147.
Beagrie R.A., Scialdone A., Schueler M., Kraemer D.C., Chotalia M., Xie S.Q., Barbieri M., de Santiago I., Lavitas L.M., Branco M.R., Fraser J., Dostie J., Game L., Dillon N., Edwards P.A., Nicodemi M., Pombo A. (2017) Complex multi-enhancer contacts captured by genome architecture mapping. Nature. 543(7646), 519‒524.
Quinodoz S.A., Ollikainen N., Tabak B., Palla A., Schmidt J.M., Detmar E., Lai M.M., Shishkin A.A., Bhat P., Takei Y., Trinh V., Aznauryan E., Russell P., Cheng C., Jovanovic M., Chow A., Cai L., McDonel P., Garber M., Guttman M. (2018) Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell. 174(3), 744‒757. e724
Chen Y., Zhang Y., Wang Y., Zhang L., Brinkman E.K., Adam S.A., Goldman R., van Steensel B., Ma J., Belmont A.S. (2018) Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J. Cell. Biol. 217(11), 4025‒4048.
Wang G., Achim C.L., Hamilton R.L., Wiley C.A., Soontornniyomkij V. (1999) Tyramide signal amplification method in multiple-label immunofluorescence confocal microscopy. Methods. 18(4), 459‒464.
Golov A.K., Gavrilov A.A., Razin S.V. (2015) The role of crowding forces in juxtaposing beta-globin gene domain remote regulatory elements in mouse erythroid cells. PLoS One. 10(10), e0139855.
Ulianov S.V., Doronin S.A., Khrameeva E.E., Kos P.I., Luzhin A.V., Starikov S.S., Galitsyna A.A., Nenasheva V.V., Ilyin A.A., Flyamer I.M., Mikhaleva E.A., Logacheva M.D., Gelfand M.S., Chertovich A.V., Gavrilov A.A., Razin S.V., Shevelyov Y.Y. (2019) Nuclear lamina integrity is required for proper spatial organization of chromatin in Drosophila. Nat. Commun. 10(1), 1176.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология