

Молекулярная биология, 2021, T. 55, № 4, стр. 531-542
SCP-фосфатазы и онкогенез
Г. А. Пузанов a, В. С. Сенченко a, *
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: senvera@yandex.ru
Поступила в редакцию 02.12.2020
После доработки 29.12.2020
Принята к публикации 30.12.2020
Аннотация
Малые SCP-фосфатазы CTDSP1, CTDSP2 и CTDSPL осуществляют специфическое дефосфорилирование остатков серина и треонина в молекулах белков. Эти ферменты участвуют в регуляции активности РНК-полимеразы II на стадии перехода от инициации транскрипции к элонгации, в регуляции экспрессии нейрон-специфичных генов и активации ключевого белка клеточного цикла pRb на границе фаз G1/S. Кроме того, субстратами SCP-фосфатаз могут быть модуляторы транскрипции SMAD, протеинкиназа AKT1 – регулятор клеточного цикла, апоптоза и ангиогенеза, факторы транскрипции TWIST1 и c-MYC, белки семейства Rаs, участвующие в сигнальных путях, регулирующих рост клеток и апоптоз, CDCA3, связанный с клеточным делением, ингибитор циклинзависимых киназ p21, белок промиелоцитарного лейкоза PML, участвующий в регуляции онкосупрессоров p53, PTEN, mTOR. Нарушение функций или инактивация SCP-фосфатаз связаны с развитием различных заболеваний, включая онкологические. Наблюдаемый в последнее время рост интереса к SCP-фосфатазам обусловлен их онкосупрессорными свойствами, а также участием в развитии злокачественных опухолей различной этиологии и локализации. В обзоре рассмотрены свойства SCP-фосфатаз и их роль в онкогенезе. Понимание функций SCP-фосфатаз и механизмов их регуляции может быть полезным при поиске эффективных мишеней для терапии опухолей.
ФУНКЦИИ SCP-ФОСФАТАЗ CTDSP1, CTDSP2, CTDSPL В КЛЕТКАХ ЧЕЛОВЕКА
Обратимое фосфорилирование белков, осуществляемое киназами и фосфатазами, представляет собой важный механизм передачи сигналов и регуляции их активности у всех живых организмов. В эукариотических клетках фосфорилированию подвергаются главным образом гидроксилсодержащие аминокислотные остатки – серин, треонин и тирозин, основным из которых является серин [1]. Выделяют три основных семейства серин/треониновых фосфатаз: фосфопротеиновые фосфатазы (PPP), металлзависимые фосфатазы (PPM) и фосфатазы на основе аспартата (семейство FCP/SCP) [2, 3]. В последнее семейство входят шесть подсемейств. Одно из них – подсемейство SCP малых сериновых фосфатаз – состоит из трех высокогомологичных фосфатаз CTDSP1, CTDSP2 и CTDSPL/CTDSP3 (они же SCP1, SCP2 и SCP3), содержащих каталитический и регуляторный домены в одной полипептидной цепи [4].
Характеристика SCP-фосфатаз
Mg2+-зависимые SCP-фосфатазы CTDSP1, CTDSP2, CTDSPL (CTDSP1/2/L), локализованные в клеточном ядре, могут участвовать в регуляции транскрипции генов, а также взаимодействовать с регуляторными белками. Значительное сходство (около 83%) аминокислотных последовательностей этих ферментов (UniProt: Q9GZU7 (CTDSP1), O14595 (CTDSP2), O15194 (CTDSPL)) [5] и трехмерных структур указывает на их функциональное сходство (рис. 1). Несмотря на высокую гомологию мотива DXDX(T/V), отвечающего за фосфатазную активность CTDSP1/2/L, эти ферменты имеют различия в N-концевых последовательностях, которые, как предполагают, принимают участие в белок-белковых взаимодействиях. Гены CTDSP1/2/L расположены на разных хромосомах: 2q35, 12q13–q15 и 3p22–p21.3 соответственно, однако профили их тканеспецифичной экспрессии различаются незначительно [6].
Рис. 1.
Множественное выравнивание аминокислотных последовательностей областей активного центра белков CTDSP1, CTDSP2 и CTDSPL (выполнено с помощью приложения T-COFFEE, версия 11) [5]. Цвет (темно-серый и светло-серый) отражает различную степень совпадения аминокислотных остатков.

Отрицательные заряды, присутствующие как на нуклеофильной, так и на основной аминокислоте (аспартат) фосфатаз CTDSP1/2/L, нейтрализуются ионами магния, необходимыми для связывания субстрата и ферментативной активности. После нуклеофильной атаки образуется промежуточное соединение аспартилфосфата, которое взаимодействует с молекулой воды с образованием дефосфорилированного серина и неорганического фосфата. Формирование фосфоаспартильного интермедиата – ключевая особенность, которая отличает ферменты семейства FCP/SCP от других металлзависимых фосфатаз [7]. Считается, что Mg2+ облегчает протекание обеих стадий реакции, нейтрализуя отрицательный заряд фосфатной группы. Определены пространственные структуры SCP-фосфатаз, а также представлены схемы механизма действия этих ферментов [2, 7–10].
Фосфорилирование C-концевого домена РНК-полимеразы II
Активность фосфатаз семейства FCP/SCP, действующих на C-концевой домен (CTD) РНК-полимеразы II, впервые обнаружена во фракции клеток HeLa [11]. Фосфатазы этого экстракта способны переводить гиперфосфорилированную РНК-полимеразу II в гипофосфорилированное состояние. Способность модифицированной таким образом РНК-полимеразы II связывать промотор аденовируса типа 2 (Ad2-MLP) стала одним из первых доказательств того, что фосфорилированная форма СTD играет важную роль в узнавании промотора [12]. Повторяющаяся область CTD самой большой субъединицы РНК-полимеразы II играет ключевую роль в регуляции экспрессии генов и служит структурным элементом, объединяющим такие различные процессы, как транскрипция, процессинг мРНК и др. [13]. Активность CTD зависит от статуса его фосфорилирования, за который ответственны CTD-фосфатазы и киназы [14]. SCP-фосфатазы СTDSP1/2/L могут катализировать дефосфорилирование Ser5 в консенсусном повторе Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7 CTD большой субъединицы РНК-полимеразы II, участвуя, таким образом, в негативной регуляции транскрипции, а также контролировать статус фосфорилирования иных cубстратов, помимо CTD-домена [15].
Киназы и фосфатазы определяют статус посттрансляционной модификации СTD РНК-полимеразы II, значимый для регуляторов транскрипции. Совокупное действие этих факторов обеспечивает нужную последовательность транскрипционных процессов [16]. Большинство CTD-киназ принадлежат к семейству циклинзависимых киназ (CDK), что подразумевает связь между регуляцией клеточного цикла и транскрипцией [17]. SCP-фосфатазы предпочтительно дефосфорилируют Ser5 в гептапептидных повторах СTD РНК-полимеразы II. На стадии элонгации транскрипции активность РНК-полимеразы II негативно регулирует не только фосфатаза FCP1, активный центр которой сходен с активными центрами других членов семейства SCP, но и другие фосфатазы. Консервативная эукариотическая фосфатаза FCP1 предпочитает в качестве субстрата Ser2 СTD РНК-полимеразы II [18–20]. SCP-фосфатазы представлены в клетках высших эукариот, они предпочтительно дефосфорилируют Ser5, а не Ser2 [15], обладают гомологией с FCP1 по аминокислотной последовательности фосфатазного домена (~20%) и сходной третичной структурой [4, 21]. Статус фосфорилирования двух наиболее хорошо изученных остатков CTD РНК-полимеразы II (Ser5 и Ser2) коррелирует со стадиями транскрипции. В зависимости от состояния фосфорилирования СTD может различать мишени, с которыми связывается, и инициировать транскрипцию [22].
Дефосфорилированный СTD связывает белки преинициаторного комплекса. Фосфорилированный остаток Ser5 СTD РНК-полимеразы II способен инициировать транскрипцию, но не может войти в фазу элонгации и транскрибировать всю мРНК. Кроме того, СTD РНК-полимеразы II, фосфорилированный по Ser5 и Ser2, распознается факторами сплайсинга и необходим для сборки сплайсосомы и эффективного протекания реакций сплайсинга в процессе элонгации [[23].
Регуляция транскрипции нейронных генов
Эволюционно консервативные регуляторы экспрессии нейронспецифичных генов – SCP-фосфатазы CTDSP1/2/L, входят в состав комплекса REST (RE-1-Silencing Transcription factor), который специфично связывается с участком ДНК репрессорного элемента RE-1 (Repressor Element 1) длиной 23 п.н., обнаруженный более чем в 1000 генов нейронов [6]. SCP-фосфатазы участвуют в подавлении экспрессии нейронных генов посредством регуляции активности комплекса REST. Профили экспрессии генов CTDSP1/2/L и REST сходны во всех клетках, кроме нейронов [24]. Согласно последним данным, REST действительно служит субстратом CTDSP1 in vivo. Коме того, CTDSP1 защищает REST от деградации [25]. Активность нейронных генов человека зависит от активности микроРНК (miR-124), подавляющей экспрессию гена CTDSP1 [26]. Низкомолекулярные ингибиторы, нацеленные на CTDSP1/2/L, могут быть полезны для изучения функции REST в различных типах клеток, а также патогенеза таких заболеваний, как глиобластома [25, 27], при которой наблюдается сверхэкспрессия REST.
Участие в регуляции клеточного цикла
Белок ретинобластомы pRb – ключевой участник сигнального пути p16ink4A–Cdk/циклин–Rb, ответственный за остановку клеточного деления на границе фаз G1/S клеточного цикла [28]. Активность рRb регулируется посттрансляционными модификациями, среди которых преобладает фосфорилирование, осуществляемое комплексами CDK4–циклин D и CDK2–циклин E, что способствует прогрессии S-фазы клеточного цикла [29, 30]. На ранней стадии фазы G1 белок pRb подвергается монофосфорилированию комплексами циклина D с CDK4/6, в то время как для выхода из клеточного цикла необходима дефосфорилированная форма pRb [31]. Известно, что фосфорилирование в нескольких сайтах экзона 23 RB, включая Ser780, Ser807/811 и Ser795, приводит к ингибированию его связывания с факторами транскрипции E2F [32, 33].
CTDSP1 взаимодействует с белком СDCA3, связанным с делением клеток и необходимым для вступления клеток в митоз. СDCA3 входит в состав комплекса убиквитин-лигазы E3, который опосредует убиквитинирование и деградацию киназы WEE1, ключевого регулятора фазы G2/M, подавляющего активность циклинзависимых киназ CDK1 и CDK2 [34, 35]. Колориметрическим методом с использованием малахитового зеленого показано, что вероятным субстратом CTDSP1 является СDCA3 [36, 37]. Повышение экспрессии CTDSP2 приводит к снижению количества клеток в S-фазе [38], а также к активации генов семейства RAS, которые, в свою очередь, регулируют экспрессию P21 [38]. Гены RAS кодируют белки сигнальных путей, регулирующих рост клеток и апоптоз [39, 40]. Белок P21, ингибитор циклинзависимых киназ, подавляет активацию комплекса циклин E/CDK2, необходимого для фосфорилирования pRb [41]. CTDSP1/2/L могут дефосфорилировать белок pRb in vitro по остаткам Ser807/811, Ser780 и Ser795 [5, 42].
Другие субстраты SCP-фосфатаз
CTDSP1/2/L регулируют не только РНК-полимеразу II, экспрессию нейронспецифичных генов с участием комплекса REST и клеточный цикл (pRb, CDCA3, и др.), они взаимодействуют и с другими субстратами. Эти фосфатазы дефосфорилируют белок SMAD1, отрицательно регулируя тем самым сигнальный путь морфогенетических белков костной ткани (BMP) [43]. SMAD1 является сигнальным белком пути TGF-β/BMP, который активирует рецепторы серин-треониновых киназ и играет важную роль в остеобластогенезе и формировании кости [44]. CTDSP1/2/L способны дефосфорилировать также N-концевые и линкерные области белков SMAD2 и SMAD3 (кроме пары CTDSP1–SMAD3), усиливая сигнальный путь TGF-β, который контролирует многие клеточные функции, включая пролиферацию и дифференцировку. Белки SMAD преобразуют сигналы и действуют как модуляторы транскрипции, вовлеченные в разнообразные сигнальные пути [45].
CTDSP1/2/L могут дефосфорилировать и стабилизировать белок SNAIL, ключевой репрессор транскрипции гена E-кадгерина, важного регулятора нескольких сигнальных путей, связанных с эпителиально-мезенхимальным переходом, клеточной адгезией и миграцией клеток [45]. Таким образом, стабилизация SNAIL, усиливая супрессию E-кадгерина, способствует миграции клеток in vitro. Известна также способность фосфатазы CTDSP1 дефосфорилировать белок TWIST1, участвующий в подавлении экспрессии E-кадгерина. При этом фосфорилирование N-концевого Ser68 способствует деградации белка TWIST1 [46].
Недавно обнаружено, что CTDSP1 локализованная преимущественно на плазматической мембране различных клеток, негативно регулирует активность белков PKB/AKT, входящих в семейство протеинкиназы В, с последующим нарушением ангиогенеза [47]. Подобные свойства CTDSP1 вызывают большой интерес, поскольку в плазматических мембранах локализованы только несколько серин/треониновых фосфатаз (из приблизительно 30 известных серин/треониновых и 107 тирозиновых протеинфосфатаз) [2, 48]. В эндотелиальных клетках сигнальные белки AKT стимулируют ангиогенез. Они перемещаются к клеточной мембране, где активируются и запускают дальнейшие сигнальные события. Активация белков AKT происходит через фосфатную группу, присоединенную к конкретному сайту [49].
Дополнительные взаимодействия белков с SCP-фосфатазами обнаружены нами с помощью приложения STRING на основе геномного контекста (соседство, слияние, совпадение генов), высокопроизводительного секвенирования (база данных NCBI Gene Expression Omnibus), коэкспрессии, интеллектуального анализа текста и известных баз данных, таких как KEGG, Reactome, BioCyc, Gene Ontology и BioCarta [50]. В группу экспериментально подтвержденных взаимодействий с SCP-фосфатазами, помимо уже описанных (SMAD1–3, REST, CDCA3 и др.), вошли еще пять белков (рис. 2). В том числе основной белок миелина (MBP), классические изоформы 4–14 которого участвуют в миелинизации и поддержании структуры миелиновой оболочки в центральной нервной системе [51]. UBLCP1, который дефосфорилирует ядерные 26S протеасомы, снижая тем самым их протеолитическую активность и предотвращая сборку “коровых” и регуляторных частиц в зрелую 26S протеасому. GTF2F1 – субъединица 1 общего фактора транскрипции IIF (TFIIF), связывается с РНК-полимеразой II и помогает рекрутировать ее в комплекс инициации с фактором транскрипции IIB (TFIIB), что способствует элонгации транскрипции [15]. Белок CTBP2 действует как корепрессор транскрипции [52]. Фактор роста GDF5 участвует в формировании костной и хрящевой ткани [53]. Взаимодействие CTDSP1 и CTDSPL выявлено в клетках HEK293T почки человека на основе анализа интерактома BioPlex, системы мультиплексного анализа, включающего данные высокопроизводительной масс-спектрометрии с аффинной очисткой [54].
Рис. 2.
Схемы белок-белковых взаимодействий CTDSP1 (а), CTDSP2 (б) и CTDSPL (в) на основе базы данных STRING (Приложение для поиска взаимодействий генов и белков). Линиями показаны связи (прямые или опосредованные). Толщина линий соответствует степени доказанности взаимодействия: жирные линии – экспериментально показанные прямые взаимодействия белков, тонкие – предсказанные ассоциации.
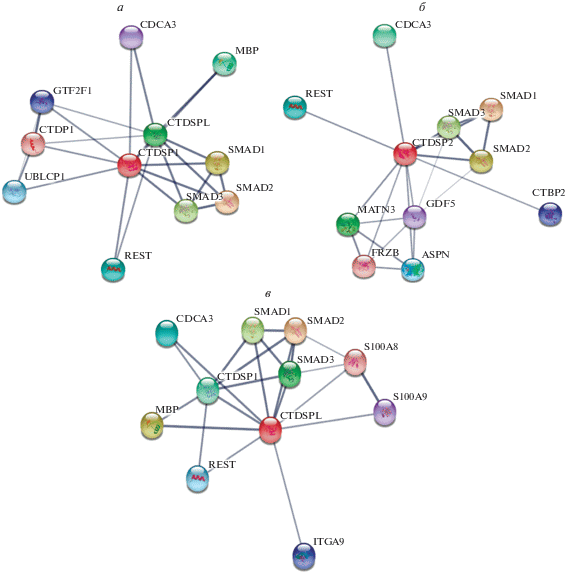
В группу предсказанных значимых функциональных ассоциаций с SCP-фосфатазами вошли семь белков: CTDP1, принадлежащий к семейству фосфатаз FCP/SCP [55]; MATN3 – матрилин-3, главный компонент внеклеточного матрикса хряща [56]; ASPN – аспорин, играющий важную роль в гомеостазе хряща и патогенезе остеоартрита [57]; FRZB, связанный с регуляцией роста и дифференцировки клеток [58]; ITGA9 – интегрин альфа-9, рецептор молекулы 1 адгезии сосудистых клеток (VCAM), цитотактин и остеопонтин [59]; S100A9 и S100A8 – кальций- и цинксвязывающие белки, играющие важную роль в регуляции воспаления и иммунного ответа [60]. Взаимодействие этих белков с SCP-фосфатазами нуждается в дальнейшем экспериментальном подтверждении.
Продолжается поиск новых субстратов фосфатаз CTDSP1, CTDSP2 и CTDSPL. На примере CTDSP1 предложены новые методические подходы к идентификации субстратов и ингибиторов фосфатаз семейства FCP/SCP [10]. Разработан новый метод фагового дисплея библиотек потенциальных субстратов [61]. Оба метода основаны на том, что молекулы фторида алюминия (AlF4, AlF3) и трифторида бериллия (BeF3) образуют комплексы с Mg2+ в каталитическом центре FCP/SCP-фосфатаз, что имитирует переходное состояние гидролиза и промежуточное соединение аспартилфосфата [62]. Сравнение аминокислотных последовательностей потенциальных субстратов CTDSP1, выявленных этим методом, позволяет предположить, что трипептиды Ser-Thr-Tyr и Pro-Phe-Glu (один или оба) могут быть связывающими мотивами, распознаваемыми CTDSP1 [61].
УЧАСТИЕ SCP-ФОСФАТАЗ В ОНКОГЕНЕЗЕ
В последние годы появляется все больше доказательств участия SCP-фосфатаз в процессах, связанных с развитием, прогрессией опухолей и способностью подавлять рост различных опухолевых клеток человека.
CTDSP1
Фосфатаза CTDSP1, как показано методом коиммунопреципитации, может прямо связываться с С-концевым доменом с основной структурой спираль-петля-спираль транскрипционного фактора и протоонкогена c-MYC. CTDSP1 взаимодействует с c-MYC в опытах in vivo и in vitro и способна подавлять пролиферацию опухолевых клеток HepG2 и SMMC-7721 путем дефосфорилирования c-MYC по Ser62, причем для фосфатазной активности CTDSP1 необходим С‑концевой Ser245. Снижение экспрессии гена CTDSP1 способствует пролиферации клеток HepG2, а его нокдаун приводит к повышению уровня белка c-MYC в клетках рака печени. Таким образом, CTDSP1 негативно регулирует пролиферацию клеток рака печени и, дефосфорилируя c-MYC по Ser62, действует как потенциальный опухолевый супрессор [63].
Экспрессия гена CTDSP1 отрицательно коррелирует с экспрессией TWIST1 в четырех клеточных линиях с морфофенотипом рака молочной железы in vitro. Сверхэкспрессия CTDSP1 ускоряет деградацию TWIST1 и подавляет опосредованную TWIST1 миграцию и инвазию клеток MDA-MB-436 и MCF7 in vitro [46]. Фосфатаза CTDSP1 дефосфорилирует Ser68, локализованный на N-конце белка TWIST1, что способствует деградации этого белка, участвующего в подавлении экспрессии E-кадгерина [46]. Известно, что инактивация E-кадгерина приводит к усилению метастазирования опухолевых клеток [64]. Таким образом, CTDSP1 может подавлять способность клеток к инвазии и метастазированию посредством дефосфорилирования TWIST1, регулирующего экспрессию Е-кадгерина.
Оценено также влияние самого распространенного метаболита холестерина и фактора риска рака молочной железы – гидроксихолестерина 27 (27-HC) – на онкобелок c-MYC, сверхэкспрессия которого характерна для рака молочной железы [65]. 27-HC подавляет экспрессию CTDSP1, что приводит к стабилизации белка c-MYC, а также транскрипцию трех ключевых негативных модуляторов стабильности белка c-MYC – PP2A, CTDSP1 и FBW7. С помощью секвенирования ДНК следующего поколения (ChIPBase) выявлено участие ряда предполагаемых факторов, включая c-MYC, в регуляции транскрипции. Полученные результаты позволяют по-новому посмотреть на механизм активации c-MYC с участием 27-HC и путем подавления транскрипции генов PP2A, CTDSP1 и FBW7 для повышения стабильности белка c-MYC в клетках рака молочной железы.
Белки семейства протеинкиназы В (PKB/AKT) участвуют в регуляции клеточного цикла, апоптозе и ангиогенезе. Значительно снижая киназную активность AKT, CTDSP1 негативно регулирует ангиогенез. Обнаружен ключевой сайт дефосфорилирования протеинкиназой – Ser473. В опытах in vivo показано, что в эндотелиальных клетках мышей с инактивированным геном CTDSP1 повышен уровень фосфорилированных белков AKT, при этом наблюдается рост новых кровеносных сосудов и быстрый рост рака легкого. Согласно этим данным, CTDSP1 действует как негативный регулятор ангиогенеза, играющего ключевую роль в онкогенезе [47].
С помощью нокдауна гена деубиквитиназы (USP29) в клетках рака желудка MGC-803 и BGC-823 показано, что, взаимодействуя с фосфатазой CTDSP1 для стабилизации белка SNAIL, USP29 способствует миграции клеток. USP29 усиливает взаимодействие SNAIL и CTDSP1, что приводит к одновременному дефосфорилированию и деубиквитинированию SNAIL и к предотвращению деградации белка SNAIL, который может способствовать эпителиально-мезенхимальному переходу и метастазированию опухолевых клеток [66].
CTDSP2
Локус 12q13-q15, в котором расположен ген CTDSP2, часто амплифицирован в образцах первичных опухолей и в клеточных линиях остеосаркомы и глиобластомы [67, 68]. Выявление ключевых генных модулей глиобластомы с применением биоинформатического анализа, учитывающего нарушение регуляции экспрессии генов в результате генетических изменений, позволило идентифицировать CTDSP2 как один из 17 генов (наряду с RB1), связанных с плохой выживаемостью пациентов [69].
Ген CTDSP2 служит мишенью транскрипционных факторов семейства FOXO (Forkhead box O ) и может вызывать остановку клеточного цикла in vitro. Промотор CTDSP2 содержит сайт связывания FOXO, который участвует во многих сигнальных путях и играет важную роль в ряде физиологических и патологических процессов, включая онкологические. Эктопическая экспрессия генов FOXO1, FOXO3 или FOXO4 повышает уровень мРНК CTDSP2, что приводит к снижению количества клеток остеосаркомы человека (U2OS) в S‑фазе [38]. Кроме того, экспрессия CTDSP2 повышается в ответ на ингибиторы PI3K или PKB/АKT, контролирующие активность эндогенных белков FOXO в клетках остеосаркомы (U2OS) и колоректальной аденокарциномы (DLD1) [70]. Фосфатаза CTDSP2, ингибируя RAS и P21, нарушение функции которых часто встречается во многих видах рака, способна участвовать в прогрессии клеточного цикла [71, 72].
CTDSPL
В ходе поиска генетических нарушений хромосомы 3 человека обнаружен локус (3p12-p2), обогащенный генами-супрессорами, вовлеченными в патогенез опухолей различного вида/типа, в котором расположен ген CTDSPL (он же SCP3, HYA22, RBSP3). Cпособность CTDSPL подавлять рост опухолевых клеток in vitro и in vivo впервые показали c помощью трансфекции клеток KRC_Y (морфофенотип светлоклеточного рака почки) и ACC-LC5 (морфофенотип мелкоклеточного рака легкого) [42]. Кроме того, экзогенную экспрессию гена CTDSPL наблюдали в ядрах клеток линии MСF-7 (морфофенотип опухоли молочной железы), трансфицированных этим геном. Экспрессия CTDSPL сопровождалась снижением содержания неактивного фосфорилированного по С-концу белка pRb за счет дефосфорилирования в сайте Ser807/811, что свидетельствовало о вкладе CTDSPL в активацию белка pRb, ключевого участника сигнального пути p16ink4A–Cdk/cyclin–Rb, ответственного за остановку клеточного деления на границе фаз G1/S [42].
Выявление соматических гипермутаций в генах-супрессорах способствует пониманию возникновения, развития, прогрессии и распространения рака. Обнаружена высокая частота соматических мутаций в генах RASSF1 и CTDSPL (RBSP3) при различных онкозаболеваниях [73]. Показано значительное и частое снижение экспрессии гена CTDSPL в опухолях легкого, шейки матки [74, 75], оценено клиническое и прогностическое значение частых нарушений CTDSPL на ранних и поздних стадиях рака молочной железы [76], в ранних дисплазиях головы и шеи [77], цервикальных интраэпителиальных неоплазиях [78], плоскоклеточном раке головы и шеи [79].
Важную роль в контроле экспрессии генов-супрессоров играет метилирование – механизм инактивации, способствующий злокачественной трансформации клеток. Предложен метод анализа метилирования и/или делеций на NotI-микрочипах, содержащих клонированные геноспецифичные фрагменты ДНК хромосомы 3, предварительно обогащенные NotI-сайтами [80]. С помощью этого метода обнаружено метилирование и/или делеции в генах, в том числе CTDSPL, при различных видах рака: яичников, легкого, шейки матки, почки, предстательной железы [81–85].
Показано, что корреляция между падением экспрессии генов CTDSPL и P16 в опухолях шейки матки (в клетках базально-парабазального слоя) и увеличением фосфорилированной формы pRb (сайты фосфорилирования Ser807/811 и Ser567)-ассоциирована с неблагоприятным прогнозом при этом заболевании. Кроме того, высокую частоту делеций CTDSPL и P16 наблюдали при дисплазии шейки матки и в ходе прогрессии заболевания. Изменение экспрессии CTDSPL и P16 оказалось синергичным, было связано с увеличением уровня фосфорилированного pRb в опухолях и коррелировало с неблагоприятным прогнозом у пациентов [75].
Согласно TargetScan (http://targetscan.org/), микроРНК miR-7 также может действовать как регулятор экспрессии CTDSPL и RB1, повышение уровня которых зависело от стадии немелкоклеточного рака легкого [86]. При остром миелоидном лейкозе CTDSPL служит мишенью miR-100, которая может регулировать переход G1/S. Сверхэкспрессия miR-100 в клетках HL60 (лейкоз человека) приводит к ингибированию CTDSPL и одновременно к повышению способности фосфорилированной формы активировать pRb и высвобождению фактора транскрипции E2F1 [87]. CTDSPL также служит мишенью miR-181b – члена семейства miR-181, активация которого индуцирует прогрессию клеточного цикла при увеальной меланоме человека. Сверхэкспрессия miR-181b ингибирует экспрессию CTDSPL, что, в свою очередь, может приводить к повышению содержания фосфорилированной формы pRb и накоплению активной формы E2F1, способствующему прогрессии клеточного цикла в клетках увеальной меланомы [88].
CTDSP1/2/L
Известно, что белок промиелоцитарного лейкоза (PML) вовлечен в сигнальные пути ряда опухолевых супрессоров, включая p53 [89], PTEN/ AKT [90] и mTOR [91]. Фосфатазы CTDSP1/2/L дефосфорилируют PML по остатку Ser518, блокируя убиквитинирование и деградацию PML, опосредованные пептидилпролилизомеразой (PIN1) и убиквитинлигазой KLHL20 [92]. Снижение уровня активности CTDSP1 и CTDSPL при светлоклеточном раке почки коррелирует с фосфорилированием PML по Ser518, метаболизмом PML и высокой степенью злокачественности опухолей. Восстановление CTDSP1-опосредованной стабилизации PML подавляет не только пролиферацию, миграцию, инвазию, ангиогенез и рост светлоклеточного рака почки, но и путь mTOR–HIF. Блокирование деградации PML при этом виде рака за счет cверхэкспрессии CTDSP1 или ингибирования PIN1 усиливало противоопухолевое действие темсиролимуса, селективного ингибитора mTOR-киназы. Таким образом, обнаружен новый путь деградации белка PML при светлоклеточном раке почки, включающий инактивацию SCP-фосфатаз, и участие этого пути в прогрессии рака почки. Обосновано применение комбинированной терапии рака почки, направленной на деградацию PML и ингибирование mTOR [92].
Недавно обнаружены супрессорные свойства CTDSP1, CTDSP2 и CTDSPL, приводящие к значительному замедлению роста и старению клеток A549 (морфофенотип аденокарциномы легкого) in vitro, которые могут реализоваться за счет увеличения доли активной формы белка pRb, дефосфорилированного по сайтам Ser807/811, Ser780 и Ser795. Кроме того, выявлено частое (86%) и высоко скоординированное (rs = 0.53–0.62, P ≤ 0.01) падение экспрессии CTDSP1/2/L и RB1 в первичных опухолях аденокарциномы и плоскоклеточного рака легкого. Показано четкое различие уровней мРНК CTDSP1, CTDSPL и RB1 в образцах аденокарциномы с метастазами в лимфатические узлы и без них, что предполагает их использование в качестве биомаркеров метастатического статуса Онкогенный кластер miR-96/182/183 может быть общим регулятором экспрессии трех фосфатаз. Эти результаты отражают функциональную связь SCP-фосфатаз и расширяют знания об их супрессорных свойствах [5].
МикроРНК (miR-26a и miR-26b), кодируемые интронами генов CTDSP1/2/L, синергически снижают долю фосфорилированной формы pRb (ppRb) и блокируют прогрессию клеточного цикла на границе фаз G1/S, подавляя CDK6 и циклин Е1 в клетках гепатоцеллюлярной карциномы [93]. Совместное действие CTDSP1/2/L и miR-26 в сигнальном пути p16INK4A/pRb может проявляться двумя независимыми способами (посредством дефосфорилирования и на уровне транскрипции), приводящими к активации белка pRb, защищающего клетки от повышенной пролиферации [93]. Таким образом, miR-26a/b и их гены-хозяева регулируют фосфорилирование pRb посредством различных молекулярных механизмов. Кроме того, показано, что c-Myc может подавлять регуляцию как членов семейства miR-26, так и SCP-фосфатаз, и в конечном итоге способствует переходу G1/S. Эти результаты дают новое представление о сложном контроле перехода G1/S и предполагают существование одного генного локуса, который содержит два независимых взаимодействующих элемента, подавляющих рост клеток, а также раскрывают новый механизм, с помощью которого c-Myc способствует прогрессии клеточного цикла путем подавления локуса гена, несущего как микроРНК, так и ген, кодирующий белок [93].
За последние годы возрос интерес к практическим приложениям SCP-фосфатаз. Селективные ингибиторы транскрипции обладают явными преимуществами в качестве противоопухолевых средств, поскольку считаются менее генотоксичными по сравнению с химио- или лучевой терапией. Известно, что опухолевым клеткам присущ высокий уровень транскрипции определенных генов, поэтому на основе ингибиторов транскрипции разработаны противоопухолевые препараты [94], в том числе ингибиторы фосфорилирования Ser2 и Ser5 РНК-полимеразы II [95]. Пристальное внимание в качестве потенциальных противоопухолевых средств, направленных на ингибирование транскрипции, привлекают SCP-фосфатазы, дефосфорилирующие Ser2 и Ser5 РНК-полимеразы II.
Сочетание прогнозирования мишеней и количественной оценки экспрессии показало, что miR-18a подавляет экспрессию генов-супрессоров PTEN, WNK2, SOX6, BTG3, CTDSPL в клетках CaSki (морфофенотип рака шейки матки). Эти гены предложены в качестве вероятных мишеней miR-18a. Прогностическую значимость экспрессии этих генов при раке шейки матки оценили на выборке из 60 пациенток. Самая низкая выживаемость отмечена у пациенток с наиболее низкими уровнями экспрессии генов, включая CTDSPL. Эти данные подтверждают возможность использования уровней экспрессии генов в опухолях шейки матки в качестве прогностических маркеров эффективности терапии [96 ] .
Иринотекан, специфически действующий на ДНК-топоизомеразу I (Торо I), применяют при различных солидных опухолях, но на терапию отвечают только 13–32% пациентов, что связано с формированием устойчивости к этому препарату, обусловленной высокой скоростью деградации Topo I. Быструю деградацию Topo I обеспечивает дерегуляция каскада ДНК-зависимых протеинкиназ. Фосфатаза CTDSP1 идентифицирована в качестве первичного регулятора ДНК-зависимых протеинкиназ в ответ на ингибиторы Topo I. Показано, что CTDSP1 способствует поддержанию чувствительности к иринотекану за счет предотвращения деградации Topo I при колоректальном раке [97].
Функции SCP-фосфатаз CTDSP1, CTDSP2 и CTDSPL, их влияние на свойства опухолевых клеток (способность к миграции, скорость роста, ангиогенез), а также роль в онкогенезе вызывают большой интерес. Выявление и изучение новых физиологических субстратов SCP-фосфатаз, а также способы их регуляции важны для разработки новых подходов и поиска эффективных мишеней для терапии различных видов опухолей.
Работа поддержана Российским фондом фундаментальных исследований (грант № 19-0400268).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликтов интересов.
Список литературы
Olsen J.V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 127, 635–648.
Shi Y. (2009) Serine/threonine phosphatases: mechanism through structure. Cell. 139, 468–484.
Seifried A., Schultz J., Gohla A. (2013) Human HAD phosphatases: structure, mechanism, and roles in health and disease. FEBS J. 280, 549–571.
Zhang Y., Kim Y., Genoud N., Gao J., Kelly J.W., Pfaff S.L., Gill G.N., Dixon J.E., Noel J.P. (2006) Determinants for dephosphorylation of the RNA polymerase II C-terminal domain by Scp1. Mol. Cell. 24, 759–770.
Krasnov G.S., Puzanov G.A., Afanasyeva M.A., Dashinimaev E.B., Vishnyakova K.S., Beniaminov A.D., Adzhubei A.A., Kondratieva T.T., Yegorov Y.E., Senchenko V.N. (2019) Tumor suppressor properties of the small C-terminal domain phosphatases in non-small cell lung cancer. Biosci. Repts. 39(12), BSR20193094. https://doi.org/10.1042/BSR20193094
Yeo M., Lee S.K., Lee B., Ruiz E.C., Pfaff S.L., Gill G.N. (2005) Small CTD phosphatases function in silencing neuronal gene expression. Science. 307, 596–600.
Zhang M., Yogesha S.D., Mayfield J.E., Gill G.N., Zhang Y. (2013) Viewing serine/threonine protein phosphatases through the eyes of drug designers. FEBS J. 280, 4739–4760.
Kamenski T., Heilmeier S., Meinhart A., Cramer P. (2004) Structure and mechanism of RNA polymerase II CTD phosphatases. Mol. Cell. 15, 399–407.
Almo S.C., Bonanno J.B., Sauder J.M., Emtage S., Dilorenzo T.P., Malashkevich V., Wasserman S.R., Swaminathan S., Eswaramoorthy S., Agarwal R., Kumaran D., Madegowda M., Ragumani S., Patskovsky Y., Alvarado J., Ramagopal U.A., Faber-Barata J., Chan-ce M.R., Sali A., Fiser A., Zhang Z.Y., Lawrence D.S., Burley S.K. (2007) Structural genomics of protein phosphatases. J. Struct. Funct. Genomics. 8, 121–140.
Rallabandi H.R., Ganesan P., Kim Y.J. (2020). Targeting the C-terminal domain small phosphatase 1. Life (Basel). 10(5), 57. https://doi.org/10.3390/life10050057
Chambers R.S., Dahmus M.E. (1994) Purification and characterization of a phosphatase from HeLa cells which dephosphorylates the C-terminal domain of RNA polymerase II. J. Biol. Chem. 269, 26243–26248.
Chesnut J.D., Stephens J.H., Dahmus M.E. (1992) The interaction of RNA polymerase II with the adenovirus-2 major late promoter is precluded by phosphorylation of the C-terminal domain of subunit IIa. J. Biol. Chem. 267, 10500–10506.
Egloff S., Murphy S. (2008) Cracking the RNA polymerase II CTD code. Trends Genet: TIG. 24, 280–288.
Palancade B., Marshall N.F., Tremeau-Bravard A., Bensaude O., Dahmus M.E., Dubois M.F. (2004) Dephosphorylation of RNA polymerase II by CTD-phosphatase FCP1 is inhibited by phospho-CTD associating proteins. J. Mol. Biol. 335, 415–424.
Yeo M., Lin P.S., Dahmus M.E., Gill G.N. (2003) A novel RNA polymerase II C-terminal domain phosphatase that preferentially dephosphorylates serine 5. J. Biol. Chem. 278, 26078–26085.
Jeronimo C., Bataille A.R., Robert F. (2013) The writers, readers, and functions of the RNA polymerase II C-terminal domain code. Chem. Rev. 113, 8491–8522.
Mayfield J.E., Burkholder N.T., Zhang Y.J. (2016) Dephosphorylating eukaryotic RNA polymerase II. Biochim. Biophys. Acta. 1864, 372–387.
Archambault J., Chambers R.S., Kobor M.S., Ho Y., Cartier M., Bolotin D., Andrews B., Kane C.M., Greenblatt J. (1997) An essential component of a C-terminal domain phosphatase that interacts with transcription factor IIF in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 94, 14300–14305.
Cho E.J., Kobor M.S., Kim M., Greenblatt J., Buratowski S. (2001) Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev. 15, 3319–3329.
Hausmann S., Shuman S. (2002) Characterization of the CTD phosphatase Fcp1 from fission yeast. Preferential dephosphorylation of serine 2 versus serine 5. J. Biol. Chem. 277, 21213–21220.
Ghosh A., Shuman S., Lima C.D. (2008) The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol. Cell. 32, 478–490.
Bataille A.R., Jeronimo C., Jacques P., Laramée L., Fortin M., Forest A., Bergeron M., Hanes S.D., Robert F. (2012) A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol. Cell. 45, 158–170.
Nojima T., Rebelo K., Gomes T., Grosso A.R., Proudfoot N.J., Carmo-Fonseca M. (2018) RNA polymerase II phosphorylated on CTD serine 5 interacts with the spliceosome during co-transcriptional splicing. Mol. Cell. 72, 369–379, e364.
Visvanathan J., Lee S., Lee B., Lee J.W., Lee S.K. (2007) The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev. 21, 744–749.
Burkholder N.T., Mayfield J.E., Yu X., Irani S., Arce D.K., Jiang F., Matthews W.L., Xue Y., Zhang Y.J. (2018) Phosphatase activity of small C-terminal domain phosphatase 1 (SCP1) controls the stability of the key neuronal regulator RE1-silencing transcription factor (REST). J. Biol. Chem. 293, 16851–16861.
Sun A.G., Wang M.G., Li B., Meng F.G. (2017) Down-regulation of miR-124 target protein SCP-1 inhibits neuroglioma cell migration. Eur. Rev. Med. Pharm. Sci. 21, 723–729.
Conti L., Crisafulli L., Caldera V., Tortoreto M., Brilli E., Conforti P., Zunino F., Magrassi L., Schiffer D., Cattaneo E. (2012) REST controls self-renewal and tumorigenic competence of human glioblastoma cells. PLoS One. 7, e38486.
Osada H., Takahashi T. (2002) Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene. 21, 7421–7434.
Rubin S.M. (2013) Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 38, 12–19.
Lundberg A.S., Weinberg R.A. (1998) Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Boil. 18, 753–761.
Narasimha A.M., Kaulich M., Shapiro G.S., Choi Y.J., Sicinski P., Dowdy S.F. (2014) Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife. 3, e02872. https://doi.org/10.7554/eLife.02872
Knudsen E.S., Wang J.Y. (1997) Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol. Cell. Biol. 17, 5771–5783.
Rubin S.M., Gall A.L., Zheng N., Pavletich N.P. 2005. Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell. 123, 1093–1106.
Watanabe N., Arai H., Nishihara Y., Taniguchi M., Watanabe N., Hunter T., Osada H. (2004) M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. USA. 101, 4419–4424.
Moiseeva T.N., Qian C., Sugitani N., Osmanbeyoglu H.U., Bakkenist C.J. (2019) WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proc. Natl. Acad. Sci. USA. 116, 23891–23893.
Maehama T., Taylor G.S., Slama J.T., Dixon J.E. (2000) A sensitive assay for phosphoinositide phosphatases. Analyt. Biochem. 279, 248–250.
Kim Y.J., Bahk Y.Y. (2014) A study of substrate specificity for a CTD phosphatase, SCP1, by proteomic screening of binding partners. Biochem. Biophys. Res. Commun. 448, 189–194.
Kloet D.E., Polderman P.E., Eijkelenboom A., Smits L.M., van Triest M.H., van den Berg M.C., Groot Koerkamp M.J., van Leenen D., Lijnzaad P., Holstege F.C., Burgering B.M. (2015) FOXO target gene CTDSP2 regulates cell cycle progression through Ras and p21(Cip1/Waf1). Biochem. J. 469, 289–298.
Barbacid M. (1987) Ras genes. Annu. Rev. Biochem. 56, 779–827.
Cox A.D., Der C.J. (2003) The dark side of Ras: regulation of apoptosis. Oncogene. 22, 8999–9006.
Stewart Z.A., Leach S.D., Pietenpol J.A. (1999) p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Mol. Cell. Biol. 19, 205–215.
Kashuba V.I., Li J., Wang F., Senchenko V.N., Protopopov A., Malyukova A., Kutsenko A.S., Kadyrova E., Zabarovska V.I., Muravenko O.V., Zelenin A.V., Kisselev L.L., Kuzmin I., Minna J.D., Winberg G., Ernberg I., Braga E., Lerman M.I., Klein G., Zabarovsky E.R. (2004) RBSP3 (HYA22) is a tumor suppressor gene implicated in major epithelial malignancies. Proc. Natl. Acad. Sci. USA. 101, 4906–4911.
Knockaert M., Sapkota G., Alarcón C., Massagué J., Brivanlou A.H. (2006) Unique players in the BMP pathway: small C-terminal domain phosphatases dephosphorylate Smad1 to attenuate BMP signaling. Proc. Natl. Acad. Sci. USA. 103, 11940–11945.
Rahman M.S., Akhtar N., Jamil H.M., Banik R.S., Asaduzzaman S.M. (2015) TGF-β/BMP signaling and other molecular events: regulation of osteoblastogenesis and bone formation. Bone Res. 3, 15005.
Wrighton K.H., Willis D., Long J., Liu F., Lin X., Feng X.H. (2006) Small C-terminal domain phosphatases dephosphorylate the regulatory linker regions of Smad2 and Smad3 to enhance transforming growth factor-beta signaling. J. Biol. Chem. 281, 38365–38375.
Sun T., Fu J., Shen T., Lin X., Liao L., Feng X.H., Xu J. (2016) The small C-terminal domain phosphatase 1 inhibits cancer cell migration and invasion by dephosphorylating Ser(P)68-TWIST1 to accelerate TWIST1 protein degradation. J. Biol. Chem. 291, 11518–11528.
Liao P., Wang W., Li Y., Wang R., Jin J., Pang W., Chen Y., Shen M., Wang X., Jiang D., Pang J., Liu M., Lin X., Feng X.H., Wang P., Ge X. (2017) Palmitoylated SCP1 is targeted to the plasma membrane and negatively regulates angiogenesis. Elife. 6, e22058. https://doi.org/10.7554/eLife.22058
Alonso A., Sasin J., Bottini N., Friedberg I., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., Mustelin T. (2004) Protein tyrosine phosphatases in the human genome. Cell. 117, 699–711.
Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 307, 1098–1101.
Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., Jensen L.J., Mering C.V. (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 47, D607–D613.
Fernandes A.O., Campagnoni C.W., Kampf K., Feng J.M., Handley V.W., Schonmann V., Bongarzone E.R., Reyes S., Campagnoni A.T. (2004) Identification of a protein that interacts with the golli-myelin basic protein and with nuclear LIM interactor in the nervous system. J. Neurosci. Res. 75, 461–471.
Ma Y., Sekiya M., Kainoh K., Matsuda T., Iwasaki H., Osaki Y., Sugano Y., Suzuki H., Takeuchi Y., Miyamoto T., Yahagi N., Nakagawa Y., Matsuzaka T., Shimano H. (2020) Transcriptional co-repressor CtBP2 orchestrates epithelial-mesenchymal transition through a novel transcriptional holocomplex with OCT1. Biochem. Biophys. Res. Commun. 523, 354–360.
Roelofs A.J., Zupan J., Riemen A.H.K., Kania K., Ansboro S., White N., Clark S.M., De Bari C. (2017) Joint morphogenetic cells in the adult mammalian synovium. Nat. Commun. 8, 15040.
Huttlin E.L., Ting L., Bruckner R.J., Gebreab F., Gygi M.P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K., Dong R., Guarani V., Vaites L.P., Ordureau A., Rad R., Erickson B.K., Wühr M., Chick J., Zhai B., Kolippakkam D., Mintseris J., Obar R.A., Harris T., Artavanis-Tsakonas S., Sowa M.E., De Camilli P., Paulo J.A., Harper J.W., Gygi S.P. (2015) The BioPlex network: a systematic exploration of the human interactome. Cell. 162, 425–440.
Ghosh A., Shuman S., Lima C.D. (2008) The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol. Cell. 32, 478–490.
Muttigi M.S., Han I., Park H.K., Park H., Lee S.H. (2016) Matrilin-3 role in cartilage development and osteoarthritis. Internat. J. Mol. Sci. 17(4), 590. https://doi.org/10.3390/ijms17040590
Ghosh A., Shuman S., Lima C.D. (2008) The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol. Cell. 32, 478–490.
Thysen S., Cailotto F., Lories R. (2016) Osteogenesis induced by frizzled-related protein (FRZB) is linked to the netrin-like domain. Lab. Invest. J. Techn. Meth. Pathol. 96, 570–580.
Vlahakis N.E., Young B.A., Atakilit A., Hawkridge A.E., Issaka R.B., Boudreau N., Sheppard D. (2007) Integrin alpha9beta1 directly binds to vascular endothelial growth factor (VEGF)-A and contributes to VEGF-A-induced angiogenesis. J. Biol. Chem. 282, 15187–15196.
Gebhardt C., Németh J., Angel P., Hess J. (2006) S100A8 and S100A9 in inflammation and cancer. Biochem. Pharmacol. 72, 1622–1631.
Otsubo K., Yoneda T., Kaneko A., Yagi S., Furukawa K., Chuman Y. (2018) Development of a substrate identification method for human scp1 phosphatase using phosphorylation mimic phage display. Protein Peptide Lett. 25, 76–83.
Schwer B., Ghosh A., Sanchez A.M., Lima C.D., Shuman S. (2015) Genetic and structural analysis of the essential fission yeast RNA polymerase II CTD phosphatase Fcp1. RNA. 21, 1135–1146.
Wang W., Liao P., Shen M., Chen T., Chen Y., Li Y., Lin X., Ge X., Wang P. (2016) SCP1 regulates c-Myc stability and functions through dephosphorylating c‑Myc Ser62. Oncogene. 35, 491–500.
Onder T.T., Gupta P.B., Mani S.A., Yang J., Lander E.S., Weinberg R.A. (2008) Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 68, 3645–3654.
Ma L.M., Liang Z.R., Zhou K.R., Zhou H., Qu L.H. (2016) 27-Hydroxycholesterol increases Myc protein stability via suppressing PP2A, SCP1 and FBW7 transcription in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 480, 328–333.
Qian W., Li Q., Wu X., Li W., Li Q., Zhang J., Li M., Zhang D., Zhao H., Zou X., Jia H., Zhang L. (2020) Deubiquitinase USP29 promotes gastric cancer cell migration by cooperating with phosphatase SCP1 to stabilize Snail protein. Oncogene. 39, 6802–6815.
Reifenberger G., Ichimura K., Reifenberger J., Elkahloun A.G., Meltzer P.S., Collins V.P. (1996) Refined mapping of 12q13–q15 amplicons in human malignant gliomas suggests CDK4/SAS and MDM2 as independent amplification targets. Cancer Res. 56, 5141–5145.
Su Y.A., Lee M.M., Hutter C.M., Meltzer P.S. (1997) Characterization of a highly conserved gene (OS4) amplified with CDK4 in human sarcomas. Oncogene. 15, 1289–1294.
Ping Y., Deng Y., Wang L., Zhang H., Zhang Y., Xu C., Zhao H., Fan H., Yu F., Xiao Y., Li X. (2015) Identifying core gene modules in glioblastoma based on multilayer factor-mediated dysfunctional regulatory networks through integrating multi-dimensional genomic data. Nucl. Acids Res. 43, 1997–2007.
Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden K.C., Blenis J., Greenberg M.E. 1999. AKT promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96, 857–868.
Shamloo B., Usluer S. (2019) p21 in cancer research. Cancers (Basel). 11(8), 1178. https://doi.org/10.3390/cancers11081178
Fernández-Medarde A., Santos E. (2011) Ras in cancer and developmental diseases. Genes Cancer. 2, 344–358.
Kashuba V.I., Pavlova T.V., Grigorieva E.V., Kutsenko A., Yenamandra S.P., Li J., Wang F., Protopopov A.I., Zabarovska V.I., Senchenko V., Haraldson K., Eshchenko T., Kobliakova J., Vorontsova O., Kuzmin I., Braga E., Blinov V.M., Kisselev L.L., Zeng Y.X., Ernberg I., Lerman M.I., Klein G., Zabarovsky E.R. (2009) High mutability of the tumor suppressor genes RASSF1 and RBSP3 (CTDSPL) in cancer. PLoS One. 4, e5231.
Senchenko V.N., Anedchenko E.A., Kondratieva T.T., Krasnov G.S., Dmitriev A.A., Zabarovska V.I., Pavlova T.V., Kashuba V.I., Lerman M.I., Zabarovsky E.R. (2010) Simultaneous down-regulation of tumor suppressor genes RBSP3/CTDSPL, NPRL2/G21 and RASSF1A in primary non-small cell lung cancer. BMC Cancer. 10, 75.
Chakraborty C., Roychowdhury A., Samadder S., Roy A., Mandal R.K., Basu P., Roychoudhury S., Panda C.K. (2016) Association of P16-RBSP3 inactivation with phosphorylated RB1 overexpression in basal-parabasal layers of normal cervix unchanged during CACX development. Biochem. J. 473, 3221–3236.
Sinha S., Singh R.K., Alam N., Roy A., Roychoudhury S., Panda C.K. (2008) Frequent alterations of hMLH1 and RBSP3/HYA22 at chromosomal 3p22.3 region in early and late-onset breast carcinoma: clinical and prognostic significance. Cancer Sci. 99, 1984–1991.
Ghosh A., Ghosh S., Maiti G.P., Sabbir M.G., Zabarovsky E.R., Roy A., Roychoudhury S., Panda C.K. (2010) Frequent alterations of the candidate genes hMLH1, ITGA9 and RBSP3 in early dysplastic lesions of head and neck: clinical and prognostic significance. Cancer Sci. 101, 1511–1520.
Mitra S., Mazumder Indra D., Bhattacharya N., Singh R.K., Basu P.S., Mondal R.K., Roy A., Zabarovsky E.R., Roychoudhury S., Panda C.K. (2010) RBSP3 is frequently altered in premalignant cervical lesions: clinical and prognostic significance. Genes, Chrom. Cancer. 49, 155–170.
Sarkar S., Panda C.K. (2018) Preferential allelic deletion of RBSP3, LIMD1 and CDC25A in head and neck squamous cell carcinoma: implication in cancer screening and early detection. Cancer Biol. Therapy. 19, 631–635.
Li J., Protopopov A., Wang F., Senchenko V., Petushkov V., Vorontsova O., Petrenko L., Zabarovska V., Muravenko O., Braga E., Kisselev L., Lerman M.I., Kashuba V., Klein G., Ernberg I., Wahlestedt C., Zaba-rovsky E.R. (2002) NotI subtraction and NotI-specific microarrays to detect copy number and methylation changes in whole genomes. Proc. Natl. Acad. Sci. USA. 99, 10724–10729.
Kashuba V., Dmitriev A.A., Krasnov G.S., Pavlova T., Ignatjev I., Gordiyuk V.V., Gerashchenko A.V., Braga E.A., Yenamandra S.P., Lerman M., Senchenko V.N., Zabarovsky E. (2012) NotI microarrays: novel epigenetic markers for early detection and prognosis of high grade serous ovarian cancer. Int. J. Mol. Sci. 13, 13352–13377.
Dmitriev A.A., Kashuba V.I., Haraldson K., Senchenko V.N., Pavlova T.V., Kudryavtseva A.V., Anedchenko E.A., Krasnov G.S., Pronina I.V., Loginov V.I., Kondratieva T.T., Kazubskaya T.P., Braga E.A., Yenamandra S.P., Ignatjev I., Ernberg I., Klein G., Lerman M.I., Zabarovsky E.R. (2012) Genetic and epigenetic analysis of non-small cell lung cancer with NotI-microarrays. Epigenetics. 7, 502–513.
Senchenko V.N., Kisseljova N.P., Ivanova T.A., Dmitriev A.A., Krasnov G.S., Kudryavtseva A.V., Panasenko G.V., Tsitrin E.B., Lerman M.I., Kisseljov F.L., Kashuba V.I., Zabarovsky E.R. (2013) Novel tumor suppressor candidates on chromosome 3 revealed by NotI-microarrays in cervical cancer. Epigenetics. 8, 409–420.
Dmitriev A.A., Rudenko E.E., Kudryavtseva A.V., Krasnov G.S., Gordiyuk V.V., Melnikova N.V., Stakhovsky E.O., Kononenko O.A., Pavlova L.S., Kondratieva T.T., Alekseev B.Y., Braga E.A., Senchenko V.N., Kashuba V.I. (2014) Epigenetic alterations of chromosome 3 revealed by NotI-microarrays in clear cell renal cell carcinoma. BioMed Res. Internat. 2014, 735292.
Dmitriev A.A., Rosenberg E.E., Krasnov G.S., Gerashchenko G.V., Gordiyuk V.V., Pavlova T.V., Kudryavtseva A.V., Beniaminov A.D., Belova A.A., Bondarenko Y.N., Danilets R.O., Glukhov A.I., Kondratov A.G., Alexeyenko A., Alekseev B.Y., Klein G., Senchenko V.N., Kashuba V.I. (2015) Identification of novel epigenetic markers of prostate cancer by NotI-microarray analysis. Dis. Markers. 2015, 241301. https://doi.org/10.1155/2015/241301
Duncavage E., Goodgame B., Sezhiyan A., Govindan R., Pfeifer J. (2010) Use of microRNA expression levels to predict outcomes in resected stage I non-small cell lung cancer. J. Thoracic Oncol. Off. Publ. Internat. Assoc. Study Lung Cancer. 5, 1755–1763.
Zheng Y.S., Zhang H., Zhang X.J., Feng D.D., Luo X.Q., Zeng C.W., Lin K.Y., Zhou H., Qu L.H., Zhang P., Chen Y.Q. (2012) MiR-100 regulates cell differentiation and survival by targeting RBSP3, a phosphatase-like tumor suppressor in acute myeloid leukemia. Oncogene. 31, 80–92.
Zhang L., He X., Li F., Pan H., Huang X., Wen X., Zhang H., Li B., Ge S., Xu X., Jia R., Fan X. (2018) The miR-181 family promotes cell cycle by targeting CTDSPL, a phosphatase-like tumor suppressor in uveal melanoma. J. Exp. Clin. Cancer Res. 37, 15.
Bernardi R., Pandolfi P.P. (2003) Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene. 22, 9048–9057.
Giorgi C., Ito K., Lin H.K., Santangelo C., Wieckowski M.R., Lebiedzinska M., Bononi A., Bonora M., Duszynski J., Bernardi R., Rizzuto R., Tacchetti C., Pinton P., Pandolfi P.P. (2010) PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 330, 1247–1251.
Bernardi R., Guernah I., Jin D., Grisendi S., Alimonti A., Teruya-Feldstein J., Cordon-Cardo C., Simon M.C., Rafii S., Pandolfi P.P. (2006) PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 442, 779–785.
Lin Y.C., Lu L.T., Chen H.Y., Duan X., Lin X., Feng X.H., Tang M.J., Chen R.H. (2014) SCP phosphatases suppress renal cell carcinoma by stabilizing PML and inhibiting mTOR/HIF signaling. Cancer Res. 74, 6935–6946.
Zhu Y., Lu Y., Zhang Q., Liu J.J., Li T.J., Yang J.R., Zeng C., Zhuang S.M. (2012) MicroRNA-26a/b and their host genes cooperate to inhibit the G1/S transition by activating the pRb protein. Nucl. Acids Res. 40, 4615–4625.
Stellrecht C.M., Chen L.S. (2011) Transcription inhibition as a therapeutic target for cancer. Cancers. 3, 4170–4190.
Morachis J.M., Huang R., Emerson B.M. (2011) Identification of kinase inhibitors that target transcription initiation by RNA polymerase II. Oncotarget. 2, 18–28.
Dong P., Xiong Y., Yu J., Chen L., Tao T., Yi S., Hanley S.J.B., Yue J., Watari H., Sakuragi N. (2018) Control of PD-L1 expression by miR-140/142/340/383 and oncogenic activation of the OCT4-miR-18a pathway in cervical cancer. Oncogene. 37, 5257–5268.
Matsuoka H., Ando K., Swayze E.J., Unan E.C., Mathew J., Hu Q., Tsuda Y., Nakashima Y., Saeki H., Oki E., Bharti A.K., Mori M. (2020) CTDSP1 inhibitor rabeprazole regulates DNA-PKcs dependent topoisomerase I degradation and irinotecan drug resistance in colorectal cancer. PLoS One. 15, e0228002.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология