

Молекулярная биология, 2020, T. 54, № 1, стр. 3-16
Комбинированная терапия рака на основе онколитической виротерапии и таргетной CAR T/NK-клеточной иммунотерапии
Г. В. Кочнева a, *, Г. Ф. Сиволобова a, А. В. Ткачева a, А. А. Горчаков b, c, С. В. Кулемзин b
a Государственный научный центр вирусологии и биотехнологии “Вектор”
630559
Новосибирская обл., Кольцово, Россия
b Институт молекулярной и клеточной биологии Сибирского отделения Российской академии наук
630090 Новосибирск, Россия
c Новосибирский государственный университет
630090 Новосибирск, Россия
* E-mail: g.v.kochneva@yandex.rukochneva@vector.nsc.ru
Поступила в редакцию 16.03.2019
После доработки 16.03.2019
Принята к публикации 19.03.2019
Аннотация
Многочисленные данные свидетельствуют о том, что иммунотерапия с использованием клеток, экспрессирующих химерный антигенный рецептор (chimeric antigen receptor, CAR), и виротерапия эффективны на иммунокомпетентных и иммунодефицитных моделях опухолей. Известны также единичные, но очень успешные попытки комбинирования этих мощных платформ. Оба подхода применяют и у людей, однако лишь при крайне ограниченном спектре патологий: CD19 CAR-T-клетки (Kymriah, Yescarta) при B-клеточных неоплазиях; онколитические вирусы при меланомах (Imlygic и Rigvir) и носоглоточных карциномах (Oncorine). Эффективность виротерапии в моноформате в значительной мере ограничена предсуществующим и быстро развивающимся иммунным ответом против вирусных эпитопов, а также необходимостью сохранения иммунной системы в относительно активном состоянии, что не характерно для онкологических больных при текущих схемах противоопухолевой терапии. Развитие CAR-клеточной терапии достаточно сильно сдерживается отсутствием у солидных опухолей опухольспецифичных поверхностных антигенов, гетерогенностью опухолей, моноспецифичным дизайном CAR, выраженным ингибированием CAR-клеток микроокружением опухолей, не говоря уже о неприемлемо высокой стоимости аутологичного формата CAR-клеточной терапии. Сочетание обоих подходов удивительным образом разрешает их “слабые места”, а именно, позволяет увеличить ассоциированные с интерфероном-β сигналы для реверсии локальной иммуносупрессии, облегчить хоминг CAR-лимфоцитов к опухоли и проникновение внутрь нее, обеспечить формирование иммуногенного опухолевого дебриса и экспонирование опухолевых неоантигенов, а также активно задействовать собственные иммунные клетки пациента в борьбе с опухолью. Таким образом, комбинирование CAR-клеточных и вирусолитических подходов выглядит более чем логичным и многообещающим.
ВВЕДЕНИЕ
Онколитические вирусы все чаще используют в научных исследованиях и медицинской практике в качестве перспективных противоопухолевых средств [1]. Онколитические вирусы обладают привлекательной комбинацией свойств, обеспечивающих опухолеспецифический лизис клеток и стимулирующих иммунитет, действуя как потенциальные противоопухолевые вакцины in situ. Онколитические вирусы могут быть генетически модифицированы для оптимизации опухолевой селективности и усиления иммунной стимуляции, они также могут легко комбинироваться с другими противоопухолевыми препаратами [2, 3]. Эффективность онколитических вирусов показана в многочисленных доклинических исследованиях и в клинике [4]. Высоким противоопухолевым потенциалом обладают рекомбинантные штаммы герпесвируса, вируса осповакцины и аденовируса, а также природные штаммы вируса кори, коксаки, реовируса, парво- и полиовирусов. В октябре 2015 года был достигнут большой прорыв ‒ FDA (США) одобрило применение препарата IMLYGIC™ на основе генетически модифицированного герпесвируса при рецидивирующей меланоме [5].
Другая активно развивающаяся технология ‒ CAR-Т-клеточная терапия. В ряде случаев с помощью этого подхода получены впечатляющие клинические результаты. В частности, применение CD19-специфических CAR-T-клеток при В-клеточном лейкозе (Kymriah ) и лимфоме (Yescarta) позволило добиться устойчивой ремиссии более чем у половины пациентов, невосприимчивых к другим видам терапии [6, 7]. В 2017 году получено разрешение на клиническое использование в США этих двух препаратов ‒ Yescarta (компания “Kite Pharma/Gilead”) и Kymriah (“Novartis”) [8, 9]. В настоящее время проводится свыше 250 клинических испытаний вариантов CAR-T-клеток, нацеленных на различные опухолевые антигены. Большинство испытаний проходят на территории США и Китая. В России подобная технология испытывается в единственном медицинском центре (NCT03467256).
Платформе CAR-T, при всех ее достоинствах и огромном потенциале, присущ ряд недостатков. Во-первых, CAR-T-терапия не является универсальной, поскольку на поверхности Т-клеток присутствуют специфические рецепторы (TCR). Чтобы избежать возникновения реакции “трансплантат против хозяина”, CAR-T-клетки необходимо получать с использованием собственных клеток пациента, проводя дорогую, сложную и не всегда безопасную процедуру адоптивного переноса в аутологичном формате. Во-вторых, активированные CAR-Т-клетки секретируют набор мощных провоспалительных цитокинов, что при определенных условиях может вызывать гиперцитокинемию (цитокиновый шторм) и нейротоксичность [10]. Наконец, поскольку CAR-T-клетки способны делиться, их популяция в организме может поддерживаться в течение нескольких лет [11, 12]. С одной стороны, это избавляет пациента от необходимости многократного введения CAR-Т-клеток и обеспечивает долговременный противоопухолевый контроль, с другой, в случае так называемой “off-target”, или нецелевой активности CAR-T уничтожению могут подвергаться нормальные клетки [13].
Использование естественных киллерных клеток (NK-клеток) в качестве носителей CAR позволяет в некоторой степени нивелировать перечисленные недостатки CAR-T-клеток. NK-клетки лишены TCR и, соответственно, не могут вызывать реакцию трансплантат против хозяина, благодаря чему возможной становится аллогенная трансплантация NK- и CAR-NK-клеток. Более того, элегантной представляется идея использования линейных NK-клеток, подвергнутых γ-облучению перед введением пациенту. Действие радиации лишает клетки способности к пролиферации при сохранении цитотоксической активности [14]. Явным преимуществом такого подхода является то, что NK- и CAR-NK-клеточные линии представляют собой гомогенный, охарактеризованный и относительно недорогой источник клеток, удовлетворяющий крайне востребованному и привычному клиницистам формату “off-the-shelf” (всегда под рукой). Важно отметить, что NK-клетки секретируют отличный от Т-клеток спектр цитокинов, которые привлекают другие иммунокомпетентные клетки, но при этом не вызывают цитокинового шторма [15]. Кроме того, использование NK-клеток в качестве носителей CAR открывает уникальную возможность применения CAR-технологии в терапии Т-клеточных лейкозов и лимфом. И действительно, использование с этой целью CAR-T-клеток сопряжено с рядом серьезных ограничений, включая присутствие мишеней CAR-T на самих CAR-T и вероятность непреднамеренной трансдукции CAR-кассетой перерожденных Т-клеток, что, в свою очередь, может делать такие клетки резистентными к уничтожению CAR-Т-клетками.
Адоптивный перенос CD19-специфических CAR-T-клеток приводит к беспрецедентной скорости развития полного ответа у больных лейкозом и лимфомой. Однако, несмотря на впечатляющие результаты при В-клеточных неоплазиях, эффект CAR-клеток при солидных опухолях пока ограничен [16]. Необходимы новые подходы, которые в дополнение к CAR-клеткам позволят преодолеть иммуносупрессорное микроокружение опухолей и гетерогенность экспрессии опухолеассоциированных антигенов. На роль синергистов CAR-клеток для усиления противоопухолевого эффекта хорошо подходят онколитические вирусы, которые к тому же можно генетически модифицировать для доставки терапевтических трансгенов с целью коррекции опухолевого микроокружения и усиления эффекторных функций опухолеспецифичных Т-клеток.
CAR-Т-КЛЕТОЧНАЯ ТЕРАПИЯ ОПУХОЛЕЙ
Для успешной терапии солидных опухолей CAR-Т-клетки должны эффективно проникать в опухоль, пролиферировать и персистировать в ней [17‒19]. В целом, Т-клетки способны быстро поступать в пораженные болезнью участки организма, однако опухоли, как правило, отличаются низким уровнем воспалительных реакций и отсутствием хемокинов, необходимых для миграции Т-клеток. Опухоли содержат также физические барьеры для Т-клеток: аберрантную васкулатуру, большую плотность ткани и высокое внутритканевое давление (рис. 1а). Пролиферация Т-клеток в опухоли ингибируется микроокружением. В ряде случаев эти ограничения можно преодолеть прямым введением CAR-Т-клеток в опухолевый узел, если он доступен [20, 21].
Рис. 1.
Комбинированная терапия солидной опухоли онколитическими вирусами и CAR-лимфоцитами. а ‒ CAR-T-клетки связываются с антигеном, соответствующим химерному рецептору, на поверхности опухолевых клеток и убивают их, но не могут продвинуться вглубь плотной опухолевой массы, а также элиминировать антигеннегативные опухолевые клетки. б ‒ CAR-NK-клетки обладают дополнительной противоопухолевой активностью по сравнению с CAR-T-клетками за счет связывания со стресс-лигандами на поверхности опухолевых клеток. в ‒ Онколитический вирус инфицирует клетки опухоли и убивает их, разрушая плотную структуру опухоли. г ‒ Последовательное введение онколитического вируса и CAR-T-клеток в опухоль обеспечивает аддитивный литический эффект.
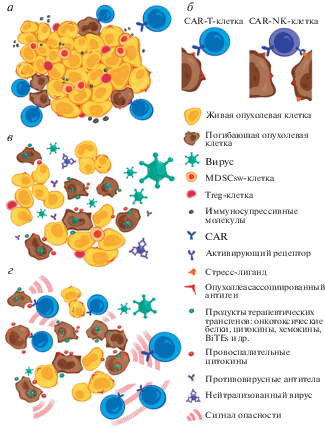
Для реализации противоопухолевого потенциала CAR-Т-клетки должны преодолеть иммуносупрессию в опухоли, которая обеспечивается: 1) миелоидными супрессорами (myeloid-derived suppressor cells, MDSCsw), опухолеассоциированными макрофагами, нейтрофилами и регуляторными Т-клетками (Treg); 2) большим количеством иммуносупрессорных молекул, например IL-10, IL-4, TGF-β, PD-L1, IDO, аргиназа-1 и др.; 3) факторами микроокружения, такими как гипоксия, низкие значения рН и недостаток питательных веществ. Для решения этих проблем разрабатывается целый ряд подходов, включающих дополнительные модификации как самих CAR [22], так и несущих их CAR-T-клеток [23].
Эволюция структуры CAR описана в ряде обзоров [22, 24]. Оказалось, что одного сигнального домена, состоящего из ζ-цепи CD3, в составе CAR первого поколения недостаточно для обеспечения значимого терапевтического эффекта CAR-T-клеток, несмотря на специфичное распознавание антигена на поверхности клетки-мишени и последующую активацию. Введение в CAR дополнительного сигнального домена (второе поколение), заимствованного у костимулирующих рецепторов, таких как CD28, 4-1ВВ или ОХ40, способствовало улучшению пролиферации, устойчивости и цитотоксичности CAR-T-клеток in vivo. Именно использование CAR второго поколения стало настоящим прорывом в адоптивной клеточной терапии и выявило гигантский потенциал этой терапии в клинических испытаниях [22]. Наиболее широко изучены терапевтические свойства CAR-T-клеток с костимулирующими доменами от CD28 и 4-1BB. Следующим шагом в эволюции CAR стало третье поколение, содержащее два костимулирующих домена. По-видимому, третье поколение CAR может быть более эффективным при отдельных патологиях [25], однако в подавляющем большинстве клинических испытаний по-прежнему используют CAR второго поколения.
Активность CAR-T-клеточной терапии повышают также с помощью модификации самих CAR-T-клеток, которая включает: введение кассет экспрессии для продукции секретируемых антител против внутриопухолевых иммуносупрессорных молекул (анти-CTLA-4/PD-1) или, напротив, стимулирующих цитокинов IL-12, IL-15, IL-18, IL-21 (TRUCKs ‒ T cells Redirected for Universal Cytokine Killing); нокаут ингибирующих рецепторов; экспрессию рецепторов хемокинов-аттрактантов (CCR2, CXCR1, CXCR2) [23]. Однако все эти модификации, хотя и улучшают миграцию и персистенцию CAR-T-клеток в солидных опухолях, не снимают проблемы неконтролируемой внеопухолевой токсичности. Экспансию CAR-T-клеток при тяжелых осложнениях останавливают с использованием индуцибельных суицидальных систем, таких как тимидинкиназа вируса простого герпеса [26] и iCasp9 [27], системы, обеспечивающей контролируемую экспрессию CAR [28, 29], и системы, основанной на конститутивной экспрессии поверхностных эпитопов, узнаваемых терапевтически одобренными моноклональными антителами, такими как ритуксимаб (RQR8/CD20) [30] и цетуксимаб (EGFRt) [31].
Усилению онкоселективности и снижению внеопухолевой токсичности способствует также правильный выбор антигена – мишени CAR. С развитием молекулярной диагностики появляется все больше данных о том, что структура опухолеассоциированных антигенов отличается от структуры аналогичных белков в нормальных клетках вследствие мутаций и нарушений посттрансляционного процессинга, в частности гликозилирования [23]. Использование новых высокоспецифичных для опухолей антигенов (неоантигенов) в качестве мишеней CAR призвано обеспечить строгую избирательность CAR-T-клеток в отношении опухолей и безопасность для нормальных тканей, но примеры применения таких CAR-T-клеточных продуктов пока не описаны.
Эффективность CAR-Т-клеточной терапии зависит от присутствия соответствующего CAR-антигена на поверхности опухолевых клеток. Однако солидные опухоли характеризуются, как правило, гетерогенностью уровня экспрессии антигена вплоть до полного его отсутствия (рис. 1а). Опухолевые клетки без антигена становятся невидимыми для CAR-Т-клеток [32‒34] и обеспечивают продолжение роста опухоли. Доклинические исследования показали, что раковые клетки с высоким уровнем экспрессии целевого антигена элиминируются преимущественно CAR-Т-клетками, в то время как клетки с низким уровнем экспрессии выживают [35‒37]. Снижение экспрессии целевого антигена, включая Her2 [38], EGFRvIII [39], IL13Rα2 [40] и мезотелин [41], наблюдали после CAR-Т-клеточной терапии в нескольких клинических исследованиях. Для преодоления проблемы гетерогенности опухолей предложено использовать новые варианты CAR, способные узнавать несколько антигенов [35, 42, 43]. Однако такой подход может привести к увеличению частоты осложнений CAR-Т-клеточной терапии, поскольку клетки здоровых тканей также могут продуцировать небольшое количество целевых антигенов, способных перекрестно реагировать с CAR [44‒46]. Чем больше мишеней охватывает CAR, тем больше репертуар здоровых клеток, которые могут подвергнуться цитотоксической активности таких CAR-Т-клеток. Позитивным фактором, участвующим в преодолении опухолевой гетерогенности, является показанная в ряде работ активация эндогенной иммунной системы, которая происходит в процессе CAR-Т-клеточной деструкции опухолевых клеток с сопутствующим высвобождением опухолевых неоантигенов [47, 48]. Это наблюдение требует дополнительных исследований, однако может объяснить феномен полной элиминации некоторых гетерогенных солидных опухолей при CAR-Т-клеточной терапии [49].
CAR-NK-КЛЕТОЧНАЯ ТЕРАПИЯ
NK-клетки ‒ уникальная разновидность лимфоцитов, отличных от В- и Т-клеток, представляют собой мостик между врожденной и адаптивной иммунными системами. NK-клетки проявляют иммунорегуляторные и цитотоксические функции в отношении трансформированных и инфицированных клеток без предварительной сенсибилизации [50]. Цитотоксическая активность NK-клеток регулируется балансом сигналов от активирующих и ингибирующих рецепторов на их поверхности. Лигандами этих рецепторов служат молекулы MHC класса I, индуцируемые стрессом белки MICA/B, белки семейства ULBP и некоторые другие молекулы [51]. В результате этого опухолевые клетки, экспрессирующие стресс-лиганды и/или снизившие экспрессию MHC-I, могут стать мишенью для NK-клеток (pис. 1б). NK-клетки также способствуют миграции дендритных клеток в опухолевый очаг, что усиливает реакции адаптивного иммунного ответа [52]. Естественно, что подобные свойства NK-клеток делают их привлекательными для терапевтического применения.
Клинические испытания выявили нетоксичность аутологичных NK-клеток, однако их противоопухолевая активность оказалась невысокой [53]. Аллогенные NK-клетки вызывали более выраженный терапевтический эффект. В частности, инфузия аллогенных NK-клеток вместе с IL-2 привела к полной ремиссия у пяти из 19 больных острым миелоидным лейкозом с плохим прогнозом [54]. Еще более успешные результаты получены при использовании гаплоидентичных NK-клеток в сочетании с химерным белком (IL-2-дифтерийный токсин), который удаляет Тreg-клетки реципиента [55].
Использование CAR способствует адресному усилению эффекторных функций NK-клеток в отношении определенных опухолей. Как уже упоминалось, CAR-Т-клетки способны оказывать достаточно сильный токсический эффект и могут вызывать синдром так называемого цитокинового шторма. Линейные аллогенные CAR-NK-клетки могут индуцировать противоопухолевый эффект и элиминироваться после этого в течение нескольких дней, что предотвращает развитие неспецифической токсичности и токсичности, связанной с синдромом лизиса опухоли [56]. К преимуществам CAR-NK-клеток относится и их универсальность, т.е. возможность использования одного и того же препарата CAR-NK-клеток разными пациентами с одним типом рака в зависимости от мишени CAR. Для конструирования CAR-NK-клеток могут использоваться те же мишени, которые уже тестируются для CAR-T-клеточной терапии [50].
Успех иммунотерапии зависит от возможности получения большого количества функциональных NK-клеток, которые могут выживать in vivo. В связи с этим были предприняты многочисленные попытки выделения NK-клеток из различных источников. Одной из возможностей является выделение клеток прямо из периферической или пуповинной крови, однако NK-клетки составляют только 10% лимфоцитов, циркулирующих в периферической крови, и 20% – в пуповинной, что повышает стоимость создания NK-клеточных продуктов для проведения множественных инфузий. Культивирование зрелых NK-клеток крови пока не получило широкого применения в клинике. Это связано с: 1) нестабильностью скорости пролиферации; 2) гетерогенностью, т.е. с присутствием различных субпопуляций в нативных NK-клетках; 3) снижением цитотоксичности клеток в процессе пролиферации in vitro [50]. Интересным представляется использование для экспрессии CAR NK-клеточных продуктов, полученных из iPS-клеток [57], однако это направление пока находится в стадии активного развития и судить о его эффективности и безопасности можно будет только после проведения клинических испытаний.
Крайне перспективным представляется использование перевиваемых культур NK-клеток в качестве носителей CAR, поскольку такие клеточные линии могут храниться в замороженном виде и представлены гомогенной популяцией. Наиболее известная линия NK-92, полученная от больного неходжкинской лимфомой, оказалась способной лизировать лейкозные, лимфомные и миеломные клетки in vitro [58]. Клинические испытания этой линии не выявили токсичности, однако явный терапевтический эффект также отсутствовал [59, 60]. Предполагается, что CAR-модифицированные клетки линии NK-92 будут сочетать в себе безопасность родительской линии и эффективность CAR-T-клеток [15].
Помимо линии NK-92, наиболее часто используемой в качестве носителя CAR, существуют и другие менее охарактеризованные NK-клеточные линии со сравнимым цитотоксическим потенциалом и отличным профилем поверхностных рецепторов [61, 62]. Различия в нюансах биологии и культивирования таких линий делают их привлекательными носителями CAR применительно к различным типам новообразований. Соответственно, показано, что CAR-модификация NK-клеточных линий KHYG-1 [63‒65], YTS [66] и YT [67‒69] приводит к перенаправлению их активности и уничтожению опухолевых клеток in vitro и/или in vivo.
ВИРОТЕРАПИЯ ОПУХОЛЕЙ
Большое разнообразие онколитических вирусов делает трудным выбор наиболее перспективного из них для проведения совместной терапии с CAR-клетками. Логичным было бы использовать в первую очередь онколитические вирусы, представленные на фармацевтическом рынке или проходящие клинические исследования. В настоящее время коммерчески доступны три онколитических вируса: вирус простого герпеса T-VEC (Imlygic), одобренный в США; аденовирус H101 (Oncorine), одобренный в Китае, и энтеровирус Rigvir, одобренный в Латвии, Армении и Грузии. Еще несколько вирусов находятся на заключительных этапах клинических испытаний [70]. По всей видимости, выбор конкретной комбинации онколитического вируса и CAR-носителя в значительной степени зависит от типа патологии и способности вируса поражать клетки-носители CAR.
Согласно результатам клинических испытаний, онколитические вирусы индуцируют терапевтический эффект, включая полную деструкцию опухоли, без тяжелых побочных эффектов [71‒74] (рис. 1в). Важно отметить, что в некоторых случаях уничтожение опухоли происходит уже после элиминации вируса из организма, что свидетельствует об активации специфического противоопухолевого иммунного ответа [72]. Вирусы являются сильными иммуногенами, но противовирусный иммунитет препятствует виротерапии опухолей, поскольку антитела нейтрализуют вирионы и не позволяют им достигать опухоли. В связи с этим возникает вопрос доставки вируса в опухолевый узел при повторных инъекциях.
Репликация вируса детектируется в опухолевых биопсиях через несколько дней после введения вируса, однако способность вируса выживать и распространяться в опухоли ограничивается активностью вирусспецифических Т-клеток [71, 72, 75]. Показано, что виротерапия усиливает инфильтрацию опухоли клетками иммунной системы, включая активированные макрофаги и цитотоксические Т‑клетки, а также приводит к локальному увеличению концентрации ряда провоспалительных цитокинов [71, 72, 75]. Опухольспецифические Т‑клетки также выявляются после виротерапии [75, 76]. Проходящие в настоящее время клинические испытания комбинации виротерапии с таргетной иммунотерапией (ингибиторы контрольных точек, check point inhibitors) демонстрируют очень обнадеживающие результаты [77‒80].
Вирусы, тестируемые как средства онкотерапии, принадлежат к различным таксономическим группам и существенно отличаются по структуре и параметрам жизненного цикла [16]. Вирусы, которые реплицируются в цитоплазме клеток (почти все вирусы с РНК-геномом), убивают клетки быстрее, чем “ядерные” вирусы, поскольку они не тратят время на достижение ядра. Однако РНК-вирусы высокочувствительны к интерферону и способны эффективно реплицироваться только в опухолях с нарушениями в механизмах индукции интерферона. Это, с одной стороны, повышает онкоселективность вирусов, а с другой, ограничивает спектр чувствительных к виротерапии опухолевых клеток. Оболочечные вирусы, такие как парамиксовирусы, вирус везикулярного стоматита, герпесвирус и вирус осповакцины, обладают в ряде случаев меньшей литической активностью, чем необолочечные, поскольку используют почкование для выхода из клетки, сохраняя ее жизнеспособность. Однако оболочечные вирусы более защищены при распространении внутри опухоли и к метастазам, так как оболочка частично защищает их от действия системы комплемента и нейтрализации антителами. Важным параметром является также размер вириона ‒ чем меньше вирус, тем легче он распространяется внутри опухоли. При этом вирусы с большим геномом обладают большей генетической емкостью и способны экспрессировать терапевтические трансгены, направленно модифицирующие их свойства. Трансгены включали в геномы таких РНК-вирусов, как вирусы везикулярного стоматита и парамиксовирусы, используемые в онколитических приложениях, а также в онколитические ДНК-вирусы ‒ аденовирусы, герпесвирусы и вирус осповакцины. Пикорнавирусы, реовирусы и парвовирусы обладают маленькими (или фрагментированными) геномами, размер которых (фрагментов) не позволяет вводить функционально значимые трансгены.
Список трансгенов, которые применяют для “вооружения” вирусов в борьбе с опухолями, достаточно большой [81]. Этот список включает: индукторы клеточной смерти и онкотоксические белки [82‒85]; цитокины [84‒89]; хемокины [90, 91]; костимуляторные белки [92‒95]; биспецифические активаторы Т-клеток (BiTE) [36, 96, 97]; блокаторы иммунных контрольных точек [98‒101]; деградирующие строму белки, которые облегчают распространение онколитических вирусов в опухоли [82]. Ряд этих трансгенов используется для усиления противоопухолевой активности CAR-Т-клеток при совместной терапии с рекомбинантными онколитическими вирусами (табл. 1).
Таблица 1.
Примеры комбинированной терапии CAR-Т-клетками и онколитическими вирусами
| Исходный вирус, штамм |
Трансген | CAR | Модельная опухоль | Ссылка |
|---|---|---|---|---|
| Аденовирус Ad5-D24 (частичная делеция гена Е1А) |
Хемокин RANTES + + цитокин IL-15 человека |
GD2 | Нейробластома, подкожные ксенографты NSG мышей | [102] |
| Аденовирус Ad5/3-D24 (частичная делеция гена Е1А и химерный 3/5 серотип) |
TNF-α + IL-2 человека |
Мезотелин | Аденокарцинома поджелудочной железы, подкожные ксенографты NSG мышей | [103] |
| Аденовирус CAd-VEC (смесь Ad5-D24 и нереплицирующегося варианта HDΔ28E4) | PD-L1-блокирующие миниантитела | aнти-HER2 | HER2 + рак предстательной железы, подкожные ксенографты NSG мышей | [104] |
| Аденовирус смесь Ad5-D24 и нереплицирующегося варианта HDΔ28E4 |
IL12p70 + + PD-L1-блокирующие миниантитела |
анти-HER2 | Рак головы и шеи (HNSCC), подкожные и ортотопические ксенографты NSG мышей | [105] |
| Аденовирус ICOVIR 15 (Ad5-D24 с модифицированной фибриллой) |
Антитела с двойной специфичностью (BiTE): анти-EGFR и анти-CD3 | Рецептор фолата-α (FR-α) |
Рак прямой кишки (НСТ116) и поджелудочной железы (Panc-1), подкожные ксенографты NSG мышей |
[36] |
| Вирус осповакцины vvDD (делеции генов тимидинкиназы и вирусного фактора роста) |
CXCL11 мыши | Мезотелин (человек) | Опухоль легкого мыши TC1, продуцирующая мезотелин человека (TC1-meso) | [106] |
КОМБИНИРОВАНИЕ ВИРОТЕРАПИИ И CAR-КЛЕТОЧНОЙ ТЕРАПИИ ОПУХОЛЕЙ
Онколитические вирусы являются идеальным партнером CAR-клеток для преодоления ими множественных внутриопухолевых барьеров (рис. 1г). Прямой лизис опухолевых клеток вирусами разрыхляет опухоль и облегчает инфильтрацию CAR-клеток. Инфицированные вирусами опухолевые клетки испускают сигналы опасности, которые снимают иммуносупрессию в опухоли, чем обеспечивают внутриопухолевый трафик, пролиферацию и персистенцию CAR-Т-клеток [107]. В процессе индуцированного вирусом лизиса клеток происходит высвобождение внутриклеточных опухолеассоциированных антигенов (неоантигенов) и формирование Т-клеточного ответа на эти антигены [75, 76], который в синергизме с CAR-Т-клетками и вирусспецифичными Т-клетками способствует элиминации гетерогенных по составу клеток опухоли.
К настоящему времени описано несколько успешных примеров объединения технологий виротерапии и CAR-Т-клеточной иммунотерапии опухолей (табл. 1). Эффективность такой двойной терапии оценивали на мышах NOD scid gamma (NSG) с тяжелыми нарушениями адаптивного и врожденного иммунитета [108]. Использование этих мышей позволяет прямо оценивать активность как введенных адоптивно модифицированных Т-клеток человека, так и онколитических вирусов в отношении ксенографтов опухолей человека. Однако эта модель не позволяет оценить противоопухолевый иммунитет, индуцированный онколитическими вирусами, а также вклад противовирусного иммунитета в подавление распространения введенного вирусного препарата.
На мышах линии NSG показано, что рекомбинантные аденовирусы, экспрессирующие трансгены IL-15 и RANTES, усиливают миграцию и длительность персистенции CAR-Т-клеток, экспрессирующих GD2-специфический CAR, в ксенографтах нейробластомы человека [102]. При этом значимо увеличивается продолжительность жизни мышей. Аналогичные результаты получены при комбинированной терапии рака поджелудочной железы онколитическим аденовирусом со вставкой трансгенов IL-2 и TNF-α и CAR-T-клетками, экспрессирующими мезотелин-специфичный CAR [103]. В этом случае выявлен не только противоопухолевый, но и антиметастатический эффект.
Использование рекомбинантного аденовируса, кодирующего EGFR-специфичный BiTE, в сочетании с FRa-специфичными CAR-T-клетками позволяет преодолеть гетерогенность антигенной экспрессии в опухоли [36]. Более того, в такой системе наблюдается активация как CAR-Т-клеток, так и нетрансдуцированных Т-клеток. Еще один сходный подход состоял в использовании рекомбинантного аденовируса, экспрессирующего блокирующие PD-L1 миниантитела, которые снимали дисфункцию Т-клеток в опухоли, предотвращая взаимодействие PD1 : PDL1 [104]. Дополнительное введение в вирус трансгена IL12p70 усиливало терапевтическую эффективность комбинации [105]. Во всех случаях сочетанной терапии аденовирус вводили первым локально в район опухоли, поскольку при системном введении его эффективность была значительно ниже.
Описана также терапия опухоли в иммунокомпетентном формате с использованием рекомбинантного вируса осповакцины в комбинации с CAR-T-клетками. Последовательное введение мышам с привитой сингенной опухолью легких рекомбинантного вируса осповакцины, экспрессирующего хемокин CXCL11 мыши, и опухолеспецифичных CAR-Т-клеток вызывало значимо лучший противоопухолевый эффект, чем монотерапия каждым из компонентов [106].
Опубликован еще один интересный подход, в котором CAR-Т-клетки используются для доставки онколитического вируса в опухоль [109]. Показано, что циркулирующие в организме клетки, такие как лимфоциты, моноциты, эритроциты и даже тромбоциты, могут сорбировать вирусы и способствовать их проникновению в опухоли [110‒113]. Введение онколитического вируса в опухолеспецифичные Т-клетки может защитить его от инактивации нейтрализующими антителами и обеспечить сохранность его противоопухолевой активности после высвобождения в микроокружении опухоли [113].
ЗАКЛЮЧЕНИЕ
Несмотря на очевидную перспективность сочетанной CAR-клеточной и вирусной терапии опухолей, остается много нерешенных вопросов. Один из них – выбор схемы и методов введения препаратов. Внутриопухолевое введение обеспечивает быстрое накопление онколитических вирусов в опухоли, однако оно неприменимо для многих опухолей и особенно метастазов. Системное введение вируса проще и потенциально эффективнее для достижения всех опухолевых узлов, включая метастазы. Однако в этом случае возрастает вероятность возникновения постинъекционных осложнений. Кроме того, высокие титры противовирусных антител, появляющихся после первого введения вируса, будут способствовать нейтрализации вирусных частиц в кровяном русле и нивелировать эффект последующих инъекций препарата. Таким образом, перспективной представляется разработка способов обеспечения временной защиты вводимых вирусных препаратов от инактивации антителами.
Необходимо также установить порядок введения онколитического вируса и CAR-клеток. Логично было бы вводить вирус первым, чтобы он изменил иммуносупрессивное микроокружение опухоли, индуцировал прямой литический эффект, облегчил инфильтрацию опухоли CAR-клетками и представлял иммунной системе опухолевые неоантигены. Однако неоантигены, высвобождаемые в процессе лизиса опухолевых клеток онколитическими вирусами, как правило, менее иммуногенны, чем собственно вирусные антигены [114‒116]. Нужны новые стратегии повышения иммуногенности опухолевых эпитопов и уменьшения иммунодоминантности вирусных антигенов с целью переадресации иммунного ответа. С этой же целью необходимо совершенствовать клеточный компонент совместной терапии. В частности, вирусспецифичные Т-клетки можно использовать для экспрессии CAR, направленных на опухоль [117]. Вирусспецифичные CAR-Т-клетки могут аккумулировать способность распознавать инфицированные вирусом и неинфицированные опухолевые клетки через их нативные и химерные рецепторы соответственно. Такие клетки представляют собой идеальный вариант носителей CAR для комбинированной терапии, так как присутствие вируса будет не только снимать иммуносупрессию внутри опухоли и обеспечивать инфильтрацию CAR-Т-клеток, но также стимулировать их амплификацию в опухоли. Очевидным недостатком этого подхода является ускорение клиренса онколитического вируса из организма.
Проведенные к настоящему времени эксперименты, в которых использована комбинированная CAR-клеточная и вирусная терапия солидных опухолей, вызывают синергичный эффект и снятие ключевых ограничений монотерапии для каждого из этих препаратов. Возможности комбинирования различных онколитических вирусов с опухолеспецифичными CAR-клетками практически безграничны, а широкая применимость этой технологии дает надежду на решение проблемы улучшения терапии при солидных опухолях поздних стадий.
Работа поддержана грантом Российского фонда фундаментальных исследований (№ 18-29-09044).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Lawler S.E., Speranza M.C., Cho C.F., Chiocca E.A. (2017) Oncolytic viruses in cancer treatment. A Review. JAMA Oncol. 3, 841‒849.
Kaufman H.L., Kohlhapp F.J., Zloza A. (2015) Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 14, 642‒662.
Swift S.L., Stojdl D.F. (2016) Big data offers novel insights for oncolytic virus immunotherapy. Viruses. 8, 45.
Choi A.H., O’Leary M., Fong Y., Chen N. (2016) From benchtop to bedside: a review of oncolytic virotherapy. Biomedicines. 4, 18.
Greig S.L. (2016) Talimogene Laherparepvec: first global approval. Drugs. 76, 147‒154.
Maude S.L., Laetsch T.W., Buechner J.S., Rives M., Boyer H., Bittencourt P., Bader M.R., Verneris H.E., Stefanski G.D., Myers M., Qayed B., De Moerloose H., Hiramatsu K., Schlis K.L., Davis K.L., Martin P.L., Nemecek E.R., Yanik G.A., Peters C., Baruchel A., Boissel N., Mechinaud F., Balduzzi A., Krueger J., June C.H., Levine B.L., Wood P., Taran T., Leung M, Mueller K.T., Zhang Y., Sen K., Lebwohl D., Pulsipher M.A., Grupp S.A. (2018) Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439‒448.
Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y., Timmerman J.M., Stiff P.J., Friedberg J.W., Flinn I.W., Goy A., Hill B.T., Smith M.R., Deol A., Farooq U., McSweeney P., Munoz J., Avivi I., Castro J.E., Westin J.R., Chavez J.C., Ghobadi A., Komanduri K.V., Levy R., Jacobsen E.D., Witzig T.E., Reagan P., Bot A., Rossi J., Navale L., Jiang Y., Aycock J., Elias M., Chang D., Wiezorek J., Go W.Y. (2017) Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531‒2544.
https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm581222.htm
https://www.fda.gov/biologicsbloodvaccines/cellulargenetherapyproducts/approvedproducts/ucm573706.htm
Lee D.W., Gardner R., Porter D.L., Louis C.U, Ahmed N., Jensen M., Grupp S.A., Mackall C.L. (2014) Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 124, 188‒195.
Kalos M., Nazimuddin F., Finklestein J.M., Gupta M., Kulikovskaya I., Ambrose D.E., Gill S., Lacey S.F., Zheng Z., Melenhorst J.J., Levine B.L., Frey N.V., Grupp S.A., Porter D.L., June C.H. (2013) Long-term functional persistence, B cell aplasia and anti-leukemia efficacy in refractory B cell malignancies following T cell immunotherapy using CAR-redirected T cells targeting CD19. Blood. 122, 163.
Porter D.L., Hwang W.T., Frey N.V., Lacey S.F., Shaw P.A., Loren A.W., Bagg A., Marcucci K.T., Shen A., Gonzalez V., Ambrose D., Grupp S.A., Chew A., Zheng Z., Milone M.C., Levine B.L., Melenhorst J.J., June C.H. (2015) Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 7, 303ra139.
Bonifant C.L. (2016) Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics. 3, 16011.
Klingemann H.G., Wong E., Maki G. (1996) A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol. Blood Marrow Transplant. 2, 68‒75.
Zhang C., Oberoi P., Oelsner S., Waldmann A., Lindner A., Tonn T., Wels W. S. (2017) Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 8, 533.
Guedan S., Alemany R. (2018) CAR-T cells and oncolytic viruses: joining forces to overcome the solid tumor challenge. Front. Immunol. 9, 2460.
Louis C.U., Savoldo B., Dotti G., Pule M., Yvon E., Myers G.D., Rossig C., Russell H.V., Diouf O., Liu E., Liu H., Wu M-F, Gee A.P., Mei Z., Rooney C.M., Heslop H.E., Brenner M.K. (2011) Antitumor activity and long-term fate of chimeric antigen receptor positive T cells in patients with neuroblastoma. Blood. 118, 6050‒6056.
Rosenberg S.A., Yang J.C., Sherry R.M., Kammula U.S., Hughes M.S., Phan G.Q., Citrin D.E., Restifo N.P., Robbins P.F., Wunderlich J.R., Morton K.E., Laurencot C.M., Steinberg S.M., White D.E., Dudley M.E. (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 17, 4550–4557.
Fraietta J.A., Lacey S.F., Orlando E.J., Pruteanu-Malinici I., Gohil M., Lundh S., Boesteanu A.C., Wang Y., O’Connor R.S., Hwang W.T., Pequignot E., Ambrose D.E., Zhang C., Wilcox N., Bedoya F., Dorfmeier C., Chen F., Tian L, Parakandi H., Gupta M., Young R.M., Johnson F.B., Kulikovskaya I., Liu L., Xu J., Kassim S.H., Davis M.M., Levine B.L., Frey N.V., Siegel D.L., Huang A.C., Wherry E.J., Bitter H., Brogdon J.L., Porter D.L., June C.H., Melenhorst J.J. (2018) Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 24, 563–571.
Adusumilli P.S., Cherkassky L., Villena-Vargas J., Colovos C., Servais E., Plotkin J., Jones D.R., Sadelain M. (2014) Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 6, 261ra151.
Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J., Kurien A., Priceman S.J., Wang X., Harshbarger T.L., D’Apuzzo M., Ressler J.A., M.D., Jensen M.C., M.D., Barish M.E., Chen M., Portnow J., Forman S.J., Badie B. (2016) Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569.
Yan L., Liu B. (2019) Critical factors in chimeric antigen receptor-modified T-cell (CAR-T) therapy for solid tumors. OncoTargets Therapy. 12, 193–204.
Knochelmann H.M., Smith A.S., Dwyer C.J., Wyatt M.M., Mehrotra S., Paulos C.M. (2018) CAR T cells in solid tumors: blueprints for building effective therapies. Front. Immunol. 9, 1740.
Salmikangas P., Kinsella N., Chamberlain P. (2018) Chimeric antigen receptor T-cells (CAR T-Cells) for cancer immunotherapy – moving target for industry? Pharm. Res. 35, 152.
http://www.bloodjournal.org/content/128/22/1851
Berger C., Flowers M., Warren E., Riddell S. (2006) Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 107, 2294–2302.
Gargett T., Brown M.P. (2014) The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 5, 235.
Gu X., He D., Li C., Wang H., Yang G. (2018) Development of inducible CD19-CAR T cells with a Tet-On system for controlled activity and enhanced clinical safety. Internat. J. Mol. Sci. 19, 3455‒3466.
Mamonkin M., Mukherjee M., Srinivasan M., Gomes-Silva D., Mo F., Krenciute G., Orange J.S., Brenner M.K. (2017) Reversible transgene expression reduces fratricide and permits 4-1BB costimulation of CAR T cells directed to T-cell malignancies. Cancer Immunol. Res. 6, 47‒58.
Philip B., Kokalaki E., Mekkaoui L., Thomas S., Straathof K., Flutter B., Marin V., Marafioti T., Chakraverty R., Linch D., Quezada S.A., Peggs K.S., Pule M. (2014) A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 124, 1277‒1287.
Paszkiewicz P.J., Fräßle S.P., Srivastava S., Sommermeyer D., Hudecek M., Drexler I., Sadelain M., Liu L., Jensen M.C., Riddell S.R., Busch D.H. (2016) Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest. 126, 4262‒4272.
Fry T.J., Shah N.N., Orentas R.J., Stetler-Stevenson M., Yuan C.M., Ramakrishna S., Wolters P., Martin S., Delbrook C., Yates B., Shalabi H., Fountaine T.J., Shern J.F., Majzner R.G., Stroncek D.F., Sabatino M., Feng Y., Dimitrov D.S., Zhang L., Nguyen S., Qin H., Dropulic B., Lee D.W., Mackall C.L. (2018) CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 24, 20–28.
Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., Mahnke Y.D., Melenhorst J.J., Rheingold S.R., Shen A., Teachey D.T. Levine B.L., June C.H., Porter D.L., Grupp S.A. (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517.
Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F., Milone M.C., Levine B.L., June C.H. (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518.
Anurathapan U., Chan R.C., Hindi H.F., Mucharla R., Bajgain P., Hayes B.C., Fisher W.E., Heslop H.E., Rooney C.M., Brenner M.K., Leen A.M., Vera J.F. (2014) Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol. Ther. 22, 623–633.
Wing A., Fajardo C.A., Posey A.D. Jr., Shaw C., Da T., Young R.M., Alemany R., June C.H., Guedan S. (2018) Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol. Res. 6, 605–616.
Song D.G., Ye Q., Poussin M., Chacon J.A., Figini M., Powell D.J. (2016) Effective adoptive immunotherapy of triple-negative breast cancer by folate receptoralpha redirected CAR T cells is influenced by surface antigen expression level. J. Hematol. Oncol. 9, 56.
Ahmed N., Brawley V.S., Hegde M., Robertson C., Ghazi A., Gerken C., Liu E., Dakhova O., Ashoori A., Corder A., Gray T., Wu M-F., Liu H., Hicks J., Rainusso N., Dotti G., Mei Z., Grilley B., Gee A., Rooney C.M., Brenner M.K., Heslop H.E., Wels W.S., Wang L.L., Anderson P., Gottschalk S. (2015) Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 33, 1688–1696.
O’Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A., Isaacs R., Mohan S., Plesa G., Lacey S.F., Navenot J.M., Zheng Z., Levine B.L., Okada H., June C.H., Brogdon J.L., Maus M.V. 2017. A single dose of peripherally infused EGFRvIII directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984.
Brown C.E., Badie B., Barish M.E., Weng L., Ostberg J.R., Chang W.C., Naranjo A., Starr R., Wagner J., Wright C., Zhai Y., Bading J.R., Ressler J.A., Portnow J., D’Apuzzo M., Forman S.J., Jensen M.C. (2015) Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 21, 4062–4072.
Beatty G.L., O’Hara M.H., Lacey S.F., Torigian D.A., Nazimuddin F., Chen F., Kulikovskaya I.M., Soulen M.C., McGarvey M., Nelson A.M., Gladney W.L., Levine B.L., Melenhorst J.J., Plesa G., June C.H. (2018) Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 155, 29–32.
Hegde M., Corder A., Chow K.K., Mukherjee M., Ashoori A., Kew Y., Zhang Y.J., Baskin D.S., Merchant F.A., Brawley V.S., Byrd T.T., Krebs S., Wu M.F., Liu H., Heslop H.E., Gottschalk S., Yvon E., Ahmed N. (2013) Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 21, 2087–101.
Hegde M., Mukherjee M., Grada Z., Pignata A., Landi D., Navai S.A., Wakefield A., Fousek K., Bielamowicz K., Chow K.K., Brawley V.S., Byrd T.T., Krebs S., Gottschalk S., Wels W.S., Baker M.L., Dotti G., Mamonkin M., Brenner M.K., Orange J.S., Ahmed N. (2016) Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Invest. 126, 3036–3052.
Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. (2010) Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851.
Lamers C.H., Sleijfer S., Vulto A.G., Kruit W.H., Kliffen M., Debets R., Gratama J.W., Stoter G., Oosterwijk E. (2006) Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol. 24, e20–e22.
Thistlethwaite F.C., Gilham D.E., Guest R.D., Rothwell D.G., Pillai M., Burt D.J., Byatte A.J., Kirillova N., Valle J.W. Sharma S.K., Chester K.A., Westwood N.B., Halford S.E.R., Nabarro S., Wan S., Austin E., Hawkins R.E. (2017) The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 66, 1425–1436.
Beatty G.L., Haas A.R., Maus M.V., Torigian D.A., Soulen M.C., Plesa G., Chew A., Zhao Y., Levine B.L., Albelda S.M., Kalos M., June C.H. (2014) Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2, 112–120.
Sampson J.H., Choi B.D., Sanchez-Perez L., Suryadevara C.M., Snyder D.J., Flores C.T., Schmittling R.J., Nair S.K., Reap E.A., Norberg P.K., Herndon J.E., Kuan C.T., Morgan R.A., Rosenberg S.A., Johnson L.A. (2014) EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin. Cancer Res. 20, 972–984.
Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J., Kurien A., Priceman S.J., Wang X., Harshbarger T.L., D’Apuzzo M., Ressler J.A., M.D., Jensen M.C.,Barish M.E., Chen M., Portnow J., Forman S.J., Badie B. (2016) Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569.
Domogala A., Madrigal J.A., Saudemont A. (2015) Natural killer cell immunotherapy: from bench to bedside. Front. Immunol. 6, 264.
Long E.O., Kim H.S., Liu D., Peterson M.E., Rajagopalan S. (2013) Controlling Natural Killer cell responses: Integration of signals for activation and inhibition. Annu. Rev. Immunol. 31, 227–258.
Böttcher J.P., Bonavita E., Chakravarty P., Blees H., Cabeza-Cabrerizo M., Sammicheli S., Rogers N.S., Sahai E., Zelenay S., Reis e Sousa C. (2018) NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 172, 1022–1037.e14.
Ljunggren H.G., Malmberg K.J. (2007) Prospects for the use of NK cells in immunotherapy of human cancer. Nat. Rev. Immunol. 7, 329–339.
Miller J.S., Soignier Y., Panoskaltsis-Mortari A., Mcnearney S.A., Yun G.H., Fautsch S.K., McKenna D., Le C., Defor T.E., Burns L.J., Orchard P.J., Blazar B.R., Wagner J.E., Slungaard A., Weisdorf D.J., Okazaki I.J., McGlave P.B. (2005) Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 105, 3051–3057.
Bachanova V., Cooley S., Defor T.E., Verneris M.R., Zhang B., Mckenna D.H., Curtsinger J., Panoskaltsis-Mortari A., Lewis D., Hippen K., McGlave P., Weisdorf D.J., Blazar B.R., Miller J.S. (2014) Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 123, 3855–3863.
Klingemann H. (2015) Challenges of cancer therapy with natural killer cells. Cytotherapy. 17, 245–249.
Li Y., Hermanson D.L., Moriarity B.S., Kaufman D.S. (2018) Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 23, 181‒192.e5.
Klingemann H.G., Wong E., Maki G. (1996) Acytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol. Blood Marrow Transplant. 2, 68–75.
Arai S., Meagher R., Swearingen M., Myint H., Rich E., Martinson J., Klingemann H. (2008) Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 10, 625–632.
Tonn T., Schwabe D., Klingemann H.G., Becker S., Esser R., Koehl U., Suttorp M., Seifried E., Ottmann O.G., Bug G. (2013) Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 15, 1563–1570.
Matsuo Y., Drexler H.G. (2003) Immunoprofiling of cell lines derived from natural killer-cell and natural killer-like t-cell leukemia-lymphoma. Leuk. Res. 27, 935–945.
Zhang J., Zheng H., Diao Y. (2019) Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 Cells in tumor immunotherapy. Int. J. Mol. Sci. 20, 317.
Kim M., Pyo S., Kang C.H., Lee C.O., Lee H.K., Choi S.U., Park C.H. (2018) Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS One. 13, e0198347.
Murakami T., Nakazawa T., Natsume A., Nishmura F., Nakamura M., Matsuda R., Omoto K., Tanaka Y., Shida Y., Park Y.-S., Motoyama Y., Nakagawa I., Yamadai S., Tamura K., Takeshima Y., Takamura Y., Wakabayashi T., Nakase H. (2018) Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. 38, 5049‒5056.
Kobayashi E., Kishi H., Ozawa T., Hamana H., Nakagawa H., Jin A., Lin Z., Muraguchi A. 2014. (2014) A chimeric antigen receptor for TRAIL-receptor 1 induces apoptosis in various types of tumor cells. Biochem. Biophys. Res. Commun. 31, 798‒803.
Topfer K., Marc Cartellieri M., Michen S., Wiedemuth R., Muller N., Lindemann D., Bachmann M., Fussel M., Schackert G., Temme A. (2015) DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 194, 3201‒3212.
Yodoi J., Teshigawara K., Nikaido T., Fukui K., Noma T., Honjo T., Takigawa M., Sasaki M., Minato N., Tsudo M. (1985) TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells). J. Immunol. 134, 1623–1630.
Matvienko D.A., Kulemzin S.V., Smagina A.S., Belovezhets T.N., Chikaev A.N., Volkova O.Y., Chikaev N.A., Koval O.A., Kuligina E.V., Taranin A.V., Gorchakov A.A. (2018) Analysis of in vitro activity of PSCA-specific CARs in the context of human NK cell line YT. Cell. Therapy Transplant. (CTT JOURNAL). 7, 70‒76.
Kulemzin S.V., Gorchakov A.A., Chikaev A.N., Kuznetsova V.V., Volkova O.Y., Matvienko D.A., Petukhov A.V., Zaritskey A.Y., Taranin A.V. (2018) VEGFR2-specific FnCAR effectively redirects the cytotoxic activity of T cells and YT NK cells. Oncotarget. 9, 9021‒9029.
Russell L., Peng K.W. (2018) The emerging role of oncolytic virus therapy against cancer. Chin. Clin. Oncol. 7, 16.
Geletneky K., Hajda J., Angelova A.L., Leuchs B., Capper D., Bartsch A.J., Neumann J.O., Schöning T., Hüsing J., Beelte B., Kiprianova I., Roscher M., Bhat R., von Deimling A., Brück W., Frehtman V., Löbhard S., Terletskaia-Ladwig E., Fry J., Jochims K., Daniel V., Krebs O., Dahm M., Huber B., Unterberg A., Rommelaere J. (2017) Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol. Ther. 25, 2620–2634.
Lang F.F., Conrad C., Gomez-Manzano C., Yung W.K.A., Sawaya R., Weinberg J.S., Prabhu S.S., Rao G., Fuller G.N., Aldape K.D., Gumin J., Vence L.M., Wistuba I., Rodriguez-Canales J., Villalobos P.A., Dirven C.M.F., Tejada S., Valle R.D., Alonso M.M., Ewald B., Peterkin J.J., Tufaro F., Fueyo J. (2018) Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus, replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 36, 1419–1427.
Russell S.J., Federspiel M.J., Peng K.W., Tong C., Dingli D., Morice W.G., Lowe V., O’Connor M.K., Kyle R.A., Leung N., Buadi F.K., Rajkumar S.V., Gertz M.A., Lacy M.Q., Dispenzieri A. (2014) Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin. Proc. 89, 926–933.
Cloughesy T.F., Landolfi J., Hogan D.J., Bloomfield S., Carter B., Chen C.C., Elder J.B., Kalkanis S.N., Ke-sari S., Lai A., Lee I.Y., Liau L.M., Mikkelsen T., Nghiemphu P.L., Piccioni D., Walbert T., Chu A., Das A., Diago O.R., Gammon D., Gruber H.E., Hanna M., Jolly D.J., Kasahara N., McCarthy D., Mitchell L., Ostertag D., Robbins J.M., Rodriguez-Aguirre M., Vogelbaum M.A. (2016) Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med. 8, 341ra75.
Garcia-Carbonero R., Salazar R., Duran I., Osman-Garcia I., Paz-Ares L., Bozada J.M., Boni V., Blanc C., Seymour L., Beadle J., Alvis S., Champion B., Calvo E., Fisher K. (2017) Phase 1 study of intravenous administration of the chimeric adenovirus enadenotucirev in patients undergoing primary tumor resection. J. ImmunoTherapy Cancer. 5, 71.
Woller N., Gurlevik E., Fleischmann-Mundt B., Schumacher A., Knocke S., Kloos A.M., Saborowski M., Geffers R., Manns M.P., Wirth T.C., Kubicka S., Kühnel F. (2015) Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol. Ther. 23, 1630–1640.
Zamarin D., Holmgaard R.B., Subudhi S.K., Park J.S., Mansour M., Palese P., Merghoub T., Wolchok J.D., Allison J.P. (2014) Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 6, 226ra32.
Rojas J.J., Sampath P., Hou W., Thorne S.H. (2015) Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin. Cancer Res. 21, 5543–5551.
Samson A., Scott K.J., Taggart D., West E.J., Wilson E., Nuovo G.J., Thomson S., Corns R., Mathew R.K., Fuller M.J., Kottke T.J., Thompson J.M., Ilett E.J., Cockle J.V., van Hille P., Sivakumar G., Polson E.S., Turnbull S.J., Appleton E.S., Migneco G., Rose A.S., Coffey M.C., Beirne D.A., Collinson F.J., Ralph C., Alan Anthoney D., Twelves C.J., Furness A.J., Quezada S.A., Wurdak H., Errington-Mais F., Pandha H., Harrington K.J., Selby P.J., Vile R.G., Grif fin S.D., Stead L.F., Short S.C., Melcher A.A. (2018) Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 10(422), eaam7577.
Bourgeois-Daigneault M.C., Roy D.G., Aitken A.S., El Sayes N., Martin N.., Varette O., Falls T., St-Germain L.E., Pelin A., Lichty B.D., Stojdl D.F., Ungerechts G., Diallo J.S., Bell J.C. (2018) Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 10(422), pii: eaao1641.
Ajina A., Maher J. (2017) Prospects for combined use of oncolytic viruses and CAR T-cells. J. ImmunoTherapy Cancer. 5, 90.
Guedan S., Grases D., Rojas J.J., Gros A., Vilardell F., Vile R., Mercade E., Cascallo M., Alemany R. (2012) GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 19, 1048–1057.
Kochneva G., Zonov E., Grazhdantseva A., Unusova A., Sivolobova G., Popov E., Taranov O., Netesov S., Chumakov P., Ryabchikova E. (2014) Apoptin enhances the oncolytic properties of vaccinia virus and modifies mechanisms of tumor regression. Oncotarget. 5, 11269‒11282.
Kochneva G.V., Sivolobova G.F., Tkacheva A.V., Grazhdantseva A.A., Troitskaya O.V., Nushtaeva A., Tkachenko A., Kuligina E., Richter V., Koval O. (2016) Engineering of double recombinant vaccinia virus with enhanced oncolytic potential for solid tumor virotherapy. Oncotarget. 7, 74171‒74188.
Koval O., Kochneva G., Tkachenko A., Troitskaya O., Sivolobova G., Grazhdantseva A., Nushtaeva A., Kuligina E., Richter V. (2017) Recombinant vaccinia viruses coding transgenes of apoptosis-inducing proteins enhance apoptosis but not immunogenicity of infected tumor cells. BioMed Res. Internat. 2017, 3620510.
Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S., Milhem M., Cranmer L., Curti B., Lewis K., Ross M., Guthrie T., Linette G.P., Daniels G.A., Harrington K., Middleton M.R., Miller W.H. Jr., Zager J.S., Ye Y., Yao B., Li A., Doleman S., VanderWalde A., Gansert J., Coffin R.S. (2015) Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 33, 2780–2788.
Carew J.F., Kooby D.A., Halterman M.W., Kim S.H., Federoff H.J., Fong Y. (2001) A novel approach to cancer therapy using an oncolytic herpes virus to package amplicons containing cytokine genes. Mol. Ther. 4, 250–256.
Stephenson K.B., Barra N.G., Davies E., Ashkar A.A., Lichty B.D. (2012) Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 19, 238–246.
Wang P., Li X., Wang J., Gao D., Li Y., Li H., Chu Y., Zhang Z., Liu H., Jiang G., Cheng Z., Wang S., Dong J., Feng B., Chard L.S., Lemoine N.R., Wang Y. (2017) Re-designing interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun. 8, 1395.
Lapteva N., Aldrich M., Weksberg D., Rollins L., Goltsova T., Chen S.Y., Huang X.F. (2009) Targeting the intratumoral dendritic cells by the oncolytic adenoviral vaccine expressing RANTES elicits potent antitumor immunity. J. Immunother. 32, 145–156.
Li J., O’Malley M., Urban J., Sampath P., Guo Z.S., Kalinski P., Thorne S.H., Bartlett D.L. (2011) Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol. Ther. 19, 650–657.
Zamarin D., Holmgaard R.B., Ricca J., Plitt T., Palese P., Sharma P., Merghoub T., Wolchok J.D., Allison J.P. (2017) Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 8, 14340.
Kim H.S., Kim-Schulze S., Kim D.W., Kaufman H.L. (2009) Host lymphodepletion enhances the therapeutic activity of an oncolytic vaccinia virus expressing 4-1BB ligand. Cancer Res. 69, 8516–8525.
Andarini S., Kikuchi T., Nukiwa M., Pradono P., Suzuki T., Ohkouchi S., Inoue A., Maemondo M., Ishii N., Saijo Y., Sugamura K., Nukiwa T. (2004) Adenovirus vector-mediated in vivo gene transfer of OX40 ligand to tumor cells enhances antitumor immunity of tumor-bearing hosts. Cancer Res. 64, 3281–3287.
Ullenhag G., Loskog A.S. (2012) AdCD40L – crossing the valley of death? Int. Rev. Immunol. 31, 289–298.
Fajardo C.A., Guedan S., Rojas L.A., Moreno R., Arias-Badia M., de Sostoa J., June C.H., Alemany R. (2017) Oncolytic adenoviral delivery of an EGFR-targeting T cell engager improves antitumor efficacy. Cancer Res. 77, 2052–2063.
Yu F., Wang X., Guo Z.S., Bartlett D.L., Gottschalk S.M., Song X.T. (2014) T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 22, 102–111.
Dias J.D., Hemminki O., Diaconu I., Hirvinen M., Bonetti A., Guse K., Escutenaire S., Kanerva A., Pesonen S., Löskog A., Cerullo V., Hemminki A. (2012) Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 19, 988–998.
Engeland C.E., Grossardt C., Veinalde R., Bossow S., Lutz D., Kaufmann J.K., Shevchenko I., Umansky V., Nettelbeck D.M., Weichert W., Jäger D., von Kalle C., Ungerechts G. (2014) CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 22, 1949–1959.
Bartee M.Y., Dunlap K.M., Bartee E. (2017) Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. 77, 2952–2963.
Kleinpeter P., Fend L., Thioudellet C., Geist M., Sfrontato N., Koerper V., Fahrner C., Schmitt D., Gantzer M., Remy-Ziller C., Brandely R., Villeval D., Rittner K., Silvestre N., Erbs P., Zitvogel L., Quéméneur E., Préville X., Marchand J.B. (2016) Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death-1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology. 5, e1220467.
Nishio N., Diaconu I., Liu H., Cerullo V., Caruana I., Hoyos V., Bouchier-Hayes L., Savoldo B., Dotti G. (2014) Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 74, 5195–5205.
Watanabe K., Luo Y., Da T., Guedan S., Ruella M., Scholler J., Keith B., Young R.M., Engels B., Sorsa S., Siurala M., Havunen R., Tähtinen S., Hemminki A., June C.H. (2018). Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. 3, pii: 99573.
Tanoue K., Rosewell Shaw A., Watanabe N., Porter C., Rana B., Gottschalk S., Brenner M., Suzuki M. (2017) Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 77, 2040–2051.
Rosewell Shaw A., Porter C.E., Watanabe N., Tanoue K., Sikora A., Gottschalk S., Brenner M.K., Suzuki M. (2017) Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol. Therapy. 25, 2440–2451.
Moon E.K., Wang L.S., Bekdache K., Lynn R.C., Lo A., Thorne S.H., Albelda S.M. (2018) Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. 7, e1395997.
Endo Y., Sakai R., Ouchi M., Onimatsu H., Hioki M., Kagawa S., Uno F., Watanabe Y., Urata Y., Tanaka N., Fujiwara T. (2008) Virus-mediated oncolysis induces danger signal and stimulates cytotoxic T-lymphocyte activity via proteasome activator upregulation. Oncogene. 27, 2375‒2381.
Morgan R.A. (2012) Human tumor xenografts, the good, the bad, and the ugly. Mol. Ther. 20, 882–884.
Van Seggelen H., Tantalo D.G., Afsahi A., Hammill J.A., Bramson J.L. (2015) Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol. Ther. Oncolytics. 2, 15014.
Agrahari V., Agrahari V., Mitra A.K. (2016) Next generation drug delivery, circulatory cells-mediated nanotherapeutic approaches. Exp. Opin. Drug Deliv. 14, 285–289.
Yotnda P. (2004) Targeted delivery of adenoviral vectors by cytotoxic T cells. Blood. 104, 2272–2280.
Jennings V.A., Ilett E.J., Scott K.J., West E.J., Vile R., Pandha H., Harrington K., Young A., Hall G.D., Coffey M., Selby P., Errington-Mais F., Melcher A.A. (2014) Lymphokine-activated killer and dendritic cell carriage enhances oncolytic reovirus therapy for ovarian cancer by overcoming antibody neutralization in ascites. Int. J. Cancer. 134, 1091–1101.
Cole C., Qiao J., Kottke T., Diaz R.M., Ahmed A., Sanchez-Perez L., Brunn G., Thompson J., Chester J., Vile R.G. (2005) Tumor targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat. Med. 11, 1073–1081.
Akram A., Inman R.D. (2012) Immunodominance: a pivotal principle in host response to viral infections. Clin. Immunol. 143, 99–115.
Frahm N., DeCamp A.C., Friedrich D.P., Carter D.K., Defawe O.D., Kublin J.G., Casimiro D.R., Duerr A., Robertson M.N., Buchbinder S.P., Huang Y., Spies G.A., De Rosa S.C., McElrath M.J. (2012) Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J. Clin. Invest. 122, 359–367.
Galivo F., Diaz R.M., Thanarajasingam U., Jevremovic D., Wongthida P., Thompson J., Kottke T., Barber G.N., Melcher A., Vile R.G. (2010) Interference of CD40L-mediated tumor immunotherapy by oncolytic vesicular stomatitis virus. Hum. Gene Ther. 21, 439–450.
Pule M.A., Savoldo B., Myers G.D., Rossig C., Russell H.V., Dotti G., Huls M.H., Liu E., Gee A.P., Mei Z., Yvon E., Weiss H.L., Liu H., Rooney C.M., Heslop H.E., Brenner M.K. (2008) Virus-specific T cells engineered to coexpress tumorspecific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 14, 1264–1270.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология