

Молекулярная биология, 2021, T. 55, № 2, стр. 243-257
Мутационный и кинетический анализ эндорибонуклеазной активности АРЕ1
А. А. Кузнецова a, А. А. Гаврилова a, b, Д. С. Новопашина a, О. С. Федорова a, *, Н. А. Кузнецов a, **
a Институт химической биологии и фундаментальной медицины Сибирского отделения
Российской академии наук
630090 Новосибирск, Россия
b Новосибирский национальный исследовательский государственный университет
630090 Новосибирск, Россия
* E-mail: fedorova@niboch.nsc.ru
** E-mail: nikita.kuznetsov@niboch.nsc.ru
Поступила в редакцию 14.09.2020
После доработки 09.11.2020
Принята к публикации 09.11.2020
Аннотация
Апуриновая/апиримдиновая эндонуклеаза 1 человека (APE1) крайне важна для нормального функционирования организма, так как участвует в системе репарации ДНК. Основной биологической функцией APE1 считается Mg2+-зависимый гидролиз АР-сайтов в ДНК. На основе структурных данных, кинетических исследований и мутационного анализа в настоящее время установлены ключевые этапы взаимодействия APE1 с поврежденной ДНК. Недавно показано, что APE1 выступает в качестве эндорибонуклеазы, которая может катализировать гидролиз мРНК по некоторым пиримидин-пуриновым сайтам и тем самым контролировать уровень определенных транскриптов. Известно, что для реализации эндорибонуклеазной активности APE1 не требуется присутствия ионов Mg2+, как в случае АР-эндонуклеазной активности, что свидетельствует о различиях в механизмах катализа в случаях РНК- и ДНК-субстратов, однако причины этих различий пока до конца не выяснены. Нами исследован эндорибонуклеазный гидролиз модельных РНК-субстратов ферментом APE1 дикого типа и мутантными формами, содержащими замены аминокислотных остатков активного центра: Y171F, R177F, R181A, D210N, N212A, T268D, M270A и D308A. Показано, что замена остатков Asn212, Asp210 и Tyr171 приводит к потере только АР-эндонуклеазной активности с сохранением эндорибонуклеазной, T268D и M270A ‒ к потере специфичности фермента к пиримидин-пуриновым последовательностям, Arg177 и Arg181 незначительно влияет на активность АРЕ1, в то время как D308A приводит к снижению эндорибонуклеазной активности фермента.
ВВЕДЕНИЕ
Апуриновая/апиримидиновая эндонуклеаза человека APE1 ‒ один из ферментов, принимающих участие в процессе эксцизионной репарации оснований ДНК. Основной биологической функцией APE1 считается гидролиз фосфодиэфирной связи с 5′-стороны от апурин/апиримидиновых сайтов (АР-сайтов), в результате чего на концах разрыва цепи образуются 5′-дезоксирибозофосфат и 3′-ОН группа, необходимая полимеразе для встраивания неповрежденного нуклеотида. Это АР-эндонуклеазная активность APE1. Однако, еще в 2004 году Gros и соавт. [1] обнаружили, что фермент может узнавать в качестве субстратов не только АР-сайты, но и поврежденные нуклеозиды (nucleotide incision repair, NIR), например 5,6‑дигидроуридин, α-аномер аденозина и другие. Также APE1 обладает 3′-фосфодиэстеразной, 3′-фосфатазной [2] и 3′-5′-экзонуклеазной [3, 4] активностями. Наряду с репарационной активностью APE1 выполняет роль окислительно-восстановительного фактора [5].
Показано, что APE1 вызывает деградацию цепи РНК в дуплексах ДНК–РНК (РНКаза H-подобная активность) [6], гидролизует РНК, содержащую AP-сайт [7], а также структурированные неповрежденные мРНК, например мРНК c-Myc, преимущественно по сайтам UA, UG и CA [8, 9]. Способность гидролизовать РНК выявлена также на микроРНК, мРНК CD44, РНК-компонентах коронавируса SARS [10]. Barnes и соавт. [8] предположили, что одной из функций цитоплазматического фермента APE1 в клетке может быть эндорибонуклеазный гидролиз мРНК, и установили, что в отличие от АР-эндонуклеазной активности, для эндорибонуклеазной активности APE1 не требуется ион металла. Из этого был сделан вывод о различных механизмах катализа APE1 на ДНК- и РНК-субстратах. Несмотря на интенсивное изучение функциональных особенностей фермента APE1, до сих пор неизвестно, каким образом фермент узнает значительно отличающиеся по своей природе и структуре поврежденные 2′-дезок-сирибонуклеотиды и неповрежденные рибо-нуклеотиды.
Из рентгеноструктурных данных [11] следует, что активный центр фермента образован аминокислотными остатками Asp308, His309, Glu96, Asp210, Asp70, Tyr171, Asn212 и Asn174. В комплексе APE1 с ДНК остатки Arg73, Ala74, Lys78 взаимодействуют с тремя последовательными фосфатами на противоположной от АР-сайта цепи ДНК. Аминокислотные остатки Tyr128, Gly127 выступают в роли клина и расширяют малую бороздку дуплекса. Остатки Met270 и Arg177 стабилизируют внеспиральное положение АР-сайта; при этом Меt270 вытесняет основание, находящееся напротив АР-сайта, внедряясь в малую бороздку ДНК, а Arg177 встраивается со стороны большой бороздки ДНК и образует водородную связь с фосфатной группой, расположенной с 3'‑стороны от AР-сайта. Координация фосфатной группы, расположенной с 5'-стороны от АP-сайта, остатками Asn174, Asn212, His309 приводит фермент-субстратный комплекс в каталитически компетентное состояние. В связывании иона металла в активном сайте APE1 участвуют Asp70, Glu96 и Asp308. С помощью сайт-направленного мутагенеза показано [12], что замена Asp219Ala снижает и ДНК-связывающую, и АР-эндонуклеазную активности фермента; при этом замена остатков Asp90, Glu96 и Asp308 на Ala приводит к снижению эндонуклеазной активности с сохранением ДНК-связывающей активности. Остатки Arg73, Ala74 и Lys78 контактируют с фосфатными группами комплементарной цепи с 5'-стороны от AP-сайта. Остатки Tyr128 и Gly127 располагаются в малой бороздке ДНК, что приводит к ее расширению приблизительно на 2 Å. Каталитическая реакция начинается с нуклеофильной атаки атома кислорода молекулы воды, координированной прямо или опосредованно через ион Mg2+ остатком Asp210 [11, 13, 14]. Предложены также альтернативные механизмы катализа [15], согласно которым остаток Tyr171 в фенолятной форме взаимодействует с гидролизуемой фосфатной группой или остаток His309 выступает в качестве нуклеофильного основания.
Мирошникова и др. показали [14], что при взаимодействии APE1 с ДНК-субстратом, содержащим F-сайт, природа двухвалентного иона металла влияет как на связывание ДНК, так и на каталитический гидролиз 5'-фосфодиэфирной связи. Так, ионы Cu2+ полностью ингибируют связывание ДНК, вероятно, вследствие сильного взаимодействия Cu2+ с дезоксирибозофосфатным остовом ДНК. Ионы Ca2+ также ингибируют АР-эндонуклеазную активность фермента, вероятно, вследствие неправильного расположения иона металла в активном сайте фермента из-за большего по сравнению с Mg2+ ионного радиуса. Другие ионы снижают ферментативную активность APE1 в следующем порядке: Mg2+ < Mn2+ < < Ni2+ < Zn2+. Кроме того, установлено, что ионы металлов оказывают влияние как на способность фермента связывать ДНК, так и на его структуру [14].
Сравнивая АР-эндонуклеазную и эндорибонуклеазную активности APE1, можно заметить ряд отличий. Для проявления АР-эндонуклеазной активности АРЕ1 с ДНК-субстратами необходимы ионы Mg2+, тогда как присутствие в реакционной смеси ионов Са2+ блокирует действие фермента. В случае РНК-субстратов АРE1 работает как в отсутствие ионов двухвалентных металлов, так и в присутствии ионов кальция. Кроме того, Kim и соавт. показали [16], что при гидролизе фосфодиэфирной связи в РНК-субстратах в качестве конечного продукта реакции образуется 3'-${\text{PO}}_{4}^{{2 - }}$, а не 3'-ОН, как в случае ДНК-субстратов. Следовательно, механизм реакции гидролиза, реализуемый АРЕ1 на разных типах субстратов, различен.
В этой же работе [16] исследована активность различных мутантных форм APE1: H309N, E96A [9], N68A, D70A, Y171F, D210N, F266A, D283N, D308A и H309S. В результате сравнительного анализа активности этих мутантных форм APE1 по отношению к модельным РНК- и ДНК-субстратам показано, что большинство перечисленных остатков, критичных для гидролиза AP-сайта в ДНК, важны также и для гидролиза РНК, за исключением остатка Asp283, замена которого приводила только к потере АР-эндонуклеазной, но не рибоэндонуклеазной активности.
Однако на сегодняшний день так и не установлено, как АРЕ1 узнает неповрежденные рибонуклеотиды, насколько важно пространственное строение субстрата, какие конкретно аминокислотные остатки активного центра АРЕ1 участвуют в узнавании РНК-субстрата и каталитическом гидролизе фосфодиэфирной связи.
Нами проведен сравнительный анализ эндорибонуклеазного гидролиза модельных РНК-субстратов ферментом APE1 дикого типа и его мутантными формами, содержащими замены аминокислотных остатков активного центра: Y171F, R177F, R181A, D210N, N212A, T268D, M270A и D308A. Впервые получены данные о связывании исследованных мутантных форм APE1 с модельным РНК-субстратом, а также о конформационных превращениях фермента и РНК при образовании фермент-субстратного комплекса. Полученные данные позволили детализировать роль проанализированных аминокислотных остатков в реализации АР-эндонуклеазной, 3'-5'-экзонуклеазной и эндорибонуклеазной активности APE1.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы. В работе использовали реактивы производства “Sigma-Aldrich” (США): акриламид, N,N'-метиленбисакриламид, 2-амино-2-(гидроксиметил)-1,3-пропандиол (Tris), тетраметилэтилендиамин (TEMED), мочевина, борная кислота, дитиотреитол (DTT), этилендиаминтетрауксусная кислота (EDTA) и ее натриевая соль, Coomassie Brilliant Blue G-250, пероксодисульфат аммония, 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота (HEPES), додецилсульфат натрия (SDS), хлорид кальция, хлорид магния, хлорид калия, глицерин. Все растворы готовили на дважды дистиллированной воде.
ДНК- и РНК-субстраты. Олигодезоксирибонуклеотиды были синтезированы на автоматическом ДНК/РНК-синтезаторе ASM-800 (“Биоссет”, Россия) с использованием коммерческих амидофосфитов 2′-дезоксирибонуклеозидов и CPG-носителей (“Glen Research”, США) (табл. 1). Синтезированные олигонуклеотиды очищали с помощью ВЭЖХ на ионообменной колонке PRP-X500 (12–30 мкм) размером 3.9 × 300 мм (“Hamilton”, Великобритания) и последующей обращенофазовой хроматографии на колонке Bondapak C18 (15–20 мкм) размером 3.9 × 300 мм (“Waters”, Ирландия).
Олигорибонуклеотиды получали твердофазным фосфитамидным методом с использованием соответствующих фосфитамидов 2'-O-трет-бутилдиметилсилильных (2'-О-TBDMS) рибонуклеотидов (“ChemGenes”, США). Для введения флуоресцеина на 5'-конец использовали фосфитамид флуоресцеина (“Glen Research”). Для получения олигонуклеотидов, содержащих на 3'-конце тушитель флуоресценции BHQ1 (Black Hole Quencher-1), использовали соответствующий модифицированный полимерный носитель 3'-BHQ-1 CPG (“Glen Research”). Деблокирование олигорибонуклеотидов проводили в стандартных условиях. Деблокированные олигорибонуклеотиды выделяли с помощью препаративного электрофореза в 15%-ном ПААГ в денатурирующих условиях (акриламид: N,N’-метиленбисакриламид (30 : 1), 8 М мочевина, 50 мМ Трис-Н3ВО3, рН 8.3, 0.1 мМ EDTA) при напряжении 50 В/см и толщине геля 0.4 мм. Полосы геля, содержащие продукт, вырезали и проводили элюцию нуклеотидного материала из ПААГ. Для этого измельченный гель помещали в 2-миллилитровую пробирку, заливали 1‒1.5 мл 0.3 М LiClO4 и выдерживали 16 ч при 25°С и перемешивании на термомиксере (Thermo-mixer Compact; “Eppendorf”, Германия). Обессоливание олигонуклеотидов проводили на колонке с С18 (“Waters”, США). Чистоту олигонуклеотидов проверяли с помощью денатурирующего электрофореза в 15%-ном ПААГ с последующим окрашиванием красителем Stains-all (“Sigma-Aldrich”).
Оптическое поглощение растворов олигонуклеотидов измеряли на спектрофотометре NanoDrop 1000 (“Thermo Fisher Scientific”, США) относительно деионизованной воды. Для расчета концентрации олигонуклеотидов в исходном растворе использовали коэффициент молярного поглощения олигорибонуклеотидов или их конъюгатов при 260 нм. Молярные коэффициенты поглощения флуоресцентно меченных производных олигорибонуклеотидов считали равными сумме значений молярных коэффициентов поглощения олигонуклеотидов и молярного коэффициента поглощения флуоресцеина и тушителя, присоединенного к олигомеру (20 900 M–1 ∙ cм–1 для FAM и 8000 M–1 ∙ cм–1 для BHQ1).
Последовательности модельных ДНК- и РНК-субстратов представлены в табл. 1.
Фермент АРЕ1 дикого типа и его мутантные формы. Фермент АРЕ1 дикого типа выделен из линии клеток Escherichia coli Rosetta 2, трансформированных плазмидой pET11а, несущей ген AP-эндонуклеазы человека, по следующей методике. Культуру клеток E. coli Rosetta 2 выращивали в среде LB (1 л), содержащей 50 мкг/мл ампициллина, при температуре 37°С до оптического поглощения среды 0.6‒0.7 при длине волны (λ) 600 нм; после чего температуру понижали до 20°С, добавляли изопропил-β-D-тиогалактопиранозид до концентрации 0.2 мМ и инкубировали в течение 16 ч при перемешивании. Клетки осаждали центрифугированием (10 мин при 4000 × g) и ресуспендировали в 30 мл буферного раствора (RB1), содержащего 20 мМ HEPES-NaOH, pH 7.8, и 40 мМ NaCl. Для разрушения клеток использовали френч-пресс. Все последующие процедуры проводили при 4°С. Полученный клеточный экстракт центрифугировали (40 мин при 40 000 × g), супернатант наносили на колонку I (30 мл, Q-Sepha-rose Fast Flow; “Amersham Biosciences”, Швеция) и промывали RB1. Фракции, содержащие белок APE1, собирали и наносили на колонку II (HiTrap-Heparin™; “Amersham Biosciences”). Хроматографию проводили в буферном растворе RB1 и линейном градиенте 40→600 мМ NaCl, оптическое поглощение раствора регистрировали на длине волны 280 нм. Степень чистоты белка APE1 определяли электрофорезом в SDS-ПААГ. Фракции, содержащие белок APE1, диализовали в буфере, содержащем 20 мМ HEPES-NaOH, pH 7.5, 1 мМ EDTA, 1 мМ DTT, 250 мМ NaCl, 50% глицерин, и хранили при ‒20°С. Концентрацию фермента рассчитывали из значения оптического поглощения белка при 280 нм и коэффициента молярной экстинкции (ε), равного 56 818 М–1 ∙ см–1.
Мутантные формы APE1, содержащие замены Y171F, R177F, R181A, D210N, N212A, T268D, M270A и D308A, выделяли таким же способом за исключением того, что клетки E. coli были трансформированы экспрессионными векторами, несущими соответствующие нуклеотидные замены, как описано ранее [17]. Мутантные формы APE1, содержащие замены Y171F, R177F и R181A, были получены впервые, для этого методом сайт-направленного мутагенеза в ген фермента были введены соответствующие мутации (QuikChange XL; “Stratagene”, США).
Анализ гидролиза модельных ДНК- и РНК-субстратов. Анализ расщепления модельных ДНК- и РНК-субстратов проводили по следующей методике. К 10 мкл раствора субстрата добавляли 10 мкл фермента в буферном растворе RB2 следующего состава: 50 мM Трис-HCl (pH 7.5), 50 мM KCl, 1 мM EDTA, 1 мM DTT, 9% глицерин ‒ при 25°С. В случае ДНК- и РНК-субстратов использовали следующие концентрации: 2 мкМ субстрат и 0.05 мкМ фермент, 2 мкМ субстрат и 4 мкМ фермент соответственно. Для определения влияния ионов металлов на эффективность гидролиза ДНК- и РНК-субстратов в реакционную смесь дополнительно добавляли 5 мМ MgCl2 или СаCl2. Через 30 с для ДНК-субстрата и через 1 ч для РНК-субстратов реакцию останавливали путем добавления 20 мкл раствора, содержащего 9 М мочевину и 25 мМ EDTA. Электрофорез проводили в 20%-ном ПААГ в денатурирующих условиях (7 М мочевина) в вертикальной термостатируемой камере Protean II xi (“Bio-Rad Laboratories, Inc.”) при напряжении 200‒300 В и температуре 55°С. Для визуализации использовали гель-документирующую систему E-Box CX.5 TS (“Vilber Lourman”, Франция).
Частичный гидролиз РНК-субстратов HP1 и HP2 РНКазой A выполняли по следующей методике. Реакционную смесь (20 мкл), содержащую 3.0 мкМ субстрат и 3.0 нМ РНКазу A (“Sigma-Aldrich”) в буферном растворе RB3 (50 мM Трис-HCl (pH 8.5), 50 мM NaCl, 1 мM EDTA, 1 мM DTT, 9% глицерин), инкубировали 5 мин при 25°С, добавляли 20 мкл раствора, содержащего 9 М мочевины и 25 мМ EDTA, и выдерживали 5 мин при 96°С.
Оценка относительной активности мутантных форм APE1. Степень гидролиза субстрата определяли с использованием программного пакета Gel-Pro Analyzer 4.0 (“Media Cybernetics”, США). Величину степени гидролиза рассчитывали как отношение площади пика продукта гидролиза к сумме площадей пиков продукта и исходного олигорибонуклеотида. Предполагаемая ошибка определения степени модификации, как правило, не превышала 20%. Активность мутантных форм определяли относительно активности фермента дикого типа, которую принимали равной 1.
Микроскопический термофорез (МСТ). Определение констант связывания исследуемых субстратов с ферментом APE1 выполнено на приборе Monolith NT.115 (“NanoTemper Technologies”, Германия) с использованием стандартных капилляров (MonolithTM NT.115 Standard Treated Capillaries). Каждая точка на кривых титрования получена путем измерения интенсивности флуоресценции отдельных растворов (10 мкл), содержащих олигонуклеотидный лиганд (1 мкМ) и фермент (0.05–18 мкМ) в буферном растворе RB2 при 25°С. Для расчета значений констант диссоциации экспериментальные данные обрабатывали в пакете программ DynaFit (“BioKin”, США) [18] с использованием модели одностадийного связывания.
Кинетические исследования методом “остановленного потока”. Кинетические кривые были зарегистрированы по изменению интенсивности флуоресценции на спектрометре “остановленного потока” SX.20 (“Applied Photophysics”, Великобритания). Кинетику взаимодействия APE1 дикого типа с флуоресцентными РНК-субстратами исследовали в буферном растворе RB2 при 25°С. В случае регистрации изменений интенсивности остатков aPu и Cpy концентрация фермента составляла 2.0 мкМ, концентрация субстрата – 1.0 мкМ. Длина волны возбуждения составляла 310 нм и 344 нм для aPu и Cpy соответственно; испускание флуоресценции детектировали при λ > 370 нм, используя светофильтр LG-370.
При анализе изменений интенсивности флуоресценции остатков Trp концентрация фермента составляла 2.0 мкМ, а концентрация РНК-субстрата изменялась в диапазоне 0.5‒4.0 мкМ. Возбуждение интенсивности флуоресцецинции остатков Trp проводили при 290 нм, а сигнал регистрировали при λ > 320 нм, используя светофильтр WG-320.
Эффективность резонансного переноса энергии флуоресценции (FRET) между парой FAM/BHQ1 регистрировали при возбуждении FAM на длине волны 494 нм и сигнала испускания FRET при λ > > 530 нм c использованием светофильтра OG-515 (“Schott”, Германия).
Мертвое время прибора составляло 1 мс. Каждую кинетическую кривую усредняли, как минимум, по четырем повторам.
Наблюдаемую константу скорости рассчитывали путем обработки кинетических кривых по уравнению (1):
(1)
${\text{f}}(t) = {{{\text{f}}}_{0}} + {{{\text{f}}}_{1}}(1 - {{e}^{{ - {{k}_{{{\text{obs}}}}} \cdot t}}}),$РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Выбор аминокислотных остатков активного центра APE1 для исследования эндорибонуклеазной активности
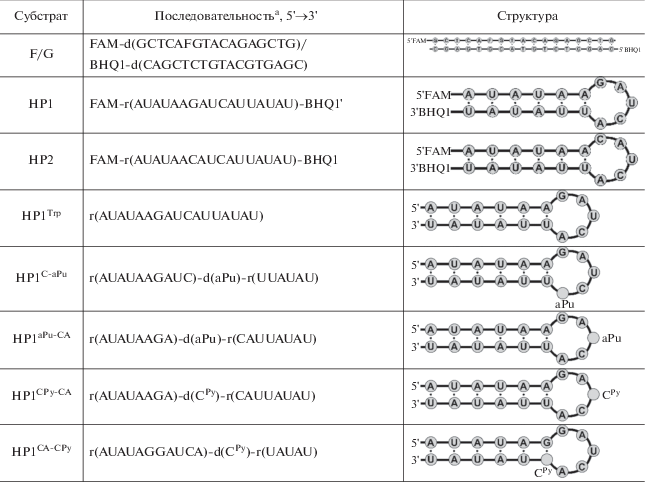
С целью идентифицировать ключевые для эндорибонуклеазной активности APE1 аминокислотные остатки активного центра фермента было исследовано расщепление модельных РНК-субстратов в присутствии APE1 дикого типа и его мутантных форм: Y171F, R177F, R181A, D210N, N212A, T268D, M270A, D308A. Выбранные для исследования аминокислотные остатки (рис. 1) можно разделить на три группы: участвующие в каталитической реакции (Tyr171, Asp210, Asn212 и Asp308), встраивающиеся в двойную спираль после выворачивания целевого нуклеотида в активный центр (Arg177 и Met270) и расположенные в субстратсвязывающем сайте (Thr268 и Arg181).
Рис. 1.
Структура комплекса АР-эндонуклеазы APE1 с F/G-субстратом (PDBID 1DE8). Каталитические остатки (Tyr171, Asp210, Asn212 и Asp308) показаны красным цветом; остатки, встраивающиеся в двойную спираль после выворачивания целевого нуклеотида в активный центр (Arg177 и Met270), – зеленым; остатки, расположенные в субстратсвязывающем сайте (Thr268 и Arg181), – синим; внеспиральное положение поврежденного нуклеотида выделено черным цветом.
Необходимо отметить, что участие этих остатков в связывании ДНК-субстратов, содержащих поврежденный нуклеотид, в узнавании F-сайта и каталитической реакции установлено ранее в ряде работ на основе рентгеноструктурных данных и данных сайт-направленного мутагенеза. Подытоживая, можно заключить, что Asp308 участвует в координации иона Mg2+ в активном центре и его замена на Ala приводит к снижению АР-эндонуклеазной активности [19]. Asn212 образует водородную связь с гидролизуемой фосфатной группой, а также участвует в координации молекулы воды и стабилизации переходного состояния. Его замена на Ala приводит к полной потере эндонуклеазной активности в отношении АР-сайта [19]. Asp210 координирует молекулу воды, участвующую в гидролизе фосфодиэфирной связи. Его замена на Asn также приводит к полной потере каталитической активности [20]. Остаток Tyr171 либо участвует в нуклеофильной атаке фосфатной группы в фенолятной форме, либо стабилизирует переходное состояние за счет образования водородной связи с фосфатной группой [21, 22].
Met270, предположительно, стабилизирует внеспиральное положение F-сайта; при этом его замена на Ala не приводит к изменению активности фермента [11]. Arg177 участвует в специфическом связывании ДНК, содержащей АР-сайт [11].
Thr268 расположен в ДНК-связывающем центре. Его замена на Asp приводит к значительному снижению активности по отношению к NIR-субстратам [23]. Arg181 расположен в ДНК-связывающем центре и участвует в связывании ДНК.
Дизайн РНК-субстратов
Согласно литературным данным [8, 10], основные сайты расщепления РНК под действием APE1 ‒ динуклеотидные последовательности CA, UA и UG в одноцепочечных или слабо спаренных областях. Кроме того, наблюдали расщепление РНК и в сайтах UC, CU, AC и AU [10].
Можно предположить, что для образования каталитического комплекса важна как последовательность пиримидин-пурин, так и структура субстрата, которая может оказывать значительное влияние на узнавание целевого сайта. Действительно, в работах группы Lee [8, 10] расщепление РНК детектировали в одноцепочечных областях вблизи стеблей шпилечных структур, что может указывать на значительный вклад именно вторичной структуры РНК в узнавание ее как субстрата APE1.
Ранее нами проанализировано взаимодействие АР-эндонуклеазы человека APE1 с модельными РНК-субстратами различной структуры [24]. Показано, что образование каталитически компетентного фермент-субстратного комплекса зависит как от конформационной напряженности петли РНК-субстрата, имеющего форму шпильки, так и от контекста и местоположения гидролизуемой фосфодиэфирной связи. Опираясь на эти данные, мы использовали два модельных РНК-субстрата длиной 17 нуклеотидов, представляющих собой короткие шпилечные структуры, в которых длина стебля была равна 6 п.о., а размер петли составлял 5 нуклеотидов. Субстрат HP1, FAM-r(AUAUAAGAAUCAUUAUAU)-BHQ1, содержал одну динуклеотидную последовательность CA в петле, а субстрат HP2, FAM-r(AUAUAACAUCAUUAUAU)-BHQ1, – две динуклеотидные последовательности CA (табл. 1).
Для анализа кинетики фермент-субстратного взаимодействия использовали как субстрат HP1Trp, не содержащий флуоресцентных меток, так и субстраты, содержащие флуоресцентные метки: HP1, HP1C-aPu, HP1aPu-CA, HP1CPy-CA, HP1CA-CPy. Олигонуклеотид HP1aPu-CA содержал остаток 2‑аминопурина с 3'-стороны от пары СА, тогда как HP1C-aPu ‒ непосредствено после цитозина. Субстраты HP1CPy-CA и HP1CA-CPy содержали остаток пироллоцитозина с 3'-стороны и с 5'-стороны от пары СА соответственно.
Влияние замен аминокислотных остатков на взаимодействие APE1 с ДНК, содержащей F-сайт
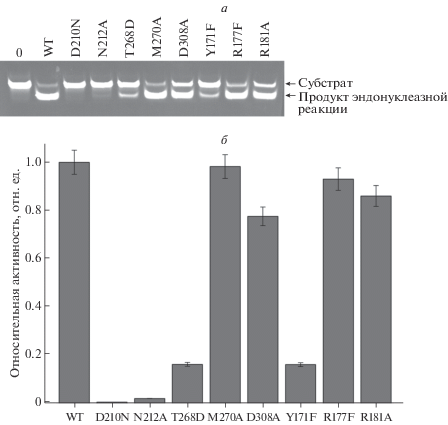
Для контроля активности мутантных форм APE1 использовали F/G-субстрат, содержащий в качестве поврежденного нуклеотида остаток 3‑гидрокси-2-гидроксиметилтетрагидрофурана (F-сайт). Как видно на рис. 2, замена каталитически значимых аминокислотных остатков: Tyr171, Asp210 и Asn212, ‒ а также Thr268, расположенного в ДНК-связывающем центре, приводит к значительной потере АР-эндонуклеазной активности фермента. В то же время замена Arg177 и Met270, встраивающихся в ДНК при образовании каталитически компетентного комплекса, а также Arg181, образующего контакт с дезоксирибозофосфатным остовом в ДНК-связывающем центре, незначительно влияют на эффективность гидролиза F-сайта. Однако замена Asp308, участвующего в координации иона Mg2+ в активном центре, приводит к снижению активности примерно на 20% по сравнению с ферментом дикого типа.
Рис. 2.
Гидролиз F/G-субстрата под действием APE1 дикого типа (WT) и мутантных форм. а ‒ Анализ продуктов реакции методом электрофореза в ПААГ. б ‒ Относительная степень гидролиза F/G-субстрата ферментом дикого типа и его мутантными формами ([фермент] = 0.05 мкМ, [F/G] = 2.0 мкM, Т = 25°C, t = 30 с).
Гидролиз РНК-субстратов
Влияние ионов Mg2+ на процессы связывания субстрата, гидролиза фосфодиэфирной связи в составе ДНК активно обсуждаются в литературе [25–32]. Более того, уже предложено два каталитических механизма действия APE1, в которых участвуют один или два катиона Mg2+ [11, 19]. В то же время, согласно литературным данным [10, 24], APE1 расщепляет РНК в отсутствие ионов двухвалентных металлов. На основании этого мы анализировали взаимодействие APE1 дикого типа и мутантных форм с модельными РНК-субстратами как в отсутствие ионов двухвалентных металлов (рис. 3), так и в присутствии ионов Ca2+ (рис. 4) и Mg2+ (рис. 5).
Рис. 3.
Гидролиз HP1 (а) и HP2 (б) под действием APE1 дикого типа (WT) и мутантных форм в присутствии 1 мM EDTA. [APE1] = 2.0 мкМ, [РНК] = 1.0 мкМ, Т = 25°C, t = 1 ч.
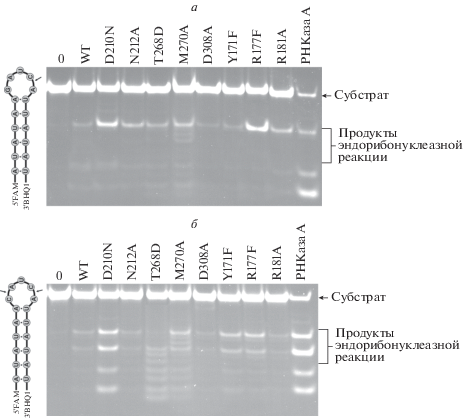
Рис. 4.
Гидролиз РНК-субстратов HP1 (а) и HP2 (б) под действием APE1 дикого типа и мутантных форм в присутствии 5 мM CaCl2. [APE1] = 2.0 мкМ, [РНК] = 1.0 мкМ, Т = 25°C, t = 1 ч.
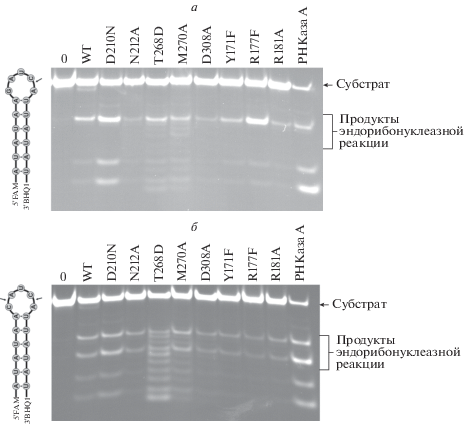
Рис. 5.
Гидролиз РНК-субстратов HP1 (а) и HP2 (б) под действием APE1 дикого типа и мутантных форм в присутствии 5 мM MgCl2. [APE1] = 2.0 мкМ, [РНК] = 1.0 мкМ, Т = 25°C, t = 1 ч.
Как показано на рис. 3, в отсутствие ионов двухвалентных металлов в случае обоих РНК-субстратов, HP1 и HP2, происходило накопление продуктов реакции; при этом расщепление происходило преимущественно по последовательности СА, расположенной в петле. Наличие продуктов эндорибонуклеазной реакции в случае мутантных форм фермента, у которых отсутствует АР-эндонуклеазная активность, свидетельствует о протекании гидролиза фосфодиэфирной связи в РНК по каталитическому механизму, отличному от такового на AP-сайтах ДНК.
Следует отметить, что в присутствии ионов Ca2+ также наблюдали накопление продуктов эндорибонуклеазной реакции (рис. 4). Здесь следует заметить, что, как показано ранее [14], при взаимодействии APE1 с ДНК-субстратом, содержащим F-сайт, присутствие ионов Ca2+ приводило к блокированию АР-эндонуклеазной реакции [33].
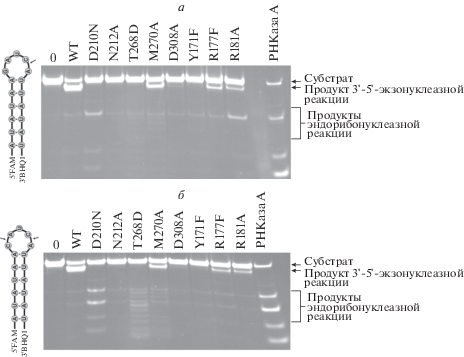
При взаимодействии APE1 с РНК-субстратами в буферном растворе, содержащем 5 мМ MgCl2, который необходим ферменту для осуществления АР-эндонуклеазной реакции, наблюдали как накопление продуктов эндорибонуклеазной активности, так и продуктов 3'-5'-экзонуклеазной реакции (отщепление тушителя BHQ1 с 3'‑конца) (рис. 5). Можно предположить, что гидролиз межнуклеотидной фосфатной группы в РНК-субстрате может проходить без участия иона Mg2+, тогда как гидролиз 3'-концевого остатка BHQ1 проходит по механизму, характерному для гидролиза ДНК-субстратов. В пользу этого заключения также свидетельствует отсутствие продуктов 3'-5'-экзонуклеазной деградации РНК-субстратов под действием мутантных форм фермента, которые лишены АР-эндонуклеазной активности (D210N, N212A, T268D, Y171F).
На основании полученных данных мы рассчитали относительную активность всех мутантных форм фермента APE1 по отношению к РНК-субстратам HP1 и HP2 в присутствии EDTA и ионов двухвалентных металлов: Mg2+ и Ca2+ (рис. 6).
Рис. 6.
Относительная активность АРЕ1 дикого типа и мутантных форм при гидролизе субстрата HP1 (а, в, д) и НР2 (б, г, е) в присутствии 1.0 мM EDTA (а, б), 5.0 мM CaCl2 (в, г) и 5.0 мM MgCl2 (д, е).
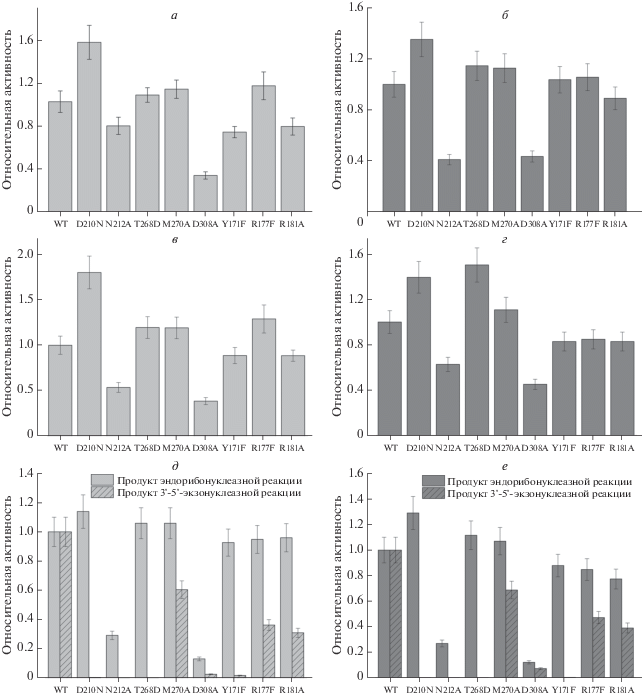
Интересно отметить, что замены D210N и N212A, которые приводят к исчезновению АР-эндонуклеазной активности в отношении ДНК-субстрата, содержащего F-сайт, не блокируют эндорибонуклеазную активность APE1. Оба остатка, Asn212 и Asp210, координируют молекулу воды в активном центре, которая действует как нуклеофил при каталитическом гидролизе ДНК. Таким образом, на основании полученных данных можно считать, что расщепление РНК зависит от присутствия Asn212 или Asp210 в гораздо меньшей степени, чем в случае ДНК-субстрата, а значит в гидролизе РНК-субстратов задействован альтернативный каталитический механизм APE1. Кроме того, замена еще одного ключевого каталитического остатка, необходимого для эффективного осуществления АР-эндонуклеазной активности – Tyr171 – также не приводит к уменьшению эффективности гидролиза РНК-субстратов.
Замена T268D оказывает существенное влияние (примерно 5-кратное снижение активности по сравнению с WT, рис. 2б) на АР-эндонуклеазную активность. Действительно, замена T268D может нарушать взаимодействия APE1 с ДНК посредством электростатического отталкивания в ДНК-связывающем центре, так как остаток Thr268 расположен в петле, которая встраивается в спираль ДНК со стороны малой бороздки рядом с AP-сайтом. В этом случае боковая группа Thr268 находится между боковой цепью Met270, встраивающегося в ДНК на место F-сайта, и фосфатной группой ДНК-субстрата. Следовательно, аспартат в положении 268 может влиять на связывание субстрата путем стерических взаимодействий с боковой цепью Met270. Интересно отметить, что замены T268D и M270A в APE1 приводят к потере специфичности фермента к пиримидин-пуриновой последовательности в обоих РНК-субстратах по сравнению с ферментом дикого типа (рис. 3‒5). Такая потеря специфичности к контексту РНК-субстратов действительно указывает на то, что эффект замены T268D не может быть обусловлен только электростатическим отталкиванием. Можно предположить, что влияние этой замены, с одной стороны, связано с взаимодействием Thr268 с боковой цепью Met270, которая встраивается в полость, образованную после выворачивания нуклеотида-мишени в активный центр фермента. С другой стороны, замена M270A теоретически должна уменьшать любые стерические затруднения при связывании APE1 как с ДНК, так и с РНК-субстратами и, следовательно, активность мутантной формы M270A практически не отличается от активности фермента дикого типа.
Действительно, замена Met270 не снижала способность APE1 расщеплять AP-сайты, из чего можно сделать вывод, что этот остаток не участвует в процессе выворачивания нуклеотидов в активный центр фермента. Таким образом, функция Met270, скорее всего, заключается в стабилизации состояния, в котором гидролизуемый нуклеотид вывернут и расположен в активном центре фермента. Потеря специфичности к СА-сайту расщепления РНК также указывает на то, что Met270 выполняет важную функцию в распознавании нуклеотида-мишени. Что касается замены остатка Arg177, который встраивается в субстрат со стороны большой бороздки, а также остатка Arg181, расположенного в ДНК-связывающем центре, то они практически не влияли ни на АР-эндонуклеазную, ни на эндорибонуклеазную активность АРЕ1.
АР-эндонуклеазная активность мутанта D308A АРЕ1 была близка таковой для фермента дикого типа, но снижена при гидролизе РНК-субстратов. Координация иона Mg2+ остатком Asp308 может быть важна для стабилизации сетевых контактов в каталитическом центре при расщеплении ДНК. Кроме того, остаток Asp308 стабилизирует положение остатка His309, который образует контакт с фосфатной группой гидролизуемого нуклеотида, тем самым способствуя достижению каталитически компетентного состояния фермента.
Связывание РНК-субстратов
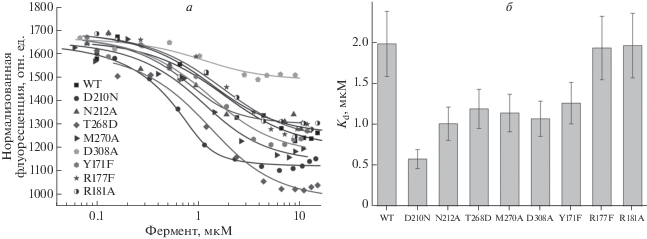
С помощью микроскопического термофореза (MCT) было исследовано связывание APE1 дикого типа и его мутантных форм с субстратом HP2 (рис. 7а). По кривым титрования рассчитаны константы диссоциации (Kd) фермент-субстратных комплексов (рис. 7б). Анализ стабильности фермент-субстратных комплексов проведен методом МСТ. В результате показано, что образование комплекса APE1, как дикого типа, так и мутантных форм, с HP2 характеризуется близкими значениями констант диссоциации в диапазоне концентраций субстрата 0.5–2.0 мкМ. Таким образом, ни одна из проанализированных мутаций не приводила к серьезным нарушениям фермент-субстратных взаимодействий. Более того, наименьшее значение Kd (0.6 ± 0.1 мкМ) зарегистрировано для D210N APE1, что хорошо согласуется с литературными данными о более прочном связывании этого мутанта с модельными ДНК-субстратами по сравнению с ферментом дикого типа [17, 20, 30, 34]. В то же время вариант D210N APE1 обладал повышенной активностью по отношению к обоим исследованным РНК-субстратам как в отсутствие, так и в присутствии ионов двухвалентных металлов. Кроме того, две наименее активные мутантные формы АРЕ1, D308A и N212A, также связывались с РНК-субстратом более эффективно, чем фермент дикого типа. Таким образом, различия в активности мутантных форм APE1, скорее всего, связаны с влиянием исследованных аминокислотных остатков на формирование каталитически компетентного состояния, а не на стадию образования фермент-субстратного комплекса.
Рис. 7.
Определение констант диссоциации (Kd) фермент-субстратных комплексов методом микроскопического термофореза с использованием в качестве субстрата HP2. а ‒ Кривые титрования субстрата HP2 под действием APE1 дикого типа и мутантных форм. б ‒ Сравнительный анализ констант диссоциации комплексов АРЕ1-HP2.
Анализ кинетики взаимодействия APE1 дикого типа с РНК-субстратом НР1
Ранее [33, 35] при исследовании связывания APE1 с АР-сайтами и гидролиза 5'-фосфодиэфирной связи в предстационарных условиях методом “остановленного потока” детектировали изменения интенсивности флуоресценции АРЕ1, вызванные присутствием в белке остатков Trp. При исследовании эндорибонуклеазной реакции нами также зарегистрированы изменения интенсивности флуоресценции остатков Trp на временах менее 0.5 с, которые, скорее всего, соответствуют образованию фермент-субстратного комплекса (рис. 8а). Следует сказать, что медленная реакция гидролиза РНК-субстрата не может быть зарегистрирована этим методом вследствие деструкции остатков Trp при длительном облучении на длине волны возбуждения флуоресценции (фотобличинга).
Рис. 8.
Кинетика взаимодействия APE1 дикого типа с РНК-субстратами. а ‒ Изменения интенсивности флуоресценции остатков Trp, характеризующие образование фермент-субстратного комплекса. б ‒ Зависимость наблюдаемой константы скорости (kobs) от концентрации РНК-субстрата HP1. в ‒ Изменения интенсивности флуоресценции остатков aPu, CPy и FRET-сигнала, характеризующие конформационные превращения РНК-субстрата.
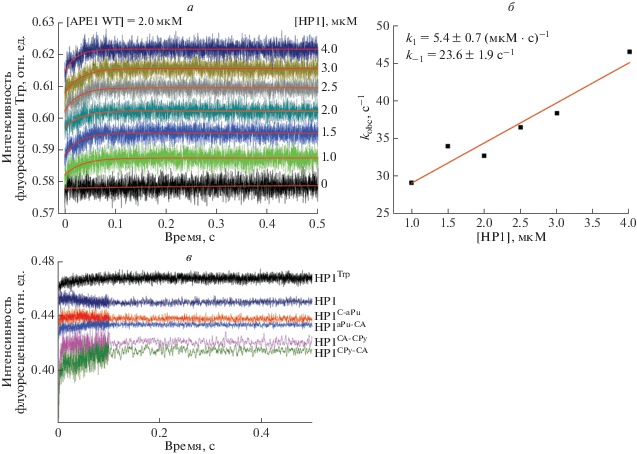
Для расчета наблюдаемых констант скорости, kobs, кинетические кривые аппроксимировали по уравнению (1). Учитывая, что на начальной стадии взаимодействия АРЕ1 с субстратами образуется фермент-субстратный комплекс, причем эта реакция обратима, для расчета kobs мы использовали уравнение (2), которое справедливо при условии избытка одного из компонентов реакции. Несмотря на то, что в проведенных нами экспериментах это не всегда было выполнимо, зависимость kobs от концентрации субстрата носила линейный характер (рис. 8б), что соответствовало одностадийному механизму связывания и позволило рассчитать константы скорости образования и распада фермент-субстратного комплекса (АРЕ1 ∙ НР1) с помощью уравнения (2), где k1 = 5.4 ± 0.7 мкM‒1 ∙ с‒1, k‒1 = 23.6 ± 1.9 с‒1. В этом случае значение Kd равно 4.7 ± 1.1 мкM. Это значение немного превышает величину, измеренную методом термофореза (см. рис. 7б), однако, учитывая принципиальное различие методов и условий реакции, использованных для получения значений Kd, такое расхождение можно считать удовлетворительным.
В проведенной нами работе использованы модельные РНК-субстраты, содержащие флуоресцирующие основания 2-аминопурин (aPu) и пирролоцитозин (CPy) в различных положениях шпильки (табл. 1, рис. 8в). Известно, что интенсивность флуоресценции aPu и CPy зависит от гидрофильности окружающей среды [36–39], поэтому, наблюдая за изменением этого параметра для аналогов азотистых оснований, можно судить о конформационных превращениях РНК-субстрата.
На основании полученных результатов можно говорить о том, что остаток aPu не чувствителен к процессам, происходящим при образовании комплекса фермента с РНК-субстратом, в то время как для CPy зарегистрирован рост интенсивности флуоресценции на начальном участке кинетических кривых (до 10 с), что может отражать процесс образования фермент-субстратного комплекса.
ЗАКЛЮЧЕНИЕ
Таким образом, нами проведен сравнительный анализ АР-эндонуклеазной и эндорибонуклеазной активности апуриновой/апиримидиновой эндонуклеазы человека APE1 дикого типа и восьми мутантных форм, содержащих замены аминокислотных остатков, входящих в состав активного и субстратсвязывающего центра. Показано, что при взаимодействии APE1 с РНК-субстратами в присутствии ионов Ca2+, а также в отсутствие ионов металлов происходит накопление продуктов только эндорибонуклеазной реакции, что свидетельствует о металлнезависимом каталитическом механизме гидролиза фосфодиэфирной связи в РНК-субстратах. В присутствии ионов Mg2+ протекают и эндорибонуклеазная, и 3'-5'-экзонуклеазная реакции. Эти два типа ферментативной активности связаны с разными каталитическими механизмами действия фермента. Так, замена аминокислотных остатков Asn212, Asp210 и Tyr171 приводит к потере АР-эндонуклеазной и 3'-5'-экзонуклеазной активностей с сохранением эндорибонуклеазной активности, из чего можно сделать вывод о том, что при гидролизе фосфодиэфирной связи в составе РНК APE1 задействует механизм катализа, альтернативный таковому на ДНК-субстратах. Установлено, что замены T268D и M270A приводят к потере специфичности фермента к пиримидин-пуриновой последовательности, а мутации Arg177 и Arg181 незначительно влияют на активность фермента. Показано, что замена D308A приводит к снижению эндорибонуклеазной активности фермента, по-видимому, за счет потери контактов с каталитически значимым остатком His309.
Экспериментальная часть работы выполнена при поддержке гранта РНФ № 19-74-10034 и частичной поддержке бюджетного финансирования для обеспечения регламентных работ на использованном оборудовании № АААА-А17-117020210022-4.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Gros L., Ishchenko A.A., Ide H., Elder R.H., Saparbaev M.K. (2004) The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Res. 32, 73–81.
Chen D.S., Herman T., Demple B. (1991) Two distinct human DNA diesterases that hydrolyze 3'-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. 19, 5907–5914.
Chou K.-M., Cheng Y.-C. (2003) The exonuclease activity of human apurinic/apyrimidinic endonuclease (APE1). Biochemical properties and inhibition by the natural dinucleotide Gp4G. J. Biol. Chem. 278, 18289–18296.
Kuznetsova A.A., Fedorova O.S., Kuznetsov N.A. (2018) Kinetic features of 3'-5' exonuclease activity of human AP-endonuclease APE1. Molecules. 23(9), 2101.
Li M., Wilson 3rd D.M. (2014) Human apurinic/apyrimidinic endonuclease 1. Antioxid. Redox Signal. 20, 678–707.
Barzilay G., Hickson I.D. (1995) Structure and function of apurinic/apyrimidinic endonucleases. Bioessays. 17, 713–719.
Berquist B.R., McNeill D.R., Wilson 3rd D.M. (2008) Characterization of abasic endonuclease activity of human Ape1 on alternative substrates, as well as effects of ATP and sequence context on AP site incision. J. Mol. Biol. 379, 17–27.
Barnes T., Kim W.C., Mantha A.K., Kim S.E., Izumi T., Mitra S., Lee C.H. (2009) Identification of apuri-nic/apyrimidinic endonuclease 1 (APE1) as the endoribonuclease that cleaves c-myc mRNA. Nucleic Acids Res. 37, 3946–3958.
Kim S.E., Gorrell A., Rader S.D., Lee C.H. (2010) Endoribonuclease activity of human apurinic/apyrimidinic endonuclease 1 revealed by a real-time fluorometric assay. Anal. Biochem. 398, 69–75.
Kim W.C., King D., Lee C.H. (2010) RNA-cleaving properties of human apurinic/apyrimidinic endonuclease 1 (APE1). Int. J. Biochem. Mol. Biol. 1, 12–25.
Mol C.D., Izumi T., Mitra S., Tainer J.A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature. 403, 451–456.
Barzilay G., Walker L.J., Robson C.N., Hickson I.D. (1995) Site-directed mutagenesis of the human DNA repair enzyme HAP1: identification of residues important for AP endonuclease and RNase H activity. Nucleic Acids Res. 23, 1544–1550.
Tsutakawa S.E., Shin D.S., Mol C.D., Izumi T., Arvai A.S., Mantha A.K., Szczesny B., Ivanov I.N., Hosfield D.J., Maiti B., Pique M.E., Frankel K.A., Hitomi K., Cunningham R.P., Mitra S., Tainer J.A. (2013) Conserved structural chemistry for incision activity in structurally non-homologous apurinic/apyrimidinic endonuclease APE1 and endonuclease IV DNA repair enzymes. J. Biol. Chem. 288, 8445–8455.
Miroshnikova A.D., Kuznetsova A.A., Vorobjev Y.N., Kuznetsov N.A., Fedorova O.S. (2016) Effects of mono- and divalent metal ions on DNA binding and catalysis of human apurinic/apyrimidinic endonuclease 1. Mol. Biosyst. 12, 1527.
Jen-Jacobson L., Engler L.E., Jacobson L.A. (2000) Structural and thermodynamic strategies for site-specific DNA binding proteins. Structure. 8, 1015–1023.
Kim W.C., Berquist B.R., Chohan M., Uy C., Wilson D.M., Lee C.H. (2011) Characterization of the endoribonuclease active site of human apurinic/apyrimidinic endonuclease 1. J. Mol. Biol. 411, 960–971.
Alekseeva I.V., Kuznetsova A.A., Bakman A.S., Fedo-rova O.S., Kuznetsov N.A. (2020) The role of active-site amino acid residues in the cleavage of DNA and RNA substrates by human apurinic/apyrimidinic endonuclease APE1. Biochim. Biophys. Acta Gen. Subj. 1864, 129718.
Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273.
Masuda Y., Bennett R.A., Demple B. (1998) Rapid dissociation of human apurinic endonuclease (Ape1) from incised DNA induced by magnesium. J. Biol. Chem. 273, 30360–30365.
Rothwell D.G., Hang B., Gorman M.A., Freemont P.S., Singer B., Hickson I.D. (2000) Substitution of Asp-210 in HAP1 (APE/Ref-1) eliminates endonuclease activity but stabilises substrate binding. Nucleic Acids Res. 28, 2207–2213.
Mundle S.T., Fattal M.H., Melo L.F., Coriolan J.D., O’Regan N.E., Strauss P.R. (2004) Novel role of tyrosine in catalysis by human AP endonuclease 1. DNA Repair (Amst.). 3, 1447–1455.
Freudenthal B.D., Beard W.A., Cuneo M.J., Dyrkheeva N.S., Wilson S.H. (2015) Capturing snapshots of APE1 processing DNA damage. Nat. Struct. Mol. Biol. 22, 924–931.
Redrejo-Rodriguez M., Vigouroux A., Mursalimov A., Grin I., Alili D., Koshenov Z., Akishev Z., Maksimenko A., Bissenbaev A.K., Matkarimov B.T., Saparbaev M., Ishchenko A.A., Moréra S. (2016) Structural comparison of AP endonucleases from the exonuclease III family reveals new amino acid residues in human AP endonuclease 1 that are involved in incision of damaged DNA. Biochimie. 128, 20–33.
Кузнецова А.А., Новопашина Д.С., Федорова О.С., Кузнецов Н.А. (2020) Влияние структуры субстрата и ионов металлов на эффективность гидролиза неповрежденной РНК апуриновой/апиримидиновой эндонуклеазой человека APE1. Acta Naturae. 12, 74–85.
Gorman M.A., Morera S., Rothwell D.G., de La Fortelle E., Mol C.D., Tainer J.A., Hickson I.D., Free-mont P.S. (1997) The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 16, 6548–6558.
Beernink P.T., Segelke B.W., Hadi M.Z., Erzberger J.P., Wilson D.M., Rupp B. (2001) Two divalent metal ions in the active site of a new crystal form of human apuri-nic/apyrimidinic endonuclease, Ape1: implications for the catalytic mechanism. J. Mol. Biol. 307, 1023–1034.
Manvilla B.A., Pozharski E., Toth E.A., Drohat A.C. (2013) Structure of human apurinic/apyrimidinic endonuclease 1 with the essential Mg2+ cofactor. Acta Crystallogr. D Biol. Crystallogr. 69, 2555–2562.
Lipton A.S., Heck R.W., Primak S., McNeill D.R., Wilson D.M., Ellis P.D. (2008) Characterization of Mg2+ binding to the DNA repair protein apurinic/apyrimidic endonuclease 1 via solid-state 25Mg NMR spectroscopy. J. Am. Chem. Soc. 130, 9332–9341.
Oezguen N., Schein C.H., Peddi S.R., Power T.D., Izumi T., Braun W. (2007) A “moving metal mechanism” for substrate cleavage by the DNA repair endonuclease APE-1. Proteins. 68, 313–323.
Erzberger J.P., Wilson D.M. (1999) The role of Mg2+ and specific amino acid residues in the catalytic reaction of the major human abasic endonuclease: new insights from EDTA-resistant incision of acyclic abasic site analogs and site-directed mutagenesis. J. Mol. Biol. 290, 447–457.
He H., Chen Q., Georgiadis M.M. (2014) High-resolution crystal structures reveal plasticity in the metal binding site of apurinic/apyrimidinic endonuclease I. Biochemistry. 53, 6520–6529.
Bulygin A.A., Kuznetsova A.A., Vorobjev Y.N., Fedo-rova O.S., Kuznetsov N.A. (2020) The role of active-site plasticity in damaged-nucleotide recognition by human apurinic/apyrimidinic endonuclease APE1. Molecules. 25, 3940.
Мирошникова A.Д., Кузнецова A.A., Кузнецов Н.A., Федорова O.С. (2016) Термодинамика конформационных переходов АP-эндонуклеазы человека APE1 при взаимодействии с ДНК. Acta Naturae. 8, 103–110.
Maher R.L., Bloom L.B. (2007) Pre-steady-state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. J. Biol. Chem. 282, 30577–30585.
Timofeyeva N.A., Fedorova O.S. (2016) A kinetic mechanism of repair of DNA containing alpha-anomeric deoxyadenosine by human apurinic/apyrimidi-nic endonuclease 1. Mol. Biosyst. 12, 3435–3446.
Jean J.M., Hall K.B. (2001) 2-Aminopurine fluorescence quenching and lifetimes: role of base stacking. Proc. Natl. Acad. Sci. USA. 98, 37–41.
Rachofsky E.L., Osman R., Ross J.B. (2001) Probing structure and dynamics of DNA with 2-aminopurine: effects of local environment on fluorescence. Biochemistry. 40, 946–956.
Rist M.J., Marino J.P. (2002) Fluorescent nucleotide base analogs as probes of nucleic acid structure, dynamics and interactions. Curr. Org. Chem. 6, 775–793.
Berry D.A., Jung K.Y., Wise D.S., Sercel A.D., Pearson W.H., Mackie H., Randolph J.B., Somers R.L. (2004) Pyrrolo-dC and pyrrolo-C: fluorescent analogs of cytidine and 2′-deoxycytidine for the study of oligonucleotides. Tetrahedron Lett. 45, 2457–2461.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология