

Молекулярная биология, 2021, T. 55, № 2, стр. 277-288
Сравнительный анализ активности полиморфных вариантов урацил-ДНК-гликозилаз человека SMUG1 и MBD4
И. В. Алексеева a, А. С. Бакман a, b, Д. А. Яковлев a, Н. А. Кузнецов a, *, О. С. Федорова a, **
a Институт химической биологии и фундаментальной медицины Сибирского отделения
Российской академии наук
630090 Новосибирск, Россия
b Новосибирский национальный исследовательский государственный университет
630090 Новосибирск, Россия
* E-mail: nikita.kuznetsov@niboch.nsc.ru
** E-mail: fedorova@niboch.nsc.ru
Поступила в редакцию 12.10.2020
После доработки 12.10.2020
Принята к публикации 26.10.2020
Аннотация
N-гликозилазы человека SMUG1 и MBD4 катализируют удаление из ДНК остатков урацила, образующихся в результате дезаминирования цитозина или ошибок репликации. Для однонуклеотидных полиморфных вариантов (SNP) SMUG1 (G90C, P240H, N244S, N248Y) и каталитического домена MBD4cat (S470L, G507S, R512W, H557D) методом молекулярной динамики получены структуры фермент-субстратных комплексов. Экспериментально установлено, что SNP-варианты SMUG1 N244S и N248Y обладают повышенной каталитической активностью по сравнению с ферментом дикого типа, по-видимому, за счет ускорения диссоциации комплекса фермента с продуктом и увеличения числа оборотов фермента. У остальных вариантов SMUG1 (G90C, P240H) и всех вариантов MBD4cat, в которых замены аминокислот нарушали область связывания с субстратом и/или активный центр, каталитическая активность была значительно ниже, чем у ферментов дикого типа.
ВВЕДЕНИЕ
Появление урацила в ДНК – одно из наиболее распространенных повреждений этой молекулы. Урацил образуется либо при дезаминировании цитозина (как спонтанном [1–3], так и ферментативном [4, 5]), либо при ошибочной вставке остатка уридина во время репликации [6]. Несмотря на то, что частота гидролитического дезаминирования цитозина на два порядка меньше, чем, например, частота апуринизации (70–200 против 104 случаев в одной клетке в сутки [3]), такие повреждения относятся к сильно мутагенным из-за того, что репликативные ДНК-полимеразы не способны отличать урацил и тимин в ДНК. Таким образом, в ходе дальнейшей репликации U/G-пара считывается как A/T, что в результате приводит к замене исходной пары C/G на T/A. Однако во всех живых организмах существует эффективный механизм поиска и удаления урацила из ДНК [7].
Урацил в ДНК человека узнается и удаляется несколькими ДНК-гликозилазами: урацил-ДНК-гликозилазой (UNG2), тимин-ДНК-гликозилазой (TDG), специфичной к одноцепочечной ДНК монофункциональной урацил-ДНК-гликозилазой (SMUG1) и метил-CpG-связывающим ферментом 4 (MBD4) [8, 9]. Первые три фермента относятся к первому, второму и третьему подсемействам структурного суперсемейства урацил-ДНК-гликозилаз (семейство UDG), а белок MBD4 – к семейству “спираль-шпилька-спираль” (семейство HhH) [8, 10]. Необходимо отметить, что четыре ДНК-гликозилазы человека, обладающие способностью удалять урацил, имеют специализированную субстратную специфичность, связанную с биологической функцией этих ферментов: UNG2 и SMUG1 в основном удаляют урацил из одно- и двухцепочечной ДНК, а TDG и MBD4 – производные урацила, возникающие в процессах эпигенетической модификации ДНК [8].
В исследованиях субстратной специфичности SMUG1 [11, 12] обнаружено, что фермент в 700 раз эффективнее работает на двухцепочечных, чем одноцепочечных урацилсодержащих ДНК-субстратах, причем преимущественно удаляет урацил из U/G-пар. Кроме того, предполагается, что SMUG1 – основной фермент, удаляющий 5‑гидроксиметилурацил (5hmU) из ДНК [11–13]. Считается, что 5hmU образуется в качестве интермедиата в многостадийном процессе активного деметилирования, где 5-метилцитозин (5mC) гидроксилируется диоксигеназами TET с образованием 5-гидроксиметилцитозина (5hmC), а последующее дезаминирование 5hmC под действием фермента AID/APOBEC приводит к образованию 5hmU.
В настоящее время трехмерная структура SMUG1 человека (hSMUG1) неизвестна, поэтому, используя в качестве матрицы структуры GmeSMUG1 из Geobacter metallireducens (PDB Acc. No 5H98 и 5H9I) [14] и xSMUG1 из Xenopus laevis (PDB Acc. No 1OE4 и 1OE5) [11], Кузнецова и др. [15] методом гомологичного моделирования получили структуру hSMUG1. При сравнении полученной модельной структуры с данными мутационного анализа [16–18] были идентифицированы аминокислотные остатки, участвующие в связывании ДНК, узнавании поврежденного нуклеотида и катализе [15]. Установлено, что аминокислотные остатки Asn85 и His239 катализируют гидролиз N-гликозидной связи. Расположенное в активном центре фермента азотистое основание образует π-π-стэкинг с Phe98, а также специфическую водородную связь с Asn163. Несколько аминокислотных остатков, Gly87–Met91, отвечают за узнавание заместителя в C5-положении пиримидинового основания. В hSMUG1 последовательность His239–Lys249, называемая интеркалирующей петлей, служит “клином”, проникающим в двойную спираль ДНК в области поврежденного нуклеотида [16]; при этом остаток Arg243 встраивается в дуплекс на место поврежденного нуклеотида.
Основной субстрат MBD4 ‒ ДНК, содержащая неканонические пары T/G и U/G [19, 20]. Также показано, что MBD4 эффективно удаляет 5hmU в двухцепочечных CpG-динуклеотидах in vitro [21, 22]. Кроме того, ферменты AID/APOBEC могут напрямую дезаминировать 5mC, превращая его в тимин и тем самым формируя мисматч T/G ‒ субстрат MBD4 [23].
ДНК-гликозилаза человека MBD4 содержит два домена: 5mC-связывающий и ДНК-гликозилазный (MBD4cat). В настоящее время известна кристаллическая структура обоих доменов [21, 24, 25]. Установлено, что каталитический домен, MBD4cat, состоит из 138 а.о., образующих 9 α-спиралей. Структура MBD4cat довольно консервативна и относится к семейству “спираль-шпилька-спираль” (HhH). Опубликованы [21, 25] структуры каталитического домена с 12-звенными ДНК-дуплексами, содержащими пары 5hmU/G, T/G и AP/G. Все комплексы и свободный белок имеют сходную кристаллическую упаковку, из чего можно сделать вывод, что при связывании ДНК домен MBD4cat не претерпевает серьезных конформационных изменений. Показано, что все дуплексы ДНК в комплексе с ферментом изогнуты в области поврежденного нуклеотида на ~38°. При анализе структурных данных показано, что MBD4cat связывается с ДНК тремя участками: петлями Leu466–Gly471 и Lue503–Leu511 и Gly-богатой шпилькой из HhH-мотива (Leu534–Gly541), взаимодействуя преимущественно с цепью ДНК, содержащей удаляемое основание. Петля Leu466–Gly471 проникает в ДНК через малую бороздку и остаток Arg468 заполняет пространство в дуплексе, освобожденное после выворачивания поврежденного нуклеотида. Гуанидиновая группа этого аминокислотного остатка взаимодействует с 5′-фосфатной группой нуклеотида, расположенного с 5′‑стороны от вырезаемого основания, и с 3′-фосфатной группой поврежденного нуклеотида. Вывернутое основание удерживается в кармане, образованном остатками Leu466, Leu447 и Lys562 [26]. Данные о функциональной роли аминокислотных остатков активных центров SMUG1 и MBD4cat были использованы при анализе базы данных NCBI dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) однонуклеотидных полиморфизмов (SNP) в соответствующих белках. Действительно, SNP-ассоциированные аминокислотные замены в ферментах репарации широко распространены в человеческой популяции [27, 28], более того роль SNP в белоккодирующей области генов ферментов системы эксцизионной репарации оснований ДНК в восприимчивости к болезням активно обсуждается в настоящее время в ряде публикаций [29–43]. Однако в настоящее время остается неизвестным, как аминокислотные замены, вызванные SNP, могут влиять на эффективность удаления повреждений из ДНК ферментами SMUG1 и MBD4cat.
В связи с этим целью работы было определение влияния некоторых SNP-ассоциированных аминокислотных замен в первичных структурах MBD4cat и SMUG1 на ферментативную активность этих белков. Для каждой ДНК-гликозилазы были выбраны четыре варианта мутантных форм, содержащих замены в области ДНК-связывающего и каталитического центров. Исследуя накопление продуктов реакции с помощью электрофореза в ПААГ, оценивали эффективность удаления урацила из модельных ДНК-субстратов ферментами дикого типа и их мутантными формами. Учитывая, что стадии связывания ДНК-субстрата, узнавания урацила и образования каталитического комплекса происходят в миллисекундном и секундном диапазонах времени, для регистрации этих процессов использовали метод “остановленного потока”. Конформационные изменения ферментов в комплексах с ДНК-субстратами регистрировали по изменению интенсивности флуоресценции остатков Trp. Анализ кинетических данных позволил выявить стадии ферментативного процесса MBD4cat и SMUG1, которые наиболее подвержены влиянию SNP-ассоциированных аминокислотных замен.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Выбор потенциально важных SNP. Используя базу данных dbSNP NCBI (http://www.ncbi.nlm. nih.gov/SNP/), мы отобрали полиморфные варианты в экзонах генов, кодирующих MBD4cat и SMUG1 человека. Последующий анализ потенциальной важности SNP был основан на принципе изменения химической природы боковой группы аминокислоты, кодируемой соответствующим триплетом нуклеотидов, и пространственным расположением аминокислотного остатка вблизи ДНК-связывающего сайта и/или активного центра фермента (рис. 1). Для каждого фермента выбрали по четыре SNP (табл. 1) для изучения их влияния на активность ДНК-гликозилаз. Потенциальная важность выбранных вариантов была проверена по базе данных BioMuta, содержащей данные по ассоциированным с онкологическими заболеваниями однонуклеотидным вариациям в геноме (https://hive.biochemistry. gwu.edu/biomuta/proteinview/Q53HV7 для hSMUG1 и https://hive.biochemistry.gwu.edu/biomuta/ proteinview/O95243 для MBD4).
Рис. 1.
Пространственное расположение аминокислотных остатков в структуре SMUG1 (а) и MBD4cat (б), подвергающихся заменам в случае отобранных SNP (см. табл. 1).
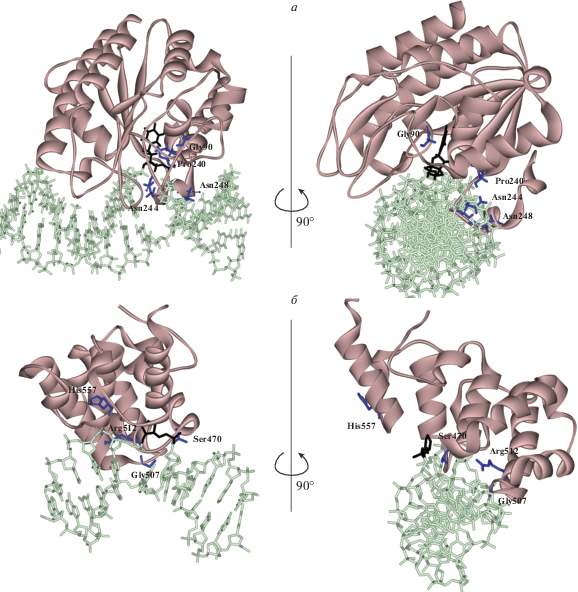
Таблица 1.
Замены аминокислотных остатков, вызванные SNP, их положение и функциональная роль в структуре фермента и встречаемость при онкологических заболеваниях
| Фермент | Замена | Положение в структуре фермента и функциональная роль | Онкологическое заболевание (по базе BioMuta) |
|---|---|---|---|
| SMUG1 | G90C | Входит в состав участка Gly87–Met91, отвечающего за узнавание заместителя в C5-положении пиримидинового основания | Рак легкого |
| P240H | Входят в состав интеркалирующей петли His239–Lys249, выступающей в роли “клина” и встраивающейся в двойную спираль ДНК в области поврежденного нуклеотида | Меланома | |
| N244S | Рак легкого | ||
| N248Y | Рак печени | ||
| MBD4cat | S470L | Входит в состав петли Leu466–Gly471, участвующей в связывании ДНК | Рак молочной железы |
| G507S | Входит в состав петли Leu503–Leu511, участвующей в связывании ДНК | Колоректальный рак | |
| R512W | Примыкает к петле Leu503–Leu511, участвующей в связывании ДНК | Колоректальный рак | |
| H557D | Расположен на поверхности и приводит к смене заряда этого участка белка | Рак молочной железы |
Моделирование структуры фермент-субстратного комплекса, содержащего полиморфные варианты белков. Для определения влияния аминокислотных замен, вызванных SNP, на структуру фермента в составе фермент-субстратного комплекса была использована программа UCSF Chimera (http://www.rbvi.ucsf.edu/chimera/). В структуре фермента дикого типа заменяли соответствующие аминокислотные остатки следующим образом: боковую цепь изучаемого остатка удаляли из исходного файла PDB и реконструировали новую боковую цепь, используя библиотеку ротамеров [44]. Затем модели фермент-субстратных комплексов с замененными аминокислотными остатками оптимизировали методом молекулярной динамики с использованием модифицированного силового поля AMBER ff99 [45–47] и программы BioPASED [47]. Для этого белки уравновешивали при 50 K с использованием неявной гауссовой модели воды [48] и затем в течение 100 пс температуру увеличивали до 300 К. После этого в течение 6.5 нс проводили оптимизацию структуры фермент-субстратного комплекса при постоянной температуре 300 K. Общая потенциальная энергия и общее среднеквадратичное отклонение (RMSD) были стабильными во время моделирования.
ДНК-субстраты. Олигодезоксирибонуклеотиды были синтезированы на автоматическом ДНК/РНК-синтезаторе ASM-800 (“Биоссет”, Россия) с использованием коммерческих амидофосфитов 2′-дезоксирибонуклеозидов и CPG-носителей (“GlenResearch”, США) (табл. 2). Синтезированные олигонуклеотиды очищали ВЭЖХ на ионообменной колонке PRP-X500 (12–30 мкм) размером 3.9 × 300 мм (“Hamilton”, США) и затем обращеннофазовой хроматографией на колонке Bondapak C18 (15–20 мкм) размером 3.9 × 300 мм (“Waters”, Ирландия).
Для расчета концентрации олигонуклеотидов в исходном растворе использовали коэффициент молярного поглощения олигонуклеотидов или их конъюгатов при длине волны 260 нм.
Ферменты дикого типа и их мутантные формы. Мутации в кодирующей последовательности генов MBD4cat и SMUG1 были созданы с использованием набора для сайт-направленного мутагенеза QuikChange XL (“Stratagene”, США). Каталитический домен ДНК-гликозилазы человека MBD4cat (аминокислотные остатки 426–580), полноразмерная гликозилаза человека SMUG1 и их полиморфные варианты были выделены из клеток Escherichia coli Rosetta 2, трансформированных плазмидой pET29b-MBD4cat или pET28c-hSMUG1 соответственно, по методикам, описанным ранее [15, 17, 21, 49, 50].
Электрофорез в ПААГ. Все эксперименты по исследованию ферментативной реакции проводили в буферном растворе (RB), содержащем 50 мМ Трис-HCl, pH 7.5, 50 мМ KCl, 1 мМ EDТА, 1 мМ дитиотреит и 7% глицерина, при 25°C. Продукты анализировали электрофорезом в 20%-ном ПААГ, содержащем 7 М мочевину, с использованием FRET-U-субстрата. Зависимость степени превращения субстрата от времени определяли следующим образом. К 20 мкл RB, содержащего 10 мкМ FRET-U-субстрат, добавляли 20 мкл 2.0 мкМ фермента в том же буфере. Реакционную смесь быстро перемешивали и через определенные промежутки времени отбирали аликвоты объемом 2 мкл, которые переносили в предварительно подготовленные пробирки, содержащие 2 мкл раствора 7 М мочевины, 25 мМ EDTA и 0.1% ксиленцианола. Затем добавляли 1 мл 2%-ного раствора LiClO4 в ацетоне, выдерживали 30 мин при ‒20°С и центрифугировали 10 мин (14 000 × g). Осадок промывали ацетоном, высушивали, растворяли в 10 мкл буферного раствора, содержащего 1.0 мкМ АР-эндонуклеазу Nfo и инкубировали при 25°C в течение 1 мин для гидролиза фосфодиэфирных связей в АР-сайтах. Реакцию останавливали добавлением раствора 7 М мочевины, 25 мкМ EDTA и 0.1% ксиленцианола. Полученные образцы наносили на ПААГ и проводили электрофорез при 50 В/см. Количество образующегося продукта определяли путем сканирования геля на приборе E-Box (“VILBER”, Франция) и обработки данных в программном пакете Gel-Pro Analyzer 4.0 (“Media Cybernetics”, США).
Ферментативную активность полиморфных вариантов определяли относительно активности фермента дикого типа, которую принимали равной 1.
Анализ предстационарной кинетики. Анализ кинетики взаимодействия ферментов с модельными ДНК-субстратами (табл. 2) проводили в RB при 25°С. Флуоресцентный сигнал регистрировали на спектрометре “остановленного потока” SX20 (“Applied Photophysics”, Великобритания). В работе использовали несколько типов флуоресцентной регистрации процесса фермент-субстратного взаимодействия. Конформационные изменения фермента в процессе взаимодействия с U-субстратом регистрировали по изменению собственной флуоресценции белка, обусловленной остатками Trp. Ферменты MBD4cat и SMUG1 содержат соответственно 8 и 4 остатка Trp. Возбуждение интенсивности флуоресценции остатков Trp проводили при длине волны 290 нм, регистрацию флуоресценции – при более 320 нм (фильтр WG-320; “Schott”, Германия). Мертвое время прибора составляло 1.0 мс. Каждую кинетическую кривую усредняли, как минимум, по четырем повторам.
Наблюдаемую константу скорости рассчитывали путем обработки кинетических кривых по уравнению (1):
где f(t) – наблюдаемая интенсивность флуоресценции в момент времени t, f0 – фоновый уровень сигнала, f1 и f2 – коэффициенты, k1 и k2 ‒ кажущиеся константы скорости.РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Влияние замен аминокислотных остатков на структуру фермент-субстратных комплексов
Модели мутантных форм построены на основе структур комплексов SMUG1 [15] и MBD4cat [25] дикого типа с ДНК. Время симуляции составляло 6.5 нс. Как видно на рис. 2, анализируемые мутации не вносили значительных изменений в общую структуру фермент-субстратного комплекса, тем не менее некоторые замены могут приводить к локальным изменениям. Так, в случае полиморфных вариантов фермента SMUG1 можно отметить, что замена G90C (рис. 2а) приводит к увеличению объема бокового радикала, что сопровождается локальным “раздвиганием” окружения, но не вызывает значительных перестроек в укладке этой области белка. При замене P240H (рис. 2б) имидазольное кольцо гистидина может образовать стэкинг с индольным кольцом Trp251 и водородную связь с Arg140, что приводит к стабилизации этой области, не вызывая в ней значительных конформационных изменений. При заменах N244S и N248Y (рис. 2в и г) остатки аспарагина, расположенные в ДНК-связывающем канале фермента, теряют сеть контактов, образованныx между карбоксамидной группой Asn с ДНК, что может сопровождаться дестабилизацией фермент-субстратного комплекса.
Рис. 2.
Сравнение структур фермент-субстратных комплексов для ферментов дикого типа (голубой) и их полиморфных вариантов (красный). В структурах показано внеспиральное положение уридина и аминокислотные остатки, подвергающиеся заменам для отобранных SNP, а именно: SMUG1 G90C (а), P240H (б), N244S (в) и N248Y (г); MBD4cat S470L (д), G507S (е), R512W (ж) и H557D (з).
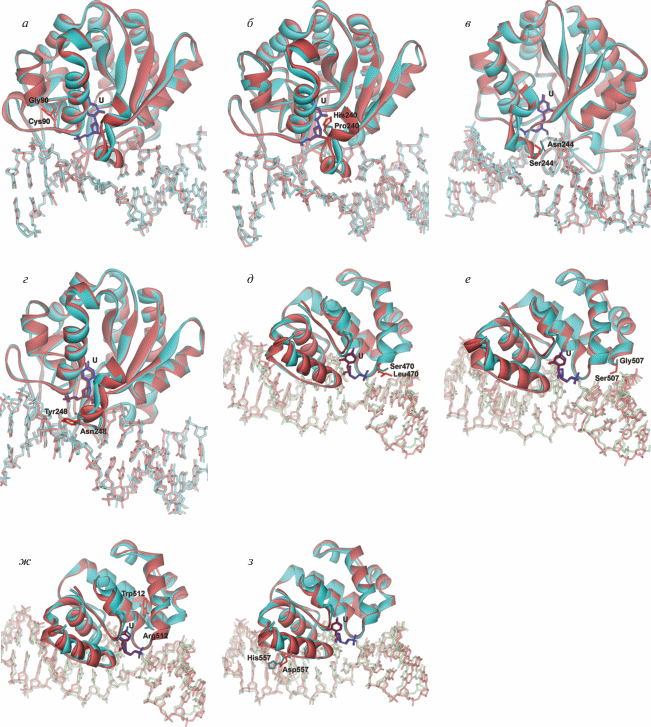
Замена S470L в MBD4cat (рис. 2д) приводит к тому, что в области контакта с сахарофосфатным остовом ДНК появляется гидрофобный аминокислотный остаток лейцина. Можно предположить, что нахождение гидрофобного остатка в заряженном окружении энергетически невыгодно и может ослаблять связывание фермента с субстратом. Замена G507S MBD4cat (рис. 2е) не вносит серьезных изменений в общую конформацию фермента, но имеет сильное локальное влияние. Так, из-за появления в ДНК-связывающей области гидроксиметильной группы серина могут возникнуть дополнительные водородные связи с ДНК, что усилит связывание фермента c ДНК и, следовательно, снизит скорость оборота фермента из-за уменьшения константы диссоциации с продуктом реакции. Замена R512W в MBD4cat (рис. 2ж) приводит, с одной стороны, к потере водородных связей с ДНК, образованных боковой цепью аргинина, а с другой стороны, к встраиванию индольного остатка триптофана в гидрофобную область белка. В этом случае происходит смещение петли, сформированной остатками Ala488–Ser520, наружу белковой глобулы. Также можно предположить, что эта замена ослабляет связь фермента и ДНК за счет уменьшения поверхностного положительного заряда в ДНК-связывающем канале. Замена положительно зараженного His557 (рис. 2з), взаимодействующего с сахарофосфатным остовом ДНК, на отрицательно заряженный аспартат приведет к “отталкиванию” петли Val550‒Asp560 от ДНК, ее деформации и, скорее всего, дестабилизации фермент-субстратного комплекса.
Эффективность удаления урацила полиморфными вариантами SMUG1 и MBD4cat
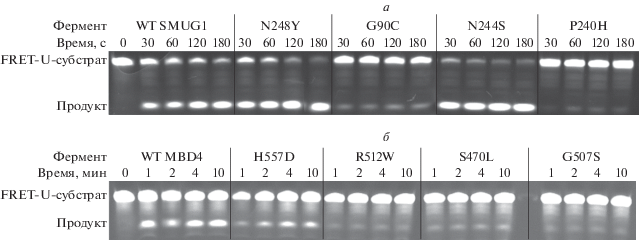
Методом электрофореза в ПААГ показано, что эффективность удаления урацила ферментами дикого типа и их полиморфными вариантами значительно отличается (рис. 3).
Рис. 3.
Ферментативная активность SMUG1 (а) и MBD4cat (б) дикого типа (WT) и их полиморфных вариантов в отношении FRET-U-субстрата. [FRET-U-субстрат] = 5.0 мкМ, [фермент] = 1.0 мкМ.
Действительно, на основании анализа кинетических кривых накопления продуктов реакции при взаимодействии SMUG1 и его вариантов с FRET-U-субстратом (рис. 4а) и сравнения относительной активности (рис. 4в) показано, что замены N244S и N248Y приводят к повышению скорости оборота фермента. Ранее сообщалось [15], что для фермента дикого типа скорость лимитирующая стадия ферментативного процесса ‒ диссоциация комплекса фермента с продуктом, поэтому нарушение прямых взаимодействий между аминокислотными остатками в позициях 244 и 248 и ДНК приводит к дестабилизации фермент-субстратного комплекса и сопровождается повышением скорости оборота фермента. Однако замена P240H ‒ остатка, играющего роль “клина” в составе интеркалирующей петли His239–Lys249, встраивающейся в двойную спираль ДНК в области поврежденного нуклеотида, ‒ приводит к значительному снижению каталитической активности фермента. Согласно модели фермент-субстратного комплекса, His240 образует сеть контактов с Arg140 и Trp251, что может приводить к ограничению конформационной подвижности всей петли His239–Lys249 и тем самым препятствовать формированию каталитически компетентного состояния фермента в комплексе с субстратом. Замена G90C, расположенная на участке Gly87–Met91, отвечающем за узнавание заместителя в C5-положении пиримидинового основания, также значительно снижает эффективность удаления урацила.
Рис. 4.
Эффективность удаления урацила из FRET-U-субстрата под действием SMUG1 (а) и MBD4cat (б) дикого типа (WT) и их полиморфных вариантов. в ‒ Сравнительный анализ полученных результатов представлен относительно активности соответствующего фермента дикого типа, принятой за 1.
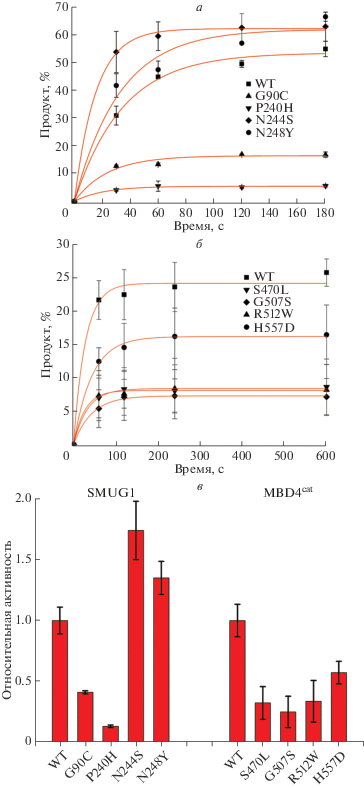
Необходимо отметить, что активность MBD4cat с заменами S470L, G507S и R512W составляла не более 30% от таковой для фермента дикого типа, а мутация H557D приводила к снижению активности до 50% (рис. 4б и в). Таким образом, в случае всех тестируемых вариантов MBD4cat наблюдается значительное снижение активности за счет нарушения локальных взаимодействий в области ДНК-связывающего канала.
Анализ кинетики образования фермент-субстратных комплексов методом “остановленного потока”
Кинетические кривые, характеризующие изменение интенсивности флуоресценции Trp при взаимодействии SMUG1 дикого типа с U-субстратом (рис. 5а), содержат фазу падения интенсивности флуоресценции до ~0.3 с и фазу роста с выходом на плато к 5 с с начала реакции. Ранее показано [15, 18], что падение сигнала на начальном участке кинетической кривой соответствует образованию первичных неспецифических контактов с ДНК, а фаза роста интенсивности флуоресценции Trp в интервале 0.5‒10 с ‒ стадиям гидролиза N-гликозидной связи и диссоциации комплекса фермента с продуктом реакции. Установлено, что диссоциация комплекса фермент-продукт ‒ скорость лимитирующая стадия ферментативного процесса. Полученные кинетические кривые аппроксимировали по уравнению (1), что позволило рассчитать значения кажущихся констант скорости обоих процессов (табл. 3).
Рис. 5.
Кинетические кривые изменения интенсивности флуоресценции остатков Trp при взаимодействии SMUG1 (а) и MBD4cat (б) дикого типа (WT) и полиморфных вариантов с U-субстратом (базовые линии искусственно смещены относительно друг друга для улучшения восприятия данных; это не сказывается на расчете значений кажущихся констант).
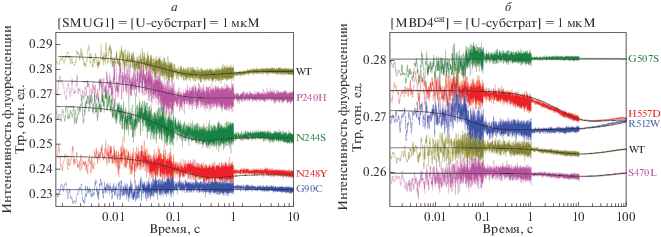
Таблица 3.
Кажущиеся константы скорости реакции SMUG1 и MBD4cat дикого типа и полиморфных вариантов с модельными ДНК-субстратами
| Фермент | k1, с–1 | k2, с–1 |
|---|---|---|
| SMUG1 | ||
| WT | 14.4 ± 0.3 | 1.2 ± 0.1 |
| G90C | ‒ | ‒ |
| P240H | 16.0 ± 0.4 | ‒ |
| N244S | 12.5 ± 0.4 | 2.5 ± 0.5 |
| N248Y | 7.0 ± 1.0 | 2.0 ± 0.8 |
| MBD4cat | ||
| WT | 0.28 ± 0.02 | 0.020 ± 0.002 |
| S470L | 0.22 ± 0.01 | 0.018 ± 0.001 |
| G507S | ‒ | ‒ |
| R512W | 15.5 ± 0.4 | 0.010 ± 0.001 |
| H557D | 0.25 ± 0.02 | 0.024 ± 0.003 |
Сравнение кинетических кривых и наблюдаемых констант скорости для SMUG1 дикого типа с таковыми для полиморфных вариантов показывает, что в случае U-субстрата изменения интенсивности флуоресценции Trp на начальном участке имеют похожий вид для всех вариантов, кроме G90C. Таким образом, в случае мутантной формы G90C нарушается прохождение начальной стадии ферментативной реакции: связывания субстрата и узнавания урацила. В то же время замена P240H, которая также приводит к снижению каталитической активности фермента, не влияет на стадию связывания ДНК, так как на кинетических кривых присутствует стадия падения интенсивности флуоресценции остатков Trp. По-видимому, блокирование ферментативной активности в случае P240H происходит на стадии перехода фермента в составе комплекса с субстратом в каталитически компетентную конформацию. Интересно отметить, что для более активных вариантов SMUG1, N248Y и N244S, кажущаяся константа фазы роста интенсивности k1 была соответственно в 1.7 и 2.1 раза выше, чем для фермента дикого типа.
На рис. 5б представлены кинетические кривые изменения интенсивности флуоресценции остатков Trp при взаимодействия MBD4cat дикого типа и его полиморфных вариантов с U-субстратом. Согласно данным Petronzelli и соавт. [51], падение интенсивности флуоресценции Trp во временном интервале 2 мс–10 с характеризует образование первичного комплекса и поиск модифицированного основания, а рост в интервале 10–100 с соответствует образованию каталитически компетентной конформации фермента и началу процесса накопления продукта. Как и в случае SMUG1, скорость лимитирующая стадия ферментативного процесса ‒ диссоциации комплекса фермент-продукт [51]. Интересно отметить, что замены S470L и H557D не оказывали влияния на значения наблюдаемых констант скорости в условиях эквимолярного соотношения фермент-субстрат. В то же время в случае избытка субстрата (рис. 4в) ферментативная активность этих мутантов MBD4cat снижается, что свидетельствует о замедлении скорость лимитирующей стадии ‒ диссоциации фермент-субстратного комплекса. В случае варианта G507S, наименее активного среди всех проанализированных (рис. 4в), изменения интенсивности флуоресценции остатков Trp не зарегистрированы (рис. 5б), что может быть связано со значительной дестабилизацией фермент-субстратного комплекса, так как остаток Gly507 входит в состав петли Leu503–Leu511, участвующей в связывании ДНК. В случае варианта R512W, содержащего дополнительный остаток Trp в области петли Leu503–Leu511, которая участвует в связывании ДНК, зарегистрирован этап взаимодействия этой петли с ДНК. Действительно, в случае R512W, в отличие от всех других вариантов, на начальном участке кинетической кривой происходило значительное уменьшение интенсивности флуоресценции Trp, что, по-видимому, характеризует конформационную перестройку петли Leu503–Leu511 при образовании фермент-субстратного комплекса; в случае всех других мутантных форм MBD4cat этот переход не удавалось зарегистрировать.
ЗАКЛЮЧЕНИЕ
Таким образом, на основании полученных данных по активности полиморфных вариантов двух урацил-ДНК-гликозилаз человека: SMUG1 и MBD4 ‒ можно сделать вывод, что природные варианты этих ферментов могут различаться по эффективности удаления остатков урацила из ДНК. Показано, что повышенная ферментативная активность полиморфных вариантов N248Y и N244S SMUG1 обусловлена ускорением лимитирующей стадии процесса ‒ диссоциации комплекса фермент-продукт. Для всех других проанализированных нами SNP-вариантов выявлена сниженная активность по сравнению с ферментами дикого типа, что может негативно отражаться на репарации геномной ДНК у носителей этих полиморфизмов.
Работа выполнена при поддержке гранта РНФ № 16-14-10038 и частичной поддержке бюджетного финансирования (№ АААА-А17-117020210022-4) для обеспечения регламентных работ на использованном оборудовании.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Lari S.-U., Chen C.-Y., Vertéssy B.G., Morré J., Bennett S.E. (2006) Quantitative determination of uracil residues in Escherichia coli DNA: contribution of ung, dug, and dut genes to uracil avoidance. DNA Repair (Amst.). 5, 1407–1420.
Lewis C.A., Crayle J., Zhou S., Swanstrom R., Wolfenden R.,Wolfenden R. (2016) Cytosine deamination and the precipitous decline of spontaneous mutation during Earth’s history. Proc. Natl. Acad. Sci. USA. 113, 8194–8199.
Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature. 362, 709–715.
Jaszczur M., Bertram J.G., Pham P., Scharff M.D., Goodman M.F. (2013) AID and Apobec3G haphazard deamination and mutational diversity. Cell. Mol. Life Sci. 70, 3089–3108.
Rebhandl S., Hümer M., Greil R., Geisberger R. (2015) AID/APOBEC deaminases and cancer. Oncoscience. 2, 320–333.
Ladner R.D. (2001) The role of dUTPase and uracil-DNA repair in cancer chemotherapy. Curr. Protein Pept. Sci. 2, 361–370.
Krokan H.E., Drabløs F., Slupphaug G. (2002) Uracil in DNA – occurrence, consequences and repair. Oncogene. 21, 8935–8948.
Jacobs A.L., Schar P. (2012) DNA glycosylases: in DNA repair and beyond. Chromosoma. 121, 1–20.
Visnes T., Doseth B., Pettersen H.S., Hagen L., Sousa M.M., Akbari M., Otterlei M., Kavli B., Slupphaug G., Krokan H.E. (2009) Uracil in DNA and its processing by different DNA glycosylases. Philos. Trans. R. Soc., B. 364, 563–568.
Dawson N.L., Lewis T.E., Das S., Lees J.G., Lee D., Ashford P., Orengo C.A., Sillitoe I. (2017) CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 45, D289–D295.
Wibley J.E.A., Waters T.R., Haushalter K., Verdine G.L., Pearl L.H. (2003) Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol. Cell. 11, 1647–1659.
Kavli B., Sundheim O., Akbari M., Otterlei M., Nilsen H., Skorpen F., Aas P.A., Hagen L., Krokan H.E., Slupphaug G. (2002) hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 277, 39926–39936.
Kavli B., Otterlei M., Slupphaug G., Krokan H.E. (2007) Uracil in DNA ‒ general mutagen, but normal intermediate in acquired immunity. DNA Repair (Amst.). 6, 505–516.
Zhang Z., Shen J., Yang Y., Li J., Cao W., Xie W. (2016) Structural basis of substrate specificity in Geobacter metallireducens SMUG1. ACS Chem. Biol. 11, 1729–1736.
Kuznetsova A.A., Iakovlev D.A., Misovets I.V., Ishchenko A.A., Saparbaev M.K., Kuznetsov N.A., Fedorova O.S. (2017) Pre-steady-state kinetic analysis of damage recognition by human single-strand selective monofunctional uracil-DNA glycosylase SMUG1. Mol. Biosyst. 13, 2638–2649.
Matsubara M., Tanaka T., Terato H., Ohmae E., Izumi S., Katayanagi K., Ide H. (2004) Mutational analysis of the damage-recognition and catalytic mechanism of human SMUG1 DNA glycosylase. Nucleic Acids Res. 32, 5291–5302.
Iakovlev D.A., Alekseeva I.V., Vorobjev Y.N., Kuznetsov N.A., Fedorova O.S. (2019) The role of active-site residues Phe98, HiS239, and Arg243 in DNA binding and in the catalysis of human uracil-DNA glycosylase SMUG1. Molecules. 24(17), 3133.
Iakovlev D.A., Alekseeva I.V., Kuznetsov N.A., Fedo-rova O.S. (2020) Role of Arg243 and His239 residues in the recognition of damaged nucleotides by human uracil-DNA glycosylase SMUG1. Biochemistry (Mosc.). 85(5), 594‒603.
Sjolund A.B., Senejani A.G., Sweasy J.B. (2013) MBD4 and TDG: multifaceted DNA glycosylases with ever expanding biological roles. Mutat. Res. 743–744, 12–25.
Turner D.P., Cortellino S., Schupp J.E., Caretti E., Loh T., Kinsella T.J., Bellacosa A. (2006) The DNA N‑glycosylase MED1 exhibits preference for halogenated pyrimidines and is involved in the cytotoxicity of 5-iododeoxyuridine. Cancer Res. 66(15), 7686–7693.
Morera S., Grin I., Vigouroux A., Couve S., Henriot V., Saparbaev M., Ishchenko A.A. (2012) Biochemical and structural characterization of the glycosylase domain of MBD4 bound to thymine and 5-hydroxymethyuracil-containing DNA. Nucleic Acids Res. 40, 9917–9926.
Hill P.W., Amouroux R., Hajkova P. (2014) DNA demethylation, Tet proteins and 5-hydroxymethylcytosine in epigenetic reprogramming: an emerging complex story. Genomics. 104, 324–333.
Rai K., Huggins I.J., James S.R., Karpf A.R., Jones D.A., Cairns B.R. (2008) DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and Gadd45. Cell. 135, 1201–1212.
Walavalkar N.M., Cramer J.M., Buchwald W.A., Scarsdale J.N., Williams D.C. (2014) Solution structure and intramolecular exchange of methyl-cytosine binding domain protein 4 (MBD4) on DNA suggests a mechanism to scan for mCpG/TpG mismatches. Nucleic Acids Res. 42, 11218–11232.
Manvilla B.A., Maiti A., Begley M.C., Toth E.A., Drohat A.C. (2012) Crystal structure of human methyl-binding domain IV glycosylase bound to abasic DNA. J. Mol. Biol. 420, 164–175.
Hashimoto H., Zhang X., Cheng X. (2012) Excision of thymine and 5-hydroxymethyluracil by the MBD4 DNA glycosylase domain: structural basis and implications for active DNA demethylation. Nucleic Acids Res. 40, 8276–8284.
Shen M.R., Jones I.M., Mohrenweiser H. (1998) Nonconservative amino acid substitution variants exist at polymorphic frequency in DNA repair genes in healthy humans. Cancer Res. 58, 604‒608.
Mohrenweiser H.W., Jones I.M. (1998) Variation in DNA repair is a factor in cancer susceptibility: a paradigm for the promises and perils of individual and population risk estimation? Mutat. Res. 400, 5‒24.
Hung R.J., Hall J., Brennan P., Boffetta P. (2005) Genetic polymorphisms in the base excision repair pathway and cancer risk: a huge review. Am. J. Epidemiol. 162, 925‒942.
Nohmi T., Kim S.R., Yamada M. (2005) Modulation of oxidative mutagenesis and carcinogenesis by polymorphic forms of human DNA repair enzymes. Mutat. Res. 591, 60‒73.
Illuzzi J.L., Harris N.A., Manvilla B.A., Kim D., Li M., Drohat A.C., Wilson D.M. (2013) Functional assessment of population and tumor-associated APE1 protein variants. PLoS One. 8, e65922.
Kim W.C., Ma C., Li W.M., Chohan M., Wilson D.M., Lee C.H. (2014) Altered endoribonuclease activity of apurinic/apyrimidinic endonuclease 1 variants identified in the human population. PLoS One. 9, 1‒9.
Kwiatkowski D., Czarny P., Galecki P., Bachurska A., Talarowska M., Orzechowska A., Bobinska K., Bielecka-Kowalska A., Pietras T., Szemraj J., Maes M., Sliwinski T. (2015) Variants of base excision repair genes MUTYH, PARP1 and XRCC1 in Alzheimer’s disease risk. Neuropsychobiology. 71, 176–186.
Czarny P., Kwiatkowski D., Toma M., Kubiak J., Sliwinska A., Talarowska M., Szemraj J., Maes M., Ga-lecki P., Sliwinski T. (2017) Impact of single nucleotide polymorphisms of base excision repair genes on DNA damage and efficiency of DNA repair in recurrent depression disorder. Mol. Neurobiol. 54, 4150–4159.
Marsden C.G., Dragon J.A., Wallace S.S., Sweasy J.B. (2017) Base excision repair variants in cancer. Methods Enzymol. 591, 119–157.
Chan K.K.L., Zhang Q.M., Dianov G.L. (2006) Base excision repair fidelity in normal and cancer cells. Mutagenesis. 21, 173‒178.
Sweasy J.B., Lang T.M., DiMaio D. (2006) Is base excision repair a tumor suppressor mechanism? Cell Cycle. 5, 250–259.
Tudek B. (2007) Base excision repair modulation as a risk factor for human cancers. Mol. Aspects Med. 28, 258‒275.
D’Errico M., Parlanti E., Dogliotti E. (2008) Mechanism of oxidative DNA damage repair and relevance to human pathology. Mutat. Res. 659, 4‒14.
Nemec A.A., Wallace S.S., Sweasy J.B. (2010) Variant base excision repair proteins: contributors to genomic instability. Semin. Cancer Biol. 20, 320‒328.
Wilson D.M., Kim D., Berquist B.R., Sigurdson A.J. (2011) Variation in base excision repair capacity. Mutat. Res. 711, 100‒112.
Wallace S.S., Murphy D.L., Sweasy J.B. (2012) Base excision repair and cancer. Cancer Lett. 327, 73‒89.
Karahalil B., Bohr V.A., Wilson D.M. (2012) Impact of DNA polymorphisms in key DNA base excision repair proteins on cancer risk. Hum. Exp. Toxicol. 31, 981‒1005.
Shapovalov M.V., Dunbrack R.L. (2011) A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure. 19, 844–858.
Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. (1995) A second generation all atom force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 117, 5179–5197.
Wang J., Cieplak P., Kollman P.A. (2000) How well does a Restrained Electrostatic Potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21, 1049–1074.
Попов А.В., Воробьев Ю.Н. (2010) Программа GUI-BioPASED для моделирования молекулярной динамики биополимеров с графическим пользовательским интерфейсом. Молекуляр. биология. 44, 735–742.
Lazaridis T., Karplus M. (1999) Effective energy function for proteins in solution. Proteins. 35, 133–152.
Kuznetsova A.A., Kuznetsov N.A., Ishchenko A.A., Saparbaev M.K., Fedorova O.S. (2014) Pre-steady-state fluorescence analysis of damaged DNA transfer from human DNA glycosylases to AP endonuclease APE1. Biochim. Biophys. Acta. 1840, 3042–3051.
Яковлев Д.А., Кузнецова А.А., Федорова О.С., Кузнецов Н.А. (2017) Поиск поврежденных участков ДНК метил-СpG-связывающим ферментом MBD4. Acta Naturae. 9, 95–105.
Petronzelli F., Riccio A., Markham G.D., Seeholzer S.H., Genuardi M., Karbowski M., Yeung A.T., Matsumoto Y., Bellacosa A. (2000) Investigation of the substrate spectrum of the human mismatch-specific DNA N-glycosylase MED1 (MBD4): fundamental role of the catalytic domain. J. Cell Physiol. 185, 473–480.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология