

Молекулярная биология, 2021, T. 55, № 3, стр. 412-421
Спектр герминальных и соматических мутаций в архивных образцах рака молочной железы с различным рецепторным статусом
И. С. Абрамов a, Ю. С. Корнева b, О. А. Шистерова c, А. Ю. Иконникова a, М. А. Емельянова a, Т. С. Лисица a, Г. С. Краснов a, Т. В. Наседкина a, *
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Смоленский государственный медицинский университет Министерства здравоохранения Российской Федерации
214019 Смоленск, Россия
c Смоленский областной онкологический клинический диспансер
214000 Смоленск, Россия
* E-mail: nased@biochip.ru
Поступила в редакцию 02.09.2020
После доработки 25.09.2020
Принята к публикации 29.09.2020
Аннотация
Молекулярное профилирование опухолей – перспективное направление персонализированной медицины. Важную роль в выборе эффективной стратегии терапии играет герминальный или соматический статус мутаций. Мы определили спектр герминальных и соматических мутаций у 23 пациенток с раком молочной железы различных молекулярных подтипов, включая опухоли, экспрессирующие рецепторы эстрогена, прогестерона и/или рецептор эпидермального фактора роста HER2/neu, и опухоли с трижды негативным подтипом – отсутствием экспрессии всех этих рецепторов. Геномную ДНК выделяли из архивных образцов опухолевой и нормальной ткани. Проведено таргетное секвенирование кодирующих участков 25 генов, ассоциированных с раком молочной железы, со средним покрытием ×1000. Эффективность биоинформатического подхода при определении герминального или соматического статуса мутаций этих генов оценивали в сравнении с результатами прямого секвенирования ДНК из образцов нормальной ткани. В группе пациенток с трижды негативным подтипом рака молочной железы выявлены патогенные герминальные мутации BRCA1 c.66_67delAG (185delAG) и BRCA1 c.3226_3227AG (3347delAG). У пациенток с позитивным рецепторным статусом опухоли обнаружена герминальная мутация BRCA2 c.658_659del (886delGT). Мутации BRCA1 представлены с высокой частотой аллельного варианта (80%), что указывает на потерю гетерозиготности в клетках опухоли. Соматические мутации в гене TP53 обнаружены у 7/10 (70%) пациенток с трижды негативным подтипом рака молочной железы и у 3/13 (23%) – в группе с положительным рецепторным статусом. В обеих группах выявлены соматические мутации генов PTEN, MSH2, MSH6, MUTYH.
Рак молочной железы (РМЖ) занимает первое место в структуре онкологических заболеваний у женщин [1]. Ежегодно в мире регистрируется более 2 млн новых случаев РМЖ, что представляет медицинскую и социальную проблему в связи с высокой смертностью (около 30%) среди женского населения [2]. Наиболее высокие показатели заболеваемости отмечены в США, несколько более низкие в странах Европы и России, еще реже РМЖ встречается в странах Азии и Африки [3, 4]. Для прогноза заболевания и выбора схемы терапии РМЖ важен целый ряд факторов, включая возраст, стадию заболевания, степень злокачественности, молекулярный фенотип опухолевых клеток [5, 6].
Заболевание с манифестацией в возрасте 40–50 лет и ранее ассоциировано с плохим прогнозом, высокой частотой рецидивов и высокой смертностью по сравнению с РМЖ, диагностированным после 50 лет. В то время как РМЖ с поздней манифестацией (после 50 лет) лучше поддается контролю и лечению, РМЖ у женщин моложе 50 лет остается серьезной проблемой с тенденцией к неуклонному росту заболеваемости и требует интенсивного изучения молекулярных характеристик опухоли [7–10].
Другой важный прогностический фактор – экспрессия рецепторов эстрогена (ER), прогестерона (PR) и рецептора эпидермального фактора роста типа 2 (HER2/neu) на поверхности опухолевых клеток [7, 11]. Трижды негативный подтип РМЖ (ТНРМЖ), который характеризуется отсутствием экспрессии ER, PR и повышенной экспрессией/амплификацией HER2/neu, это крайне агрессивное и плохо поддающееся лечению заболевание [12, 13]. ТНРМЖ встречается в 10–20% вновь диагностируемых случаев РМЖ и чаще у молодых женщин. К ТНРМЖ относятся, как правило, низкодифференцированные опухоли с высокой пролиферативной активностью, склонностью к отдаленному метастазированию и плохим ответом на терапию [14].
Ответ на противоопухолевую терапию у пациенток с РМЖ сильно различается, поэтому все большее значение для диагностики и лечения РМЖ приобретают биологические маркеры, определяемые непосредственно в опухолевой ткани [11, 15]. Молекулярно-генетический анализ опухолевых клеток широко используется в настоящее время для прогноза и выбора схемы терапии у пациентов с онкологическими заболеваниями [16]. Наиболее полную информацию о молекулярном профиле опухоли безусловно предоставляют полноэкзомное секвенирование и анализ транскриптома, но это дорогостоящие и затратные по времени методы, поэтому на сегодняшний день наибольшее практическое применение находят целевые (таргетные) панели генов, ассоциированных с раком [17, 18]. Поскольку в опухолевой ткани присутствуют и наследуемые, и вновь приобретенные генетические варианты, крайне важно точно дифференцировать соматические и герминальные мутации. “Золотым стандартом” считается парное тестирование образцов ДНК опухоли и клеток зародышевой линии (кровь или нормальная ткань), однако интерес представляет возможность идентификации наследуемых генетических вариантов на основе секвенирования только опухолевой ДНК без анализа ДНК зародышевой линии [19–21]. С одной стороны, это позволяет минимизировать стоимость и длительность генетического тестирования, с другой – дает возможность анализировать архивный материал в тех случаях, когда в наличии имеется только опухолевая ткань.
В настоящей работе проанализированы кодирующие последовательности 25 наиболее изученных генов, ассоциированных с РМЖ. Исследованы архивные образцы опухолей молочной железы в группах с ТНРМЖ и с положительным рецепторным статусом (люминальный А, люминальный В, HER2-позитивный подтип РМЖ). На момент манифестации заболевания подавляющее большинство пациенток были моложе 50 лет. Для определения герминального или соматического статуса мутаций использовали биоинформатические подходы, а также прямое секвенирование ДНК из образцов нормальной ткани, если они были доступны. В результате выявлены редкие наследственные мутации в генах BRCA1/2, а также клинически значимые соматические мутации в генах TP53, PTEN, MSH2, MSH6, MUTYH.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пациенты. В исследование вошли 23 пациентки с диагнозом РМЖ в возрасте от 27 до 68 лет (средний возраст 44 года). Возраст 21 пациентки на момент постановки диагноза не превышал 50 лет, три пациентки с ТНРМЖ были старше 60 лет (63, 66 и 68 лет). Все пациентки постоянно проживали в Смоленске или Смоленской области. Хирургическое удаление первичной опухоли выполнено в период с 2013 по 2016 гг. в Смоленском областном онкологическом диспансере. У 10 пациенток выявлен ТНРМЖ. У 13 пациенток в клетках опухоли обнаружена экспрессия ER и/или PR, и/или HER2/neu – эти опухоли отнесены к люминальному А, люминальному B и HER2/neu-позитивному подтипам РМЖ.
Выделение ДНК. Геномную ДНК выделяли из архивных образцов опухолевой или нормальной ткани молочной железы с использованием набора QIAamp DNA FFPETissueKit (“Qiagen”, Германия). Концентрацию ДНК измеряли с помощью флуориметра Qubit 2 (“Invitrogen”, США) и набора реагентов Qubit® ds DNA HS (“Invitrogen”). Степень фрагментации и чистоту ДНК оценивали с использованием микроспектрофотометра NanoDrop™ 3300 (“ThermoScientific”, США), а также с помощью электрофореза в агарозном геле.
Массовое параллельное секвенирование. Целевые последовательности ДНК отбирали с использованием панели жидких зондов NimbleGenIDP (“Roche”, Швейцария). Библиотека олигонуклеотидных зондов включала последовательности, комплементарные кодирующим участкам следующих генов: APC, ATM, BRCA1, BRCA2, CDH1, CDH3, CDK4, CHEK2, MET, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RET, STK11, TP53, VHL. Подготовку образцов проводили по стандартному протоколу для приготовления быстрых библиотек KAPA LibraryPreparationKit (“Roche”) согласно инструкциям производителя. Образцы ДНК фрагментировали ультразвуком (технология Covaris). Качество библиотек оценивали с помощью системы капиллярного электрофореза (“Agilent”, США). Секвенирование проводили на платформе MiSeq (“Illumina”, США) со средним покрытием ×1000.
Анализ результатов секвенирования и использованные базы данных. Биоинформатический анализ проводили, как описано ранее [22]. В качестве референсного генома использовали версию GRCh38 (Ensembl). Полученные VCF-файлы анализировали с помощью GATK FilterMutectCalls. В дальнейшем анализировали только варианты, прошедшие фильтры. Полученный список вариантов аннотировали с помощью сервера Annovar (http://wannovar.wglab.org). Варианты с популяционной частотой более 1% по базам данных ExAC, 1000 Genomes и dbSNP из анализа исключали. Также исключали мотивы polyN, в частности GGGTG> GGGGG, CCCCG> CCCCC.
К значимым герминальным мутациям-кандидатам относили следующие варианты: 1) мутации в генах BRCA1, BRCA2, CDH1, NF1, PTEN, STK11 и TP53; 2) частота вариантного аллеля более 30% при достаточной глубине покрытия; 3) варианты, зарегистрированные в базе данных ClinVar как патогенные или вероятно патогенные (а); или варианты с потерей функции (nonsense или frameshift мутации) (б); или варианты в генах BRCA1/2, не зарегистрированные в ClinVar или HGMD, но имеющие уровень выше 15 согласно предиктору патогенности CADD (Combined Annotation Dependent Depletion) (https://cadd.gs.washington.edu) (в). Соматические мутации-кандидаты удовлетворяли следующим требованиям: 1) любые несинонимичные замены, включая мутации в сайтах сплайсинга; 2) частота аллельного варианта 10–50% при достаточной глубине покрытия; 3) число прочтений альтернативного аллеля не менее 10; 4) аннотация в базе данных COSMIC c оценкой патогенности по FATHMM (Functional Analysis through Hidden Marcov Models) (http:// fathmm.biocompute.org.uk).
Верификация мутаций методом секвенирования по Сэнгеру. Выявленные мутации определяли в образцах опухолевой и нормальной ткани с помощью прямого секвенирования по Сэнгеру c использованием специфичных праймеров, подобранных для каждой мутации. Результаты анализировали с помощью программы ChromasLite.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В архивных образцах опухолевой ткани 23 пациенток с РМЖ методом таргетного секвенирования геномной ДНК исследованы кодирующие участки генов APC, ATM, BRCA1, BRCA2, CDH1, CDH3, CDK4, CHEK2, MET, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RET, STK11, TP53, VHL. Клинико-генетические характеристики пациенток представлены в табл. 1 и 2.
Таблица 1.
Клинические характеристики и мутационный профиль пациенток с ТНРМЖ
| Возраст, лет | Диагноз | Ген | Замена нуклеотида | Замена АА | Тип мутации | ID | Тип опухоли (по Cosmic database) |
VAF, % | |
|---|---|---|---|---|---|---|---|---|---|
| биоинформатический анализ | анализ по Сэнгеру | ||||||||
| 31 | РМЖ | TP53 | c.503A>C | p.H168P | S | S | COSM1480071 | РМЖ | 26 |
| 46 | То же | TP53 | c.536A>C | p.H179P | S | S | COSM44218 | РМЖ | 65 |
| MUTYH | c.725G>A | p.R242H | S | Н.о. | COSM681164 | РЛ | 12 | ||
| 44 | » | TP53 | c.659A>G | p.Y220C | G (rs121912666) | S | COSM99718 | РМЖ | 57 |
| 34 | » | BRCA1 | c.68_69delAG | p.E23fs | G | G | rs80357914 | – | 77 |
| 63 | » | BRCA1 | c.3226_3227AG | p.G1077fs | G | G | rs80357635 | – | 80 |
| 66 | » | TP53 | c.723del | p.C242Afs*5 | S | S | COSM3718845 | РМЖ | 84 |
| 49 | ДМРМЖ | TP53 | c.733G>A | p.G245S | G (rs28934575) | S | COSM6932 | РТК | 44 |
| PTEN | c.286C>A | p.P96T | S | S | COSM921085 | РЭ | 19 | ||
| 49 | РМЖ | MSH6 | c.2633T>C | p.V878A | S | Н.о. | COSM4985848 | РП | 25 |
| TP53 | c.396G>T | p.K132N | S | S | COSM213186 | РЛ, РМЖ | 53 | ||
| MSH2 | c.965G>A | p.G322D | S | Н.о. | COSM26086 | РП | 47 | ||
| 44 | РМЖ, ММ, Ф | TP53 | c.818G>T | p.R273L | S | S | COSM318169 | РЛ | 16 |
| 68 | РМЖ | – | – | – | – | – | – | – | |
Примечание. ДМРМЖ – двусторонний метахронный РМЖ, ММ – миома матки, Ф – дерматофиброма; S, somatic – соматическая, G, germline – герминальная мутация; VAF, variant allele frequency – частота аллельного варианта в образце опухоли, РМЖ – рак молочной железы, РЛ – рак легкого, РП – рак предстательной железы, РЭ – рак эндометрия.
Таблица 2.
Клинические характеристики и спектр герминальных и соматических мутаций у пациенток с позитивным рецепторным статусом РМЖ*
| Возраст, лет | Рецепторный статус | Ген | Замена нуклеотида | Замена АА | Тип мутации | ID | Тип опухоли (по Cosmic database) |
VAF, % | |
|---|---|---|---|---|---|---|---|---|---|
| биоинформатический анализ | Анализ по Сэнгеру | ||||||||
| 33 | ER+/PR+/HER2– | MSH2 | c.1204C> A | p.Q402K | S | Н.о. | COSM7340167 | М, РЩЖ | 19 |
| 40 | ER+/PR+/HER2+ | BRCA2 | c.658_659del | p.Val220fs | G | G | rs80359604 | 54 | |
| MSH2 | c.965G> A | p.G322D | S | Н.о. | COSM26086 | РПР | 98 | ||
| 45 | ER+/PR+/HER2+ | – | – | – | – | – | – | – | |
| 44 | ER+/PR+/HER2– | TP53 | c.1136G> A | p.R379H | S | S | COSM44189 | ПКР | 18 |
| 27 | ER+/PR+/HER2– | CHEK2 | c.470T> C | p.I157T | G | G | rs17879961 | 42 | |
| PTEN | c.640C> T | p.Q214* | G (rs121909227) | S | COSM5150 | РЭ | 47 | ||
| 44 | ER+/PR+/HER2– | – | – | – | – | – | – | – | |
| 39 | ER+/PR+/HER2– | PTEN | c.511C> T | p.Q171* | S | Н.о. | COSM5149 | РМЖ | 15 |
| 44 | ER–/PR+/HER2– | TP53 | c.637C> T | p.R213* | S | S | COSM99618 | РТК, РМЖ | 14 |
| 27 | ER–/PR–/HER2+ | – | – | – | – | – | – | ||
| 42 | ER+/PR+/HER2– | – | – | – | – | – | – | ||
| 46 | ER+/PR+/HER2+ | TP53 | c.751A> C | p.I251L | S | S | COSM10931 | РЖ | 25 |
| TP53 | c.731G> A | p.G244D | S | S | COSM1646854 | РТК | 25 | ||
| 41 | ER+/PR+/HER2– | – | – | – | – | – | – | – | |
| 45 | ER–/PR–/HER2+ | – | – | – | – | – | – | – | |
* Все пациентки имели диагноз РМЖ с односторонним поражением молочных желез. Примечание. S, somatic – соматическая, G, germline – герминальная мутация; VAF, variant allele frequency – частота аллельного варианта в образце опухоли, М –меланома, РЩЖ – рак щитовидной железы, РП – рак предстательной железы, ПКР – рак почки, РЭ – рак эндометрия, РТК – рак толстой кишки, РЖ – рак желудка.
Первичная оценка герминальных и соматических вариантов в образцах опухоли проведена с использованием алгоритмов, предложенных ранее [20], а также согласно рекомендациям NCCN (National Comprehensive Cancer Network) [23].
В группе пациенток с ТНРМЖ выявлены две герминальные мутации гена BRCA1. Мутация c.68_69del (185delAG) относится к известным в России мутациям с эффектом основателя (founder mutation) [24]. Напротив, мутация BRCA1 c.3226_3227AG (3347delAG) описана как мутация с эффектом основателя в популяциях Италии [25] и Норвегии [26]. Герминальная мутация в гене BRCA2 c.658_659del (886delGT) обнаружена у пациентки с гормоноположительным, HER2-положительным РМЖ. Ранее эта мутация была описана как наиболее частая мутация с эффектом основателя в популяции Литвы [27] и Польши [28]. Мутации в гене BRCA1 представлены в опухолях РМЖ с частотой 77 и 80%, что указывает на утрату аллеля дикого типа. Секвенирование по Сэнгеру образцов опухолевой и нормальной ткани подтвердило этот факт (рис. 1).
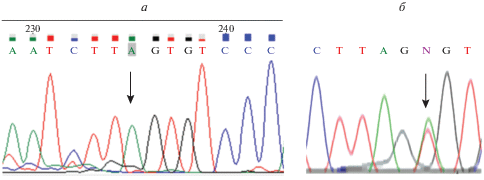
Рис. 1.
Результаты секвенирования ДНК из образцов опухолевой (а) и нормальной (б) тканей пациентки с герминальной мутацией BRCA1 c.66_67delAG (p.Glu23fs). Мутация присутствует в гомозиготном состоянии в опухоли и в гетерозиготном – в нормальной ткани (указаны стрелкой).
Наибольшее количество мутаций в обеих группах выявлено в гене TP53. Большинство мутаций отнесено к соматическим, однако у двух пациенток с ТНРМЖ обнаружены мутации, которые предположительно относятся к герминальным вариантам. Мутации p.Y220C (rs121912666) и p.G245S (rs28934575) зарегистрированы в базе данных ClinVar как патогенные генетические варианты, ассоциированные с синдромом Ли–Фраумени. Частота вариантного аллеля составила 57 и 44% соответственно. Так как был доступен архивный материал нормальной ткани молочной железы, мы провели сравнительный анализ ДНК, выделенной из парных образцов (опухоль–норма), методом секвенирования по Сэнгеру. В обоих случаях мутации обнаружены только в опухолевой ткани, но не в нормальных клетках, что позволило однозначно отнести их к соматическим (рис. 2).
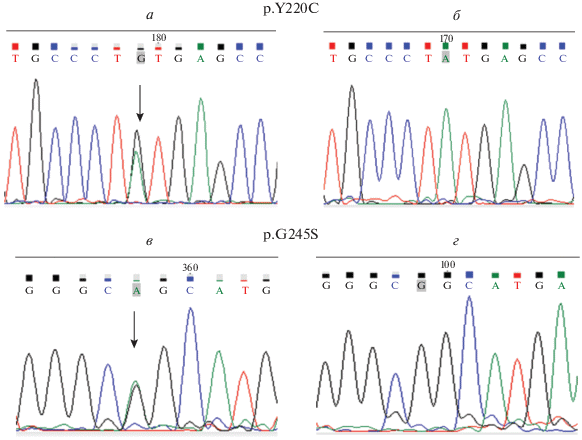
Рис. 2.
Результаты секвенирования ДНК из образцов опухолевой и нормальной ткани пациенток с мутациями в гене TP53. Мутации p.Y220C и p.G245S выявлены только в опухоли (а, в) и отсутствуют в нормальной ткани (б, г).
Около 55% соматических мутаций, выявленных в нашем исследовании в гене TP53, описаны при РМЖ ранее, в то время как 45% мутаций TP53, а также мутации в генах MSH2, MSH6, PTEN, MUTYH обнаружены и при других типах опухолей. Частота вариантного аллеля варьировала от 12 до 98%. Различные соматические мутации в гене TP53 представлены на рис. 3.
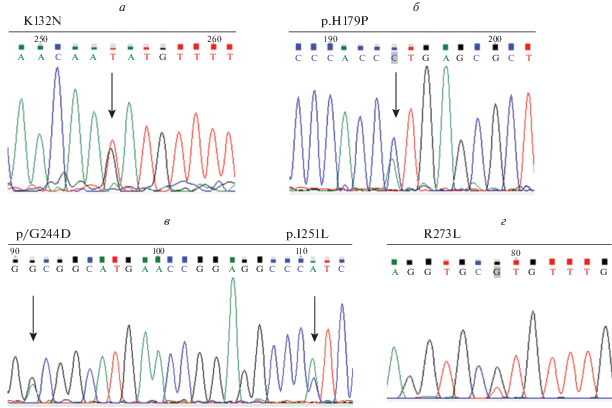
Рис. 3.
Верификация соматических мутаций гена TP53, обнаруженных в архивных образцах опухолевой ткани пациенток с РМЖ (а–г). В одном образце одновременно выявлены две мутации гена TP53 (в).
Сравнительный анализ спектра герминальных и соматических мутаций у пациенток с ТНРМЖ и пациенток с позитивным рецепторным статусом представлен в табл. 3. Мутации в генах BRCA1/2 обнаружены у 20% пациенток с ТНРМЖ и у 8% – в объединенной подгруппе с позитивным рецепторным статусом (p = 0.56). Соматические мутации в гене TP53 при ТНРМЖ выявлены у 70% пациенток по сравнению с 23% при других подтипах РМЖ, различия статистически значимы (p = 0.04).
Таблица 3.
Сравнительный анализ спектра клинически значимых герминальных и соматических мутаций в группах с ТНРМЖ и с позитивным статусом хотя бы одного из рецепторов (Р + РМЖ)
| Ген | ТНРМЖ (n = 10) | Р + РМЖ (n = 13) | p | ||
|---|---|---|---|---|---|
| пациентки, n | % | пациентки, n | % | ||
| Герминальные мутации | |||||
| BRCA1 | 2 | 20 | – | – | 0.56 |
| BRCA2 | – | – | 1 | 8 | |
| CHEK2 | – | – | 1 | 8 | 1.00 |
| Соматические мутации | |||||
| TP53 | 7 | 70 | 3 | 23 | 0.04 |
| PTEN | 1 | 10 | 2 | 15 | – |
| MSH2 | 1 | 10 | 2 | 15 | – |
Эти данные согласуются с результатами, полученными другими авторами. Герминальные мутации BRCA1/2 выявляли в 10–20% случаев ТНРМЖ, и более 30% приходилось на пациенток старше 60 лет [29, 30]. Нами также выявлена мутация BRCA1 у пациентки 63 лет. Важным следствием этого наблюдения стал вывод о необходимости тестирования мутаций BRCA1/2 при ТНРМЖ независимо от возраста манифестации заболевания [30], тогда как согласно рекомендациям NCCN, проведение генетического анализа наиболее актуально для пациенток моложе 45–50 лет. В ряде случаев – при онкологическом отягощении семейного или персонального анамнеза, генетический анализ показан и для пациенток с ТНРМЖ моложе 60 лет [31]. Определение мутаций в генах BRCA1/2 имеет большое значение для выбора схемы терапии, так как опухоли с дефектом системы репарации двухцепочечных разрывов ДНК чувствительны к действию ингибиторов поли-(АDP-рибоза)-полимеразы (ингибиторы PARP) [32]. Повышенная частота мутаций BRCA1/2 при ТНРМЖ делает их важной молекулярной мишенью при этом агрессивном виде рака [33]. Потеря гетерозиготности наблюдается примерно в 10% случаев РМЖ с герминальными мутациями гена BRCA1 и ассоциирована с повышенной чувствительностью к ДНК-повреждающим агентам, в том числе к соединениям платины (цисплатин, карбоплатин) [34]. В нашем исследовании у обеих пациенток с мутациями BRCA1 отмечена потеря гетерозиготности в опухоли.
Ген TP53 относится к генам, наиболее часто мутирующим при РМЖ. Мутации TP53 ассоциированы с плохим прогнозом [35]. Суммарная частота соматических мутаций в гене TP53 при РМЖ составляет около 30%, однако частота и спектр мутаций, а также их клиническое значение различаются между подтипами РМЖ [36]. Наиболее высокая частота соматических мутаций наблюдается при ТНРМЖ (до 70–80%). В нашей работе частота соматических мутаций при ТНРМЖ, представленных в большинстве случаев миссенс-мутациями и в одном – делецией нуклеотида со сдвигом рамки считывания, составила 70%. Показано, что мутации TP53 повышают риск возникновения метастазов [37], однако это более характерно для подтипов РМЖ с позитивным рецепторным статусом [15]. Таким образом, вопрос о прогностической роли соматических мутаций TP53, особенно в случае ТНРМЖ, требует дальнейшего изучения. Следует отметить принципиальную важность определения терминального или соматического статуса мутаций TP53, так как герминальные мутации ассоциированы с наследственными онкопатологиями – синдромом Ли–Фраумени и Ли–Фраумени-подобным синдромом, что требует специфичных подходов к ведению пациентов [38].
Таким образом, молекулярно-генетический анализ архивных образцов опухолевой и нормальной ткани 23 пациенток с РМЖ позволил выявить как герминальные, так и соматические мутации, которые служат важными прогностическими маркерами, а кодируемые мутантными генами белки могут быть потенциальными молекулярными мишенями для терапии различных подтипов РМЖ, в первую очередь ТНРМЖ.
Работа выполнена при поддержке Федеральной целевой программы “Исследования и разработки по приоритетным направлениям развития научно-технологического комплекса России на 2014–2020 годы” (соглашение № 05.604.21.0234, уникальный идентификатор проекта RFMEFI60419X0234).
Все процедуры, выполненные в данной работе, соответствуют этическим стандартам институционального комитета по исследовательской этике и Хельсинкской декларации 1964 года и ее последующим изменениям или сопоставимым нормам этики. От пациентов получено письменное добровольное информированное согласие на использование результатов исследования в обезличенной форме в научных целях.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Каприн А.Д., Старинский В.В., Петрова Г.В. (2019) Злокачественные новообразования в России в 2018 году (заболеваемость и смертность). Москва: МНИОИ им. П.А. Герцена, 250 с.
Любченко Л.Н., Батенева Е.И. (2014) Медико-генетическое консультирование и ДНК-диагностика при наследственной предрасположенности к раку молочной железы и раку яичников. Москва: ИГ РОНЦ, 75 с.
Ahmad A. (2019) Breast cancer statistics: recent trends. Adv. Exp. Med. Biol. 1152, 1–7.
Li N., Deng Y., Zhou L., Tian T., Yang S., Wu Y., Zheng Y., Zhai Z., Hao Q., Song D., Zhang D., Kang H., Dai Z. (2019) Global burden of breast cancer and attributable risk factors in 195 countries and territories, from 1990 to 2017: results from the Global Burden of Disease Study 2017. J. Hematol. Oncol. 12(1), 140.
Hunter C.P. (2000) Epidemiology, stage at diagnosis, and tumor biology of breast carcinoma in multiracial and multiethnic populations. Cancer. 88(S5), 1193–1202.
Stevanovic L., Choschzick M., Moskovszky L., Varga Z. (2019) Variability of predictive markers (hormone receptors, Her2, Ki67) and intrinsic subtypes of breast cancer in four consecutive years 2015–2018. J. Cancer Res. Clin. Oncol. 145(12), 2983–2994.
González-Reymúndez A., de los Campos G., Gutiérrez L., Lunt S. Y., Vazquez A. I. (2017) Prediction of years of life after diagnosis of breast cancer using omics and omic-by-treatment interactions. Eur. J. Hum. Genet. 25(5), 538–544.
Yankaskas B.C. (2006) Epidemiology of breast cancer in young women. Breast Disease. 23(1), 3–8.
Anders C.K., Johnson R., Litton J., Phillips M., Bleyer A. (2009) Breast cancer before age 40 years. Seminars Oncol. 36(3), 237–249.
Narod S.A. (2012) Breast cancer in young women. Nat. Rev. Clin. Oncol. 9(8), 460.
Perou C.M., Sørlie T., Eisen M.B., Van De Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A., Fluge O., Pergamenschikov A., Williams C., Zhu S.X., Lønning P.E., Børresen-Dale A.L., Brown P.O., Botstein D. (2000) Molecular portraits of human breast tumours. Nature. 406(6797), 747–752.
Foulkes W.D., Smith I.E., Reis-Filho J.S. (2010) Triple-negative breast cancer. N. Eng. J. Med. 363(20), 1938–1948.
Ahn S.G., Kim S.J., Kim C., Jeong J. (2016) Molecular classification of triple-negative breast cancer. J. Breast Cancer. 19(3), 223–230.
Dent R., Trudeau M., Pritchard K.I., Hanna W.M., Kahn H.K., Sawka C.A., Lickley L.A., Rawlinson E., Sun P., Narod S.A. (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin. Cancer Res. 13(15), 4429–4434.
Cancer Genome Atlas Network. (2012) Comprehensive molecular portraits of human breast tumours. Nature. 490(7418), 61.
Damodaran S., Sember Q.C., Arun B.K. (2020) Clinical implications of breast cancer tumor genomic testing. Breast J. 26(8), 1565–1571.
Easton D.F., Pharoah P., Antoniou A.C., Tischkowitz M., Tavtigian S.V., Nathanson K.L., Devilee P., Meindl A., Couch F.J., Southey M., Goldgar D.E., Evans D., Chenevix-Trench G., Rahman N., Robson M., Domchek S.M., Foulkes W.D. (2015) Gene-panel sequencing and the prediction of breast-cancer risk. N. Eng. J. Med. 372(23), 2243–2257.
Xuan J., Yu Y., Qing T., Guo L., Shi L. (2013) Next-generation sequencing in the clinic: promises and challenges. Cancer Lett. 340(2), 284–295.
Raymond V.M., Gray S.W., Roychowdhury S., Joffe S., Chinnaiyan A.M., Parsons D.W., Plon S.E. (2016) Germline findings in tumor-only sequencing: points to consider for clinicians and laboratories. J. Natl. Cancer Inst. 108(4), djv351. https://doi.org/10.1093/jnci/djv351
Kamio T., Kamio H., Aoki T., Ondo Y., Uchiyama T., Yamamoto-Shimojima K., Watanabe M., Okamoto T., Kanno H., Yamamoto T. (2020) Molecular profiles of breast cancer in a single institution. Anticancer Res. 40(8), 4567–4570.
Meric-Bernstam F., Brusco L., Daniels M., Wathoo C., Bailey A.M., Strong L., Shaw K., Lu K., Qi Y., Zhao H., Lara-Guerra H., Litton J., Arun B., Eterovic A.K., Aytac U., Routbort M., Subbiah V., Janku F., Davies M.A., Kopetz S., Mendelsohn J., Mills G.B., Chen K. (2016) Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann. Oncol. 27(5), 795–800.
Абрамов И.С., Емельянова М.А., Рябая О.О., Краснов Г.С., Заседателев А.С., Наседкина Т.В. (2019) Соматические мутации, вносящие вклад в метастазирование акральной меланомы. Молекуляр. биология. 53(4), 648–653.
Daly M.B., Pilarski R., Berry M., Buys S.S., Farmer M., Friedman S., Garber J.E., Kauff N.D., Khan S., Klein C., Kohlmann W., Kurian A., Litton J.K., Madlensky L., Merajver S.D., Offit K., Pal T., Reiser G., Shannon K.M., Swisher E., Vinayak S., Voian N.C., Weitzel J.N., Wick M.J., Wiesner G.L., Dwyer M., Darlow S. (2017) NCCN guidelines insights: genetic/familial high-risk assessment: breast and ovarian, version 2.2017. J. Natl. Comprehensive Cancer Network. 15(1), 9–20.
Наседкина Т.В., Громыко О.Е., Емельянова М.А., Игнатова Е.О., Казубская Т.П., Портной С.М., Заседателев А.С., Любченко Л.Н. (2014) Определение герминальных мутаций в генах BRCA1, BRCA2 и CHEK2 с использованием биочипов у больных раком молочной железы. Молекуляр. биология. 48 (2), 243–250.
Papi L., Putignano A.L., Congregati C., Zanna I., Sera F., Morrone D., Falchetti M, Turco M.R., Ottini L., Palli D., Genuardi M. (2009) Founder mutations account for the majority of BRCA1-attributable hereditary breast/ovarian cancer cases in a population from Tuscany, Central Italy. Breast Cancer Kes. Treat. 117(3), 497–504.
Heimdal K., Maehle L., Apold J., Pedersen J.C., Møller P. (2003) The Norwegian founder mutations in BRCA1: high penetrance confirmed in an incident cancer series and differences observed in the risk of ovarian cancer. Eur. J. Cancer. 39(15), 2205–2213.
Janavičius R., Rudaitis V., Mickys U., Elsakov P., Griškevičius L. (2014) Comprehensive BRCA1 and BRCA2 mutational profile in Lithuania. Cancer Genet. 207(5), 195–205.
Jakubowska A., Scott R., Menkiszak J., Gronwald J., Byrski T., Huzarski T., Górski B., Cybulski C., Debniak T., Kowalska E., Starzyńska T., Ławniczak M., Narod S., Lubinski J. (2003) A high frequency of BRCA2 gene mutations in Polish families with ovarian and stomach cancer. Eur. J. Hum. Genet, 11(12), 955–958.
Gonzalez-Angulo A.M., Timms K.M., Liu S., Chen H., Litton J.K., Potter J., Lanchbury J.S., Stemke-Hale K., Hennessy B.T., Arun B.K., Hortobagyi G.N., Do K.-A., Mills G.B., Meric-Bernstam F. (2011) Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 17(5), 1082–1089.
Wong-Brown M.W., Meldrum C.J., Carpenter J.E., Clarke C.L., Narod S.A., Jakubowska A., Rudnicka H., Lubinski J., Scott R.J. (2015) Prevalence of BRCA1 and BRCA2 germline mutations in patients with triple-negative breast cancer. Breast Cancer Res. Treat. 150(1), 71–80.
National Comprehensive Cancer Network. (2010) NCCN Clinical Practice Guidelines in OncologyTM: genetic/familial high risk assessment: breast and ovarian VI 2010.
Fong P.C., Boss D.S., Yap T.A., Tutt A., Wu P., Mergui-Roelvink M., Mortimer P., Swaisland H., Lau A., O’Connor M.J., Ashworth A., Carmichael J., Kaye S.B., Schellens J.H.M., de Bono J.S. (2009). Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Eng. J. Med. 361(2), 123–134.
Geenen J.J., Linn S.C., Beijnen J.H., Schellens J.H. (2018) PARP inhibitors in the treatment of triple-negative breast cancer. Clin. Pharmacokinet. 57(4), 427–437.
Maxwell K.N., Wubbenhorst B., Wenz B.M., De Sloover D., Pluta J., Emery L., Barrett A., Kraya A.A., Anastopoulos I.N., Yu S., Jiang Y., Chen H., Zhang N.R., Hackman N., D’Andrea K., Daber R., Morrissette J.J.D., Mitra N., Feldman M., Domchek S.M., Nathanson K.L. (2017). BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 8(1), 1–11.
Olivier M., Langerød A., Carrieri P., Bergh J., Klaar S., Eyfjord J., Charles Theillet, Rodriguez C., Lidereau R., Bièche I., Varley J., Bignon Y., Uhrhammer N., Winqvist R., Jukkola-Vuorinen A., Niederacher D., Kato S., Ishioka C., Hainaut P., Børresen-Dale A.-L. (2006). The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 12(4), 1157–1167.
Silwal-Pandit L., Vollan H.K., Chin S.F., Rueda O.M., McKinney S., Osako T., Quigley D.A., Kristensen V.N., Aparicio S., Børresen-Dale A.L., Caldas C., Langerød A. (2014) TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin. Cancer Res. 20(13), 3569–3580.
Meric-Bernstam F., Zheng X., Shariati M., Damodaran S., Wathoo C., Brusco L., Demirhan M.E., Tapia C., Eterovic A.K., Basho R.K., Ueno N.T., Janku F., Sahin A, Rodon J., Broaddus R., Kim T.B., Mendelsohn J., Mills Shaw K.R., Tripathy D., Mills G.B., Chen K. (2018) Survival outcomes by TP53 mutation status in metastatic breast cancer. JCO Precision Oncol. 2, 1–15.
Nandikolla A.G., Venugopal S., Anampa J. (2017) Breast cancer in patients with Li–Fraumeni syndrome – a case-series study and review of literature. Breast Cancer (Dove Med. Press). 9, 207–215.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология