

Молекулярная биология, 2021, T. 55, № 4, стр. 543-561
Основные подходы к контролируемой деградации белков в клетке
М. А. Капитонова a, О. А. Шадрина a, b, С. П. Королев b, М. Б. Готтих a, b, *
a Химический факультет Московского государственного университета им. М.В. Ломоносова
119991 Москва, Россия
b Научно-исследовательский институт физико-химической биологии им. А.Н. Белозерского Московского государственного университета им. М.В. Ломоносова
119991 Москва, Россия
* E-mail: gottikh@belozersky.msu.ru
Поступила в редакцию 24.11.2020
После доработки 22.01.2021
Принята к публикации 22.01.2021
Аннотация
Изменение внутриклеточной концентрации определенного белка в клетке считается одним из наиболее информативных подходов к изучению роли этого белка в жизнедеятельности клетки. Чаще всего c этой целью применяют методы генетического нокаута или нокдауна. Однако в некоторых случаях более информативным или удобным представляется воздействие непосредственно на уже синтезированный белок, причем это воздействие в идеале должно быть контролируемым во времени и обратимым. В настоящем обзоре проанализированы системы, позволяющие осуществлять контролируемую деградацию белков как путем их убиквитинирования с последующей протеасомной деградацией, так и за счет иных механизмов.
ВВЕДЕНИЕ
Одним из наиболее информативных подходов к изучению роли отдельных белков в жизнедеятельности клетки считается изменение внутриклеточной концентрации этого белка. Уровень белка в клетке чаще всего снижают с помощью генетического нокаута или нокдауна. Однако полное “выключение” синтеза ключевых белков метаболических путей может вести к гибели клеток или существенному изменению их метаболизма, что значительно затрудняет изучение функциональной роли данных белков. Эту проблему можно решить с использованием методов, позволяющих в определенный момент времени обратимо снижать концентрацию исследуемого белка в клетке, например, путем его деградации. Для достижения контролируемой деградации часто используют модификацию эндогенных белков ауксин-индуцибельным дегроном (Auxin-Inducible Degron, AID). Принцип работы этого дегрона позаимствован у растений и приспособлен к исследованию механизмов процессов, протекающих в клетках нерастительного происхождения. Однако за последнее время появились и другие методы, позволяющие обратимо снижать внутриклеточный уровень различных белков в клеточных линиях и даже в модельных животных.
В настоящем обзоре рассмотрены возможности как системы AID, так и некоторых других систем, позволяющих осуществлять контролируемую деградацию белков путем их убиквитинирования и последующей протеасомной деградации либо за счет иных механизмов.
УБИКВИТИНИРОВАНИЕ
Убиквитинирование – одна из важнейших посттрансляционных модификаций в клетках эукариот, в ходе которой происходит ковалентное присоединение одного или нескольких остатков убиквитина (белок 8.5 кДа) к аминокислотным остаткам в составе белков-мишеней. Убиквитин связывается с ε-аминогруппами остатков лизина в молекулах большинства белков, но описано также присоединение убиквитина через их α-NН2 концевые группы, а также остатки серина или треонина [1–3]. Возможно как моно-, так и полиубиквитинирование белков. При полиубиквитинировании молекулы убиквитина присоединяются друг к другу с образованием изопептидной связи между карбоксильной группой С-концевого остатка глицина одной молекулы и аминогруппой другой, уже связанной с белком-мишенью. Наличие остатков убиквитина способно определять внутриклеточную локализацию белка, усиливать или подавлять активность, препятствовать белок-белковым взаимодействиям, но самое главное – активировать протеолитическую деградацию белка при участии протеасомы 26S [1]. Долгое время считалось, что субстрат для его распознавания протеасомой должен быть полиубиквитинирован [4], а обнаруживаемые в клетке моноубиквитинированные белки участвуют в процессах, не связанных с протеолизом. Однако в последнее время выяснилось, что для гидролиза некоторых белков протеасомой достаточно их моноубиквитинирования или множественного моноубиквитинирования нескольких остатков лизина в составе одного белка [5]. Так, регулятор дифференцировки мышечных клеток PAX3 подвергается протеолизу после моноубиквитинирования одного определенного остатка лизина [6], а для расщепления протеасомой предшественника фактора транскрипции NF-κB – белка p105 – до активного фактора транскрипции (p50) необходимо моноубиквитинирование нескольких остатков лизина [7]. В состав убиквитина входят семь остатков лизина в положениях 6, 11, 27, 29, 33, 48 и 63. Показано, что положение лизина, участвующего в образовании связи между молекулами убиквитина в цепочке, определяет дальнейшую судьбу белка. Так, полиубиквитинирование за счет образования связей через К48 приводит к протеасомной деградации белка, в то время как лизин в положении 63 играет ключевую роль в координации таких процессов, как направленный эндоцитоз, воспаление, трансляция и репарация ДНК [1, 8]. Функции полиубиквитиновых цепочек, связанных через другие остатки лизина, смешанных и разветвленных цепочек, N-концевых линейных цепочек активно изучаются, но до сих пор остаются не до конца понятными [9, 10]. Интересно, что в некоторых случаях сигналами к протеасомной деградации оказались остатки убиквитина, присоединенные к остаткам, отличным от К48 [5]. Так, протеасома способна разрушать белки, содержащие разветвленные полиубиквитиновые цепи, присоединенные к K11/K48 или K48/K63 [11–13].
Убиквитинирование в клетках млекопитающих происходит в три стадии – стадии активации, конъюгации и лигирования (рис. 1) [1, 14].
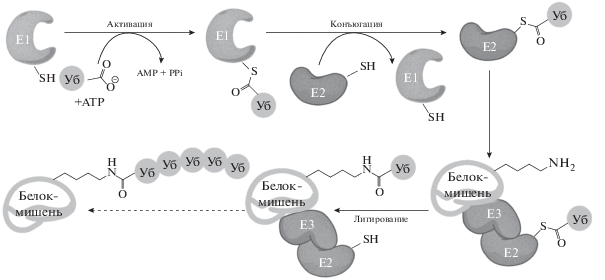
1) Активация. Убиквитин-активирующий фермент (Е1) последовательно связывает ATР и убиквитин. После этого фермент Е1 катализирует нуклеофильную атаку С-концевой карбоксильной группой на α-фосфат в АТР с образованием соответствующего ацилфосфата, а затем ацилирование этим ацилфосфатом остатка цистеина фермента E1 с образованием тиоэфирной связи между Е1 и убиквитином.
2) Конъюгация. Посредством тио-переэтерификации убиквитин переносится на остаток цистеина убиквитин-конъюгирующего фермента (Е2).
3) Лигирование. Убиквитинлигазы (E3) обеспечивают образование изопептидной связи между лизином белка-мишени и С-концевым остатком глицина убиквитина.
Существуют два основных семейства убиквитинлигаз Е3 [1, 10]. Наиболее многочисленно семейство убиквитинлигаз, содержащих домен RING-finger (Really Interesting New Gene), – мультисубъединичных комплексов, выполняющих роль платформы, на которой в непосредственной близости связываются убиквитин-конъюгирующий фермент E2 и белок-мишень, что позволяет эффективно переносить активированный фрагмент убиквитина от E2 к мишени (именно этот вариант приведен на рис. 1). Второе семейство – это убиквитинлигазы HECT-типа, содержащие так называемый домен HECT (Нomologous to E6-associated protein (E6-AP) Сarboxyl Тerminus). Для этих убиквитинлигаз характерен перенос активированного убиквитина с фермента E2 на их внутренний остаток Cys, а затем уже сама убиквитинлигаза Е3 катализирует убиквитинирование белковой мишени.
Один из представителей RING-убиквитинлигаз – комплекс SCF (SKP, Cullin, F-box containing complex), состоит из вариабельного белка с F-box (FBP) и трех коровых субъединиц: куллина (CUL1), белка RBX1, содержащего цинксвязывающий RING-домен, и адаптерного белка SKP1 (S-phase kinase-associated protein 1) [15]. Куллин образует основной структурный каркас комплекса SCF и связывает белок SKP1 с белком RBX1. Различные комбинации куллина и FBP могут генерировать порядка 100 типов убиквитинлигаз E3, которые нацелены на разные субстраты. Для обеспечения субстратной специфичности комплекса SCF важен белок FBP, поскольку именно он связывает белок-мишень независимо от комплекса [16]. Каждый FBP (например, белок SKP2) может распознавать несколько различных субстратов, причем характер распознавания зависит от их посттрансляционных модификаций, таких как фосфорилирование или гликозилирование. После связывания целевого белка-мишени белок FBP за счет своего структурного мотива F-box связывается с белком SKP1 (компонент комплекса SCF), обеспечивая сближение белка-мишени и убиквитин-конъюгирующего фермента E2. Связывание фермента Е2 с комплексом SCF осуществляется через RING-домен. В результате остаток убиквитина переносится от фермента Е2 к белку-мишени.
В заключение хотелось бы отметить, что процессы, аналогичные убиквитинированию в эукариотических клетках, протекают и в клетках прокариот. Так, обнаружен прокариотический убиквитин-подобный белок Pup (prokaryotic ubiquitin-like protein), присоединение которого к белкам служит сигналом к протеолизу протеасомами Mycobacterium tuberculosis [17].
УБИКВИТИНИРОВАНИЕ В РАСТЕНИЯХ
В клетках растений полиубиквитинирование и последующая протеасомная деградация белков, содержащих специфические домены – дегроны, осуществляются с помощью гормонов ауксинов, из которых наиболее распространены гетероауксин (индолил-3-уксусная кислота, IAA), и жасмонат изолейцина ((+)-7-iso-jasmonoyl-L-isoleucine, JA-Ile). Эти гормоны связываются с F-box-доменами белков TIR1 (Тransport Inhibitor Response 1) и COI1 (Соronatine-insensitive protein 1), соответственно, и способствуют их ассоциации с белками-мишенями, содержащими специфические мотивы дегрона. Белки TIR1 и COI1 входят в состав E3 убиквитинлигазного комплекса SCF, который вместе с убиквитин-конъюгирующим ферментом E2 катализирует полиубиквитинирование и последующую протеасомную деградацию содержащих дегрон белков [18–22]. Мишенями TIR1, связанного с ауксином, служат белки, содержащие AID, в то время как белок COI1, связанный с JA-Ile, нацелен на белки, содержащие дегроны JAZ [21, 23].
Для контролируемой деградации белков в нерастительных клетках чаще используется система AID, поэтому остановимся на ней более подробно. Гормон гетероауксин (рис. 2а) способен регулировать транскрипцию в растениях, стимулируя деградацию белков Aux/IAA, негативных регуляторов генной экспрессии [24]. Ауксин стимулирует связывание белков Aux/IAA с белком TIR1, который входит в состав комплекса Е3 убиквитинлигазы SCFTIR1. Изучение 29 белков Aux/IAA Arabidopsis, деградирующих в присутствии ауксина, показало, что все они состоят из четырех консервативных доменов. За связывание с убиквитинлигазой SCFTIR1 отвечает домен II, который содержит консервативную аминокислотную последовательность, называемую AID [25]. Сродство комплекса SCFTIR1 к AID-содержащим белкам резко возрастает именно в результате связывания ауксина с белком TIR1. Помимо F‑box-домена белок TIR1 содержит также лейцин-богатые участки (LRR), по форме напоминающие подковообразные соленоиды. Карман связывания ауксина в белке TIR1 сформирован тремя протяженными петлями, входящими в состав трех LRR (LRR 2, 12 и 14), которые названы Loop 2, Loop 12 и Loop 14 соответственно (рис. 2б) [19]. Кроме того, в белке TIR1 в непосредственной близости от кармана связывания ауксина находится сайт связывания фитиновой (D-мио-инозитол-1,2,3,4,5,6-гексакисдигидрофосфорной) кислоты [19], которая, вероятнее всего, служит структурным кофактором. Интересно, что сайт связывания фитиновой кислоты в TIR1 консервативен у белков AFB (auxin signaling F-box), которые, как и TIR1, принадлежат к семейству F-box-белков Arabidopsis [21], что свидетельствует о важной роли фитиновой кислоты в формировании структуры белков этого семейства.
Рис. 2.
Структура гетероауксина (а) и кармана для его связывания в белке TIR1 (б), петли (Loop) 2, 12 и 14 выделены желтым; PDBID 2P1P.
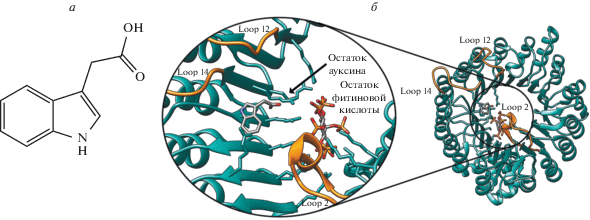
Показано, что связывание ауксина с комплексом SCFTIR1 не приводит к значительным изменениям конформации белка TIR1, а лишь усиливает взаимодействие комплекса с AID [19]. Однако такое взаимодействие возможно и в отсутствие фитогормона, что влечет за собой ауксиннезависимую деградацию белков, содержащих дегрон.
От ауксиннезависимой деградации могут предохранять белки семейства ARF (Auxin Response Transcription Factor) – факторы транскрипции, узнающие последовательность TGTCTC, встречающуюся в промоторах генов белков, участвующих в ауксинзависимой регуляции [26]. Растения Arabidopsis содержат более 10 различных белков ARF, каждый из которых распознает последовательность TGTCTC и связывается с ней за счет консервативного ДНК-связывающего домена (120 аминокислот). Также в белках ARF есть домен PB1 (Phox and Bem1), который взаимодействует с доменами III и IV белков Aux/IAA. В результате белки Aux/IAA и ARF образуют гетеродимерный комплекс, в котором Aux/IAA защищены от взаимодействия с TIR1 и последующей ауксиннезависимой деградации [27, 28].
ПРИМЕНЕНИЕ СИСТЕМЫ АУКСИН-ИНДУЦИБЕЛЬНОГО ДЕГРОНА ДЛЯ КОНТРОЛИРУЕМОЙ ДЕГРАДАЦИИ БЕЛКОВ В КЛЕТКАХ НЕРАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ
В 2009 году систему AID успешно модифицировали для контролируемой деградации белков в нерастительных клетках [29]. Предположили, что добавление AID к белку-мишени (в качестве дегрона использовали полную последовательность белка IAA17/AXR3 из семейства Aux/IAA A. thaliana), а также экспрессия растительного белка TIR1 в клетках позволят запустить деградацию интересующего белка под действием фитогормона ауксина [29]. Из генома A. thaliana был выделен ген белка TIR1 (AtTIR1), который экспрессировали в клетках дрожжей Saccharomyces cerevisiae. С помощью иммунопреципитации показано, что полученный в результате белок TIR1 взаимодействует с белком CDC53, входящим в состав SCF-комплекса дрожжей. В эти клетки вводили последовательность гена зеленого флуоресцентного белка (GFP), модифицированного AID, а затем клетки обрабатывали ауксином. В результате интенсивность свечения GFP снижалась уже спустя 30 мин. Выяснилось также, что деградация GFP не зависит от того, к какому концу белка (С- или N-) присоединен дегрон. Огромным преимуществом этой системы является обратимость деградации: спустя 30 мин после удаления ауксина из среды свечение GFP начало восстанавливаться. Однако известно, что взаимодействие между AtTIR1 и белком, содержащим AID, стабильно при температуре от 4 до 25°С, а при 37°С сильно ослабевает [21], что связано с понижением стабильности AtTIR1 при температуре выше 25°С. Следовательно, чтобы применять AID-систему для деградации белков в клетках млекопитающих, культивируемых при температуре 37°С, необходимо было повысить ее термостабильность. Для решения этой проблемы из растений, растущих в более жарком климате, выделили гены белков TIR1, а для использования в клетках млекопитающих выбрали продукт гена TIR1 из Oryza sativa (osTIR1), взаимодействие которого с дегроном было устойчивым в широком интервале температур [29]. В результате получили систему индуцируемой деградации белков в клетках млекопитающих, состоящую из белка-мишени, меченного дегроном IAA17, белка osTIR1 и фитогормона ауксина (рис. 3).
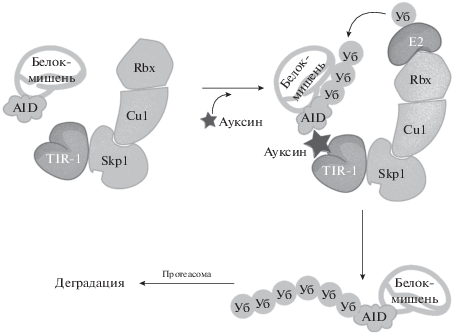
Полученную систему опробовали для контролируемой деградации белка GFP-AID-NLS (NLS – nuclear localization signal, сигнал ядерной локализации), экспрессируемого в нескольких клеточных линиях: HeLa (клетки, полученные из раковой опухоли шейки матки), COS1 (клетки зеленой мартышки), CHOK1 (клетки китайского хомячка), NIH3T3 (эмбриональные клетки мыши), HEK 293T (эмбриональные клетки человека) и DT40 (B-клетки курицы) [29]. Концентрация белка-мишени при экспрессии в клетках osTIR1 и добавлении ауксина снижалась вплоть до 3% от концентрации в клетках дикого типа. В свою очередь, замена среды на среду без ауксина приводила к полному восстановлению уровня интересующего белка. Кроме того, экспрессия osTIR1 никак не сказывается на клеточном росте. В этой же работе система с использованием AID показала свою эффективность при снижении содержания эндогенного белка CENP-H (Centromere protein H) в клетках DT40 курицы.
Разработанную систему в дальнейшем успешно использовали на модельных организмах: дрожжах (Schizosaccharomyces pombe [30]), малярийном плазмодии (Plasmodium falciparum [31]), токсоплазме (Toxoplasma gondii [32]), дрозофиле (Drosophila melanogaster [33]) и нематодах (Caenorhabditis elegans [34]). В 2012 г. была подтверждена эффективность AID-системы для контролируемой деградации эндогенных белков (как ядерных, так и цитоплазматических) в клетках млекопитающих [35].
Модификация системы AID-дегрона
Уменьшение длины AID-дегрона IAA17. Длина дегрона IAA17, предложенного в [29], составляет 228 а.к., что затрудняет его встраивание в клеточные белки. Кроме того, присоединение достаточного большого белка IAA17 к белку-мишени может препятствовать его правильному фолдингу и нарушать функциональность. Как сказано выше, белки семейства Aux/IAA, к которому относится IAA17, состоят из четырех доменов, однако только второй домен абсолютно необходим для узнавания комплексом SCFTIR1 и запуска деградации [20, 21]. Предприняты попытки определить минимальную длину AID, необходимую для успешной деградации целевого белка-мишени.
Проанализирована эффективность работы дегрона при укорачивании белка IAA17 до фрагментов 1–114 (115 а.к.), 31–114 (84 а.к.) и 71–114 (44 а.к., назван авторами AID*) [36]. Модификация выбранного в качестве мишени белка RAD53 любым из указанных укороченных дегронов вызывала деградацию этого белка в клетках дрожжей, экспрессирующих osTIR1, при добавлении ауксина. Более короткие последовательности из IAA17 не приводили к деградации RAD53. Однако оказалось, что белок RAD53, меченный любым из дегронов, серьезно деградирует даже в отсутствие ауксина, причем деградация была сильнее в случае укороченных дегронов. Проблему нежелательной ауксиннезависимой деградации RAD53 удалось решить только путем модификации укороченного дегрона AID* эпитопом 9myc. Поскольку белок RAD53 отвечает за правильное протекание репликации в клетках, снижение его уровня должно повышать чувствительность клеток к агентам, приводящим к сбою репликации, например, к гидроксимочевине и УФ-излучению. Оказалось, что только в случае RAD53AID*–9myc чувствительность клеток к обоим агентам в отсутствие ауксина была сравнимой с чувствительностью клеток дикого типа, а при добавлении ауксина клетки вели себя как клоны с делетированным RAD53. В результате проделанной работы сделан вывод, что последовательность AID*–9myc может использоваться для индукции ауксинзависимой деградации белков в культурах клеток [36].
Предложен и другой вариант укороченного дегрона – mini-AID 65–132 (68 а.к.) [37]. Присоединение нескольких конструкций mini-AID к белку Elg1 приводило к его деградации в клетках S. cerevisiae в присутствии ауксина. Интересно, что триплет такого мини-дегрона (3×mini-AID) вызывал даже более эффективную деградацию белка-мишени, чем полноразмерный дегрон IAA17. При этом не выявлено ауксиннезависимой деградации модифицированного дегроном белка Elg1. Возможно, этот нежелательный эффект, наблюдаемый в случае RAD53 [36], характерен не для всех белков, содержащих AID.
В 2016 г. укороченный дегрон впервые использовали для деградации белков в клетках млекопитающих [38]. Валидацию системы проводили на клетках НЕК 293Т, в которых дополнительно экспрессировали оsTIR1, в качестве белка-мишени использовали GFP, к которому присоединяли либо полноразмерный дегрон IAA17, либо его укороченный вариант – AID47 (63–109, 47 а.к.). Интересно, что в клетках оба дегрона спонтанно отщепляются от GFP, однако полноразмерный дегрон IAA17 отщепляется значительно быстрее, чем AID47, разработанный в [38]. Однако поскольку о подобном эффекте другие группы, использующие AID-систему, не сообщали, этот эффект сочли связанным исключительно с особенностями выбранной системы. Оказалось также, что скорость отщепления дегрона зависит от локализации белка. Клетки трансфицировали векторами, кодирующими GFP-AID47 и NLS-GFP-AID47. Отщепление AID47 происходило одинаково эффективно в обоих случаях, но NLS-GFP-AID47, локализующийся в ядре, расщеплялся быстрее. Тем не менее, далее AID47 успешно использовали для деградации белков NANOG, CHK1 (Human checkpoint kinase 1), p53 и NOTCH1 (Neurogenic locus notch homolog protein 1) в стволовых клетках млекопитающих. Модификация белков AID47 позволяла добиться практически полной их деградации уже через 20 мин после добавления ауксина, причем деградация была обратимой: через 2 ч после удаления ауксина из среды концентрация белка в клетках восстанавливалась до первоначального уровня.
Недавно был предложен еще более короткий вариант AID на основе белка IAA17 – smAID (74–115, 42 а.к.) [39]. Присоединение smAID к эндогенному белку Ku70 человека приводило к его деградации в клетках НЕК 293Т, экспрессирующих osTIR1. Однако в этом случае для полной деградации белка требовалось 2 ч после добавления ауксина, что может быть связано как с дополнительным уменьшением последовательности дегрона, так и с очень высоким содержанием белка Ku70 в клетках человека [40]. Надо отметить, что, как и в некоторых других исследованиях [36, 38], наблюдали ауксиннезависимую деградацию Ku70, хотя уровень ее был не очень значительным.
Подходы по предотвращению ауксиннезависимой деградации белков-мишеней, содержащих дегрон
Ауксиннезависимая деградация белков, модифицированных AID, может быть достаточно серьезной проблемой. Как сказано выше, TIR1 способен взаимодействовать с AID даже в отсутствие ауксина [19]. Несмотря на то, что это взаимодействие значительно слабее, чем при участии ауксина, оно достаточно для фоновой деградации белка. Так, в клеточных линиях HEK 293T, MCF-7 и DLD-1, экспрессирующих экзогенный TIR1, уровень AID-модифицированных белков даже в отсутствие ауксина составлял от 3 до 50% (в зависимости от белка) от уровня соответствующего белка в клетках дикого типа [41].
Один из вариантов решения данной проблемы – использование индуцируемых промоторов для контролируемой экспрессии гена белка TIR1. Так, уже в пионерской работе K. Nishimura и соавт. [29] белок TIR1 продуцировали с использованием индуцируемого галактозой промотора GAL. Однако для этого дрожжи необходимо выращивать на средах, содержащих галактозу в качестве основного ресурса углерода, что меняет метаболизм клеток, а потому может сказываться на результатах изучения роли тех или иных белков в клеточном метаболизме [42]. Для решения этой проблемы разработана система β-est AID, в которой синтез белка osTIR1 контролируется промотором Z4EVpr, не требующим кардинальных изменений в среде, в которой растут клетки [42]. В этой системе использовали сконструированный фактор транскрипции ATF (Artificial Transcription Factor), состоящий из вирусного активатора транскрипции VP16, рецептора эстрадиола и четырех доменов типа цинковый палец. При добавлении в среду β-эстрадиола ATF проникает в клеточное ядро и активирует транскрипцию гена osTIR, связываясь с промотором Z4EVpr. В этой системе концентрация белка-мишени DCP1 (mRNA-Decapping enzyme 1) после инкубации с β-эстрадиолом, а затем с ауксином снижается до 13% от его уровня в необработанных клетках. Кроме того, были подобраны оптимальные условия добавления индукторов (предварительная инкубация с β-эстрадиолом в течение 60 мин и последующая 15-минутная инкубация с ауксином).
Для борьбы с ауксиннезависимой деградацией используют также белки ARF, которые регулируют деградацию в клетках растений, связываясь с белками Aux/IAA, содержащими AID, и препятствуя взаимодействию белка TIR1 с дегроном в отсутствие ауксина [26]. Наиболее интересными оказались факторы ARF16 и ARF25 из O. sativa [41]. Трансфекция клеток, экспрессирующих модифицированный дегроном белок-мишень и osTIR1, плазмидой, кодирующей домен PB1 белков ARF16 или ARF25, приводила к частичному восстановлению уровня белка-мишени. Напомним, что домен PB1 взаимодействует с доменами III и IV белков Aux/IAA и за счет этого мешает взаимодействию TIR1 с доменом II этих белков и последующей ауксиннезависимой деградации [27]. Важно отметить, что домен PB1 белка ARF16 не только более эффективно препятствует деградации белка-мишени в отсутствие ауксина, но и ускоряет ее в присутствии ауксина. При экспрессии домена PB1 белка ARF16 количество белка-мишени спустя 1 ч после добавления ауксина снижается до уровня, недетектируемого вестерн-блотингом, в то время как полная деградация того же белка в отсутствие PB1-ARF16 не наступала даже спустя 3 ч после обработки ауксином. В этой же работе [27] показано, что для эффективного предотвращения фоновой ауксиннезависимой деградации необходимо сначала экспрессировать в клетках PB1-ARF16, а затем модифицировать белок-мишень AID и экспрессировать osTIR. Однако система с использованием PB1-ARF16 неприменима для укороченных AID, поскольку факторы ARF связываются с третьим и четвертым доменами полноразмерного AID [27, 28], а все применяемые укороченные дегроны не содержат этих доменов в своем составе.
Предложено и другое решение проблемы ауксиннезависимой деградации [43]. Можно заменить osTIR1 другим белком, содержащим домен F-box и эффективно связывающим AID, но при этом не вызывающим ауксиннезависимой деградации белка-мишени. В качестве такого белка выбрали AtAFB2 из A. thaliana, который, как и osTIR1, образует устойчивый комплекс с AID при 37°С. Установлено, что при использовании AtAFB2 вместо osTIR1 белки-мишени в гораздо меньшей степени подвержены ауксиннезависимой деградации. Подобран также новый укороченный дегрон (miniIAA7, 37–104, 67 а.к.), который лучше всего связывается с AtAFB2 и приводит к снижению концентрации белка-мишени уже спустя 1 ч после добавления ауксина. При этом неважно, на каком конце белка, C- или N-, расположен miniIAA7. Выяснилось, однако, что снижение уровня ядерных белков в присутствии AtAFB2 проходит менее эффективно, чем в случае osTIR1. Решить данную проблему предложено с помощью слабого сигнала ядерной локализации, который вводится на C-конец белка AtAFB2. Такая конструкция доступна в банке плазмид Addgene (#129717).
К преимуществам системы AtAFB2-miniIAA7 относятся ее применимость к различным линиям клеток, достаточно высокая скорость деградации (значительное снижение концентрации белка уже спустя 1 ч после добавления ауксина), отсутствие ауксиннезависимой деградации, а также возможность использовать укороченный AID [43].
Комбинирование AID-системы с другими системами регуляции экспрессии белков
Применение AID-системы не всегда позволяет добиться полной деградации исследуемого белка. Так, например, AID-модификация белков MCM10 и DPB11, деградация которых должна приводить к остановке роста клеток дрожжей, поскольку они участвуют в инициации репликации ДНК и критически важны для перехода клетки в S-фазу, не приводила к желаемому эффекту при добавлении ауксина [44]. Соответственно сделан вывод о том, что только AID-системы недостаточно для полной деградации этих белков: для инициации репликации хватало той доли белков, которая оставалась интактной. Для дополнительного снижения уровня белка-мишени систему 3×mini-AID [37] скомбинировали с системой TET-Off [45]. Полученную систему Tet-OFF-AID назвали iAID (improved auxin-inducible degron) [44].
TET-Off – это система регуляции экспрессии белка-мишени, состоящая из трех компонентов: оператора TetO (последовательность, расположенная до промотора гена белка-мишени), транскрипционного активатора tTA и репрессора TetR. В отсутствие тетрациклина или его аналога доксициклина tTA связан с TetO и тем самым активирует транскрипцию с расположенного далее промотора, добавление же антибиотика нарушает связывание активатора с оператором и способствует связыванию репрессора, что останавливает транскрипцию гена белка-мишени [45].
Разработанную систему iAID применили к белкам MCM10 и DPB11. С этой целью перед промоторами генов белков MCM10 и DPB11, модифицированных AID c N- или С-конца, расположили оператор TetO [44]. Клетки дрожжей с такими модифицированными генами трансфицировали плазмидой, содержащей последовательности tTA, TetR и osTIR1, а затем обрабатывали ауксином и тетрациклином. В ходе отбора клеток, в которых действие системы iAID настолько снижало уровень белков MCM10 и DPB11, что приводило к полной и необратимой остановке перехода клеток в S-фазу, выяснилось, что полное нарушение роста клеток происходило только при расположении дегрона на N-конце белка DPB11. Клеточные линии, содержащие либо модифицированный белок MCM10, либо модифицированный по С-концу белок DBP11, все-таки входили в S-фазу.
Другой подход к повышению эффективности деградации белков-мишеней предложен в работе [46], в которой исследовали влияние белка MCM4 на инициацию репликации ДНК в клетках дрожжей. Добавление ауксина приводило к снижению уровня белка, модифицированного полноразмерным AID, но рост клеток при этом не останавливался. По-видимому, причиной недостаточной эффективности системы могла быть низкая концентрация белка AtTIR1 в ядрах клеток или непрочный контакт между AtTIR1 и компонентом SCF-комплекса – белком SKP1. Чтобы определить истинную причину, сконструировали два вектора – один для экспрессии белка AtTIR1, слитого с сигналом ядерной локализации, а второй – AtTIR1, слитого с белком SKP1. Этими плазмидами трансфицировали клетки, в которых белок MCM4 был модифицирован AID, и наблюдали более эффективную остановку клеточного роста при использовании плазмиды SKP1-AtTIR1. Таким образом, основной причиной отсутствия нарушения репликации был слабый контакт между белками SCFTIR1-комплекса, приводящий к нарушению убиквитинирования и последующей деградации белка MCM4. Тем не менее, введение сигнала ядерной локализации увеличивало, пусть и менее эффективно, деградацию белка. Поэтому для экспрессии белка TIR1 было решено использовать конструкцию SKP1-AtTIR1-NLS, чтобы повысить эффективность работы AID, скомбинировав два подхода: усилить взаимодействие между SKP1 и AtTIR1 и увеличить концентрацию AtTIR1 в ядре. Эта система, протестированная на нескольких десятках белков, в большинстве случаев оказалась эффективной, но в случае белка MCM10 и еще нескольких она не позволяла снижать концентрацию мишени настолько, чтобы это сказывалось на клеточном росте. Предположили, что эта проблема может быть обусловлена синтезом нового белка в период активации деградации. Поэтому систему решили доработать, заменив эндогенный промотор генов исследуемых белков-мишеней на промотор Pnmt81, репрессируемый тиамином. Добавление тиамина приводит к остановке транскрипции, и введение ауксина запускает деградацию уже синтезированного белка. В такой системе концентрация исследуемых белков снижалась до уровня, не детектируемого Вестерн-блотингом, и рост клеток прекращался, что свидетельствует об эффективности полученной системы, названной off-AID [46].
ДРУГИЕ СИСТЕМЫ ДЛЯ КОНТРОЛИРУЕМОЙ ДЕГРАДАЦИИ БЕЛКОВ
Система с использованием дегрона JAZ
Как уже отмечено, помимо ауксинов в процессе индуцируемой гормонами деградации белков в растениях принимает участие JA-Ile (рис. 4а) [18, 22, 47]. Сигнальный путь с участием JA-Ile включает убиквитинирование специфических белков-мишеней комплексом SCFCOI1 и их последующую деградацию 26S протеасомой. В данном случае в состав E3 убиквитин-лигазного комплекса SCF в качестве белка, содержащего домен F-box, входит COI1. Мишенью комплекса SCFCOI1 служат так называемые JAZ-белки (jasmonate ZIM-domain proteins) [22, 48]. Все JAZ-белки содержат центральный высококонсервативный ZIM-домен и С-концевую консенсусную последовательность SLX2FX2KRX2RX5PY, называемую Jas-мотив. Показано, что именно Jas-мотив важен для связывания JAZ-белков с доменом F-box белка COI1, причем замена всего двух аминокислот – R205 и R206, в Jas-мотиве белка JAZ1 нарушает это взаимодействие [49]. Действием, аналогичным действию JA-Ile, обладает фитотоксин коронатин, продуцируемый патогенной бактерией Pseudomonas syringae [50], который, как и JA-Ile, стимулирует связывание JAZ-белков с COI1 [49]. Коронатин обладает структурным сходством с JA-Ile (рис. 4б), оба эти соединения связываются в одном структурном кармане белка COI1 и их связывание стабилизируется в присутствии Jas-мотива JAZ-белков. Подчеркнем, что в отличие от ауксина, который взаимодействует только с доменом F-box белка TIR1, коронатин и JA-Ile связываются уже с комплексом COI1/JAZ. Причем для образования стабильного комплекса JA-Ile(коронатин)/JAZ/COI1 достаточно пептида, соответствующего последовательности (Glu200–Val220) из Jas-мотива белка JAZ1 [51].
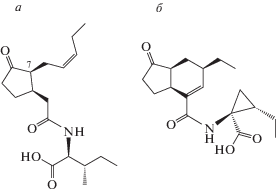
Показано, что JA-Ile-зависимое взаимодействие белков COI1 и JAZ1 возможно и в клетках дрожжей, т.е. для него не нужны другие растительные белки [49]. Тем не менее, эта система не нашла столь широкого применения для направленной деградации белков в нерастительных клетках, как система AID. Cравнение двух систем показало, что система JAZ-дегрона может использоваться для направленной деградации белков в ядрах клеток млекопитающих под действием коронатина [38]. Однако для этого необходимо модифицировать белок-мишень 33-звенным пептидом из JAZ-дегрона риса (OsJAZ33), а в качестве белка F-box использовать химерный белок оsTIR1F-box-оsCOI1BLRR. Таким образом, эта система оказалась более сложной в использовании, чем AID-система.
Система dTAG
Система dTAG (degradation tag) деградации белка-мишени также основана на процессе убиквитинирования, но при этом использует механизм инициации убиквитинирования, существующий в клетках животных [52]. Помимо SCF-комплекса, функции лигазы E3 способен исполнять белковый комплекс, состоящий из цереблона (CRBN), куллина-4А (CUL4A), белка DDB1 (DNA Damage-Binding Protein), связывающего поврежденную ДНК, и белка RBX1/ROC1 (RING-box protein 1/regulator of cullins 1), содержащего домен RING-box для связывания с куллином [53]. Для привлечения этого комплекса к белку-мишени разработана специальная метка, представляющая собой белок FKBP12F36V, полученный методами белковой инженерии из FKBP12. Белки семейства FKBP (FK506 Вinding Рrotein) обладают пролилизомеразной активностью и выполняют функцию шаперонов, способствуя правильному фолдингу пролинсодержащих белков. Кроме того, получен синтетический низкомолекулярный лиганд dTAG-13 (рис. 5а), с которым одновременно могут связаться FKBP12F36V и белок цереблон. Предполагалось, что добавление dTAG-13 к культуре клеток, в которых белок-мишень слит с белком FKBP12F36V, должно способствовать связыванию с данным белком E3 лигазы, в состав которой входит цереблон, и убиквитинированию мишени с последующей деградацией (рис. 5б). Предварительно показано, что в присутствии dTAG-13 мутант FKBP12F36V деградирует, а FKBP12 дикого типа деградации не подвергается.
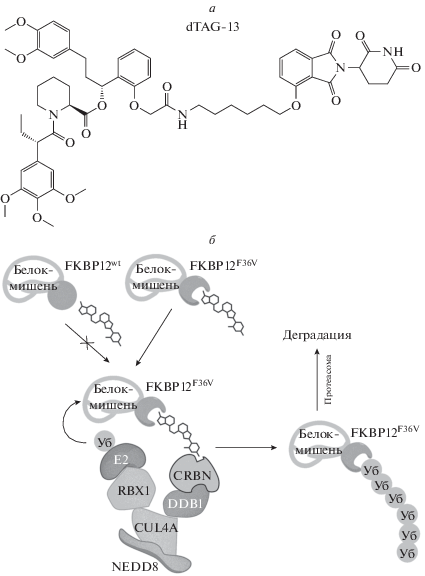
Систему dTAG протестировали на культуре клеток HEK 293T и показали, что деградация бромодоменсодержащего белка 4 (BRD4), слитого c белком FKBP12F36V, наблюдалась уже через 1 ч после добавления 500 нМ dTAG-13. Кроме того, эта система проверена на нескольких десятках белков в линии клеток MV4-11 миелоидного лейкоза. Деградация всех исследуемых белков происходила через 2–4 ч, причем в некоторых случаях ее эффективность была достаточной для подавления клеточного роста [52].
К преимуществам системы dTAG по сравнению с AID-системой относится отсутствие необходимости экспрессии в животных клетках компонентов растительной убиквитинлигазы, таких как белок TIR1, поскольку система dTAG изначально основана на процессе убиквитинирования в клетках животных. Важно, что эффективность системы dTAG показана и на мышах, на которых AID-систему не применяли. В костный мозг мышей вводили клетки, экспрессирующие люциферазу, слитую с белком FKBP12F36V (luc-FKBP12F36V), и уже через 4 ч после введения dTAG-13 наблюдали значительное снижение уровня биолюминесценции, что указывает на эффективное разрушение белка luc-FKBP12F36V. Через 28 ч после обработки dTAG-13 биолюминесценция восстанавливалась до уровня в контроле [52].
Системы HaloTag и HaloPROTAC
Система HaloTag отличается от описанных выше тем, что в ней используется принцип убиквитиннезависимой деградации белков. HaloTag-метка была разработана для визуализации белков-мишеней в клетке, их иммобилизации, а также выявления клеточных партнеров [54]. Эта метка представляет собой модифицированную галоалкандегалогеназу, способную реагировать с синтетическим лигандом, содержащим хлоралкильный фрагмент, присоединенный к флуоресцентным красителям, биотину, аффинным меткам и т.д. Образование ковалентной связи между белком и хлоралкильным фрагментом происходит быстро и в физиологических условиях, приводя в результате к образованию белка, содержащего необходимую метку (HaloTag). Использовать HaloTag для деградации белков предложили в работе [55]. Предположили, что модификация поверхности белка-мишени HaloTag-меткой, содержащей гидрофобный лиганд, может привести к его деградации, поскольку клетка будет воспринимать белок, модифицированный такой гидрофобной меткой, как неправильно свернутый и подлежащий уничтожению [56, 57] (рис. 6а). И действительно, 5 из 30 синтезированных гидрофобных лигандов [55] вызывали разрушение модельной мишени (люциферазы, слитой с HaloTag (Luc-HaloTag), экспрессированной в клетках НЕК 293Т), причем наиболее эффективным оказался лиганд НуТ13 (рис. 6б). Изучение кинетики деградации Luc-HaloTag показало, что белок не детектировался в клетках через 8 ч после добавления к ним 100 нМ лиганда НуТ13.
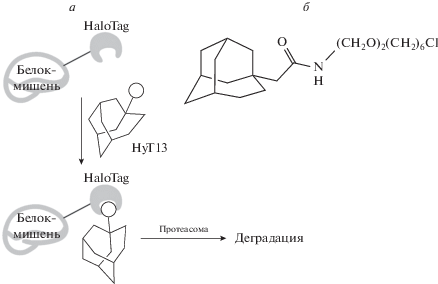
Систему HaloTag:HyT13 также успешно применяли для деградации мембранных белков, белков эмбрионов Danio rerio, а также мутантного белка H-RasG12V в злокачественной опухоли, привитой мышам. Показано, что лиганд НуТ13 может эффективно проникать в клетки и ткани организма и направлять меченные HaloTag белки на деградацию [55]. Позднее систему HaloTag с другими лигандами успешно использовали для направленной деградации белков CREB1 (responsive element binding protein 1) и c-Jun в культурах клеток [58].
В дальнейшем в системе HaloTag предложили использовать молекулы PROTAC (PROteolysis TArgeting Chimera) – бифункциональные низкомолекулярные лиганды, которые могут рекрутировать определенные E3 лигазы к нужному белку (подробно система PROTAC будет рассмотрена ниже) [59]. Синтезирована серия хлоралкилсодержащих лигандов PROTAC (HaloPROTAC), способных связываться с усовершенствованной версией HaloTag – HaloTag7 и опухолевым супрессором VHL, который в комплексе с элонгинами C и B и куллином выполняет роль E3 лигазы и способен активировать деградацию белка путем его убиквитинирования. Таким образом, полученные лиганды, связываясь одновременно с белками, меченными HaloTag7, и VHL, могут активировать деградацию меченых белков (рис. 7а). Самым эффективным оказался HaloPROTAC3 (рис. 7б), добавление которого к клеткам НЕК 293Т, экспрессирующим модельный белок-мишень GFP-HaloТag7, приводило к снижению его концентрации на 90% от начального уровня.
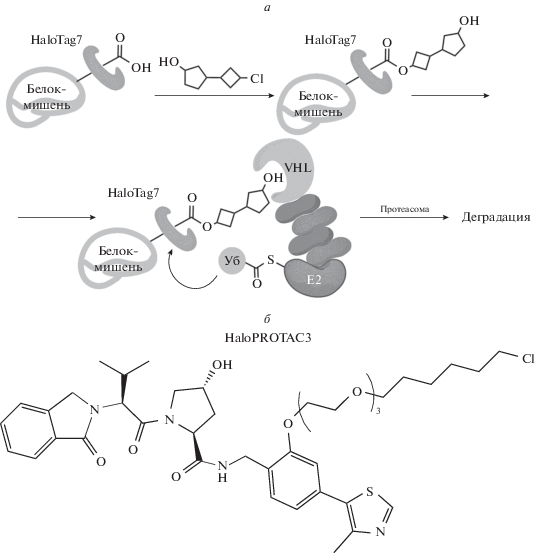
Сравнение уровня деградации белка GFP-HaloТag7 под действием HaloPROTAC3 и HyT36 (триплет наиболее эффективного гидрофобного лиганда HyT13 [55]) показало более высокую эффективность системы HaloPROTAC (при концентрации HaloPROTAC3 19 ± 1 нM максимальная степень деградации равна 90 ± 1%, а при концентрации HyT36 134 ± 7 нM – 56 ± 1%) [59].
Систему HaloPROTACs успешно применили для деградации эндогенных белков SGK3 и VPS34, модифицированных меткой HaloTag7, с помощью технологии CRISPR/Cas9. В этом случае использовали усовершенствованную молекулу HaloPROTAC-E – хлоралкилсодержащий высокоаффинный лиганд белка VHL (VH298), вызывающий ∼50%-ную деградацию белка SGK3, меченного HaloTag7, уже за 30 мин и ∼95%-ную деградацию через 48 ч [60].
Некоторые компании (“Promega”, “GeneCopoeia”) предлагают коммерческие наборы и векторы, позволяющие быстро и эффективно вводить метку HaloTag на C- и N-концы различных белков-мишеней. Однако стоит учитывать, что любой хлоралкилсодержащий лиганд ковалентно присоединяется к метке HaloTag, а значит, деградацию модифицированного им белка невозможно остановить, просто удалив этот лиганд из среды, как в случае деградации, индуцируемой ауксином. Именно по этой причине появление белков-мишеней детектируют не раньше, чем через 24 ч после удаления лигандов, когда в клетках успевают синтезироваться новые порции этих белков [55, 59]. Кроме того, как в системе HaloTag, так и HaloPROTAC к эндогенным белкам для их деградации необходимо присоединить достаточно большой белок (FKBP12F36V или HaloTag), длина которого значительно превышает длину укороченных вариантов AID, и который может повлиять на фолдинг и функциональность исследуемых белков-мишеней.
Система PROTAC
Как отмечено выше, при разработке системы HaloTag использовали молекулы PROTAC (PROteolysis TArgeting Chimera), которые состоят из лиганда (чаще всего низкомолекулярного ингибитора) белка-мишени и лиганда убиквитинлигазы E3, которые ковалентно соединены линкером длиной 5–15 атомов. В результате связывания PROTAC одновременно с белком-мишенью и убиквитинлигазой E3 эти два белка сближаются, что обеспечивает убиквитинирование мишени и ее последующую деградацию 26S протеасомой [61]. Систему PROTAC впервые предложили в работе [62], в которой показана возможность направленного убиквитинирования метионин-аминопептидазы-2 (MetAP-2) комплексом SCF (Skp1-Cullin-F-box), содержащим белок Hrt1 (RING-H2 protein Hrt1). Белок MetAP-2 способен ковалентно связывать ингибитор ангиогенеза овалицин, поэтому синтезировали бифункциональное соединение Protac-1, один домен которого состоял из овалицина, а другой содержал фосфопептид IkBa, распознаваемый F-box-белком b-TRCP. Показано, что молекула Protac-1 успешно связывает белки SCF/b-TRCP и MetAP-2, что приводит к убиквитинированию и протеасомной деградации последнего. При этом деградации подвергается только белок MetAP-2, содержащий остаток Protac-1.
Значительным прогрессом в развитии технологии PROTAC стало создание в 2008 г. первого бифункционального соединения, содержащего в качестве обоих лигандов (мишени и убиквитинлигазы Е3) только низкомолекулярные соединения [63]. Новая молекула PROTAC состояла из лиганда рецептора андрогенов (SARM) и нутлина – лиганда белка MDM2 (mouse double minute 2 homolog), связанных линкером на основе полиэтиленгликоля. В этой системе белок MDM2 функционирует как убиквитинлигаза E3. Новый PROTAC эффективно привлекал рецептор андрогенов к белку MDM2, что приводило к убиквитинированию рецептора и его последующей деградации протеасомой.
К настоящему времени разработаны молекулы PROTAC, в которых используются низкомолекулярные лиганды не только MDM2, но и таких убиквитинлигаз Е3, как онкосупрессор VHL (Von Hippel-Lindau) [64], белки-ингибиторы апоптоза IAP (inhibitor of apoptosis proteins) [65], цереблон [66]. Технология PROTAC активно развивается: разрабатываются новые лиганды белков-мишеней разного типа, оптимизируется структура линкеров, определяются условия, позволяющие наиболее эффективно формировать тройной комплекс белка-мишени/PROTAC/убиквитинлигазы в различных клетках и животных моделях. Для оптимизации структуры PROTAC активно используют данные рентгеноструктурного анализа комплексов белков с их лигандами [67, 68], а также компьютерный докинг [69].
Первый положительный эффект от использования молекул PROTAC in vivo получили в 2013 г., когда с помощью контролируемой деградации меченных PROTAC белков удалось подавить развитие раковой опухоли в мышиной модели [70]. С тех пор получены многочисленные данные об успешном использовании in vitro и in vivo молекул PROTAC, нацеленных на различные мишени, причем надо особо отметить, что технология PROTAC оказалась особенно удобным инструментом разрушения белков, вовлеченных в развитие онкологических заболеваний [71, 72]. К настоящему времени удалось создать молекулы PROTAC, способные не только эффективно индуцировать протеолиз мишеней, но и обладающие хорошей фармакокинетикой и фармакодинамикой. В результате две молекулы проходят сейчас клинические испытания в качестве противоопухолевых препаратов [72, 73]. Необходимо отметить некоторые достаточно серьезные преимущества молекул PROTAC по сравнению с традиционными низкомолекулярными ингибиторами ферментов, вовлеченных в развитие онкологических заболеваний. Во-первых, молекула PROTAC способна катализировать деградацию нескольких молекул белка-мишени. Благодаря такому каталитическому механизму действия, для достижения желаемого фармакологического эффекта требуются значительно более низкие концентрации PROTAC, чем обычных ингибиторов. Еще одно преимущество PROTAC состоит в том, что они могут использоваться для преодоления лекарственной устойчивости, возникающей из-за мутаций в белках-мишенях в результате стандартной химиотерапии [74]. Более широкое распространение этой технологии ограничивается главным образом тем, что для каждого нового белка-мишени необходимо подбирать свой лиганд. Кроме того, содержание убиквитинлигаз Е3 может сильно варьировать в разных клетках, что необходимо учитывать при создании молекул PROTAC, нацеленных на деградацию определенного белка в клетках определенного типа.
Система SMASh
Еще одна система, позволяющая осуществлять контролируемую деградацию белков, названа SMASh (Small Molecule-Assisted Shutoff) [75]. Эта система, как и система HaloTag, использует принцип убиквитиннезависимой деградации белков. Суть ее состоит в том, что к белку-мишени присоединяется дегрон, который в отсутствие соответствующего ингибитора сам отщепляет себя, оставляя в клетке интактный белок. В качестве такого дегрона использован протеазный домен NS3pro неструктурного белка 3 (NS3) вируса гепатита С, слитый с белком NS4A этого же вируса (NS3pro-NS4A). Если в среде отсутствуют ингибиторы протеазной активности NS3, то модуль NS3pro-NS4A сразу по окончании трансляции отщепляется от белка-мишени и подвергается деградации, а сам белок-мишень остается в интактном состоянии (рис. 8). В присутствии же любого ингибитора сериновой протеазы NS3, например асунапревира, отщепление блокируется, и исследуемый белок, слитый с дегроном NS3pro-NS4A, подвергается деградации.
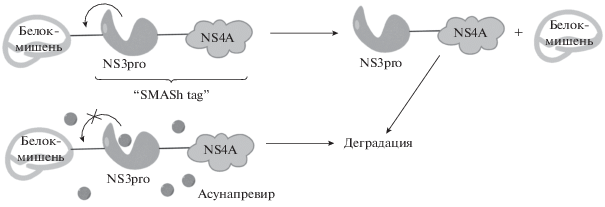
Для объяснения такого необычного свойства дегрона NS3pro-NS4A необходимо обратиться к функциональной роли белка NS4A в репликации вируса гепатита C. В ходе репликации синтезируемый полипептид NS3-4A расщепляется на два отдельных белка благодаря активности протеазного домена NS3. В результате N-конец белка NS4A формирует гидрофобную α-спираль, которая встраивается в мембрану эндоплазматического ретикулума. Позиционирование полипептида NS3-4A в активном центре протеазы NS3 обеспечивается за счет хеликазного домена NS3 [76]. Однако в конструкции NS3pro-NS4A, использованной в системе SMASh, отсутствует хеликазный домен NS3, поэтому NS3pro-NS4A не расщепляется, и свободный N-конец NS4A не образуется. Из-за этого NS4A не может встроиться в мембрану, при этом его гидрофобная структура распознается клетками как дегрон и уничтожается вместе со слитым с ним белком.
Доработка системы SMASh позволила модифицировать белки-мишени как с N-, так и с C‑конца, а также повысить эффективность отщепления дегрона в отсутствие асунапpевира. При использовании в качестве мишени желтого флуоресцентного белка (YFP), слитого с дегроном NS3pro-NS4A, 50%-ная эффективная концентрация (ЕС50) асунапревира, измеренная по уменьшению флуоресценции YFP, составляла приблизительно 1 нМ, что сравнимо с его ЕС50 в тестах по ингибированию репликона вируса. Этот результат показывает, что связывание асунапревира с протеазой NS3 не изменяется в конструкции SMASh. Примечательно, что YFP не детектировался при концентрации асунапревира 1.5 мкМ [75], при которой он не проявляет активности против клеточных протеаз и не обладает цитотоксичностью [77]. Кроме того, полученная система деградации белка действительно обратима: уже спустя 1 ч после удаления асунапревира из среды концентрация белка-мишени восстанавливалась до исходного уровня. Система показала свою эффективность в снижении уровня белка-мишени как в животных клетках (HEK 293T, клетки кортико-гиппокампальной системы крыс и кортикальные нейроны мышей), так и в клетках дрожжей [75].
Преимущество системы SMASh перед системой AID заключается в отсутствии необходимости продуцировать дополнительные белки. Система лишена и такого недостатка, как описанная индукторнезависимая деградация, которая часто наблюдается в случае AID. К преимуществам этой системы относится и то, что метка NS3pro-NS4A удаляется сразу после синтеза белка в отсутствие блокаторов протеаз, и никак не влияет на функции и фолдинг белка-мишени. Возможно, именно по этим причинам систему SMASh начали активно использовать для направленной деградации вирусных и клеточных белков [78–80]. Тем не менее, возможности этой системы, по-видимому, ограничены активно экспрессируемыми белками. Если исходить из механизма деградации белков с меткой NS3pro-NS4A, то становится понятным, что белок можно разрушить, только если он синтезируется в присутствии ингибитора протеазы NS3pro, если же трансляция уже завершилась и дегрон отщепился, то добавление ингибитора не приведет к разрушению белка.
Оптогенетический подход к деградации белков
Все описанные выше методы основаны на использовании химических соединений, индуцирующих деградацию соответствующим образом меченых белков (ауксин, dTAG, PROTAC) или, наоборот, препятствующих ей (асунапревир). Принципиально иной подход основан на индукции деградации белка, содержащего специально сконструированный светочувствительный домен, при облучении светом определенной длины волны. Этот подход назван оптогенетическим по аналогии с методом изучения работы нервных клеток, основанным на внедрении в их мембрану белков-опсинов, реагирующих на возбуждение светом [81]. В большинстве работ в качестве светочувствительного домена используется домен LOV (Light-Oxygen-Voltage) из фоторецепторных белков растений [82–86]. Этот домен состоит из корового домена, связывающего FMN, и Ja-спирали на С-конце, которая в темноте ассоциирована с коровым доменом. При облучении синим светом остаток цистеина из корового домена образует ковалентный аддукт с возбужденным FMN. Это вызывает конформационные изменения в коре, за которыми следует диссоциация и разворачивание Ja-спирали [87]. Если к С-концу Ja-спирали присоединен какой-либо дегрон, то весь домен LOV может подвергаться протеасомной деградации. Если же домен LOV присоединен к исследуемому белку-мишени, то облучение синим светом вызывает деградацию этого белка (рис. 9).
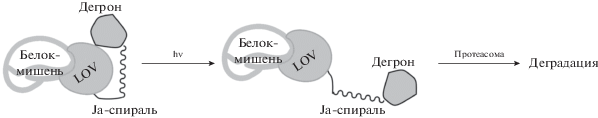
В первой работе, в которой предложено использовать оптогенетический подход к деградации белков, в качестве дегрона была выбрана последовательность орнитиндекарбоксилазы (ODC) мыши [82]. Этот дегрон индуцирует убиквитиннезависимую протеасомную деградацию, если он присоединен к С-концу белка. Он состоит из неструктурированного пептида длиной 37 а.к., содержащего цистеин-аланиновый мотив, который участвует в распознавании протеасомой и поэтому важен для деградации [88]. Структурно-функциональные исследования показали, что для эффективной деградации красного флуоресцентного белка (RFP), к С-концу которого присоединен домен LOV с ODC-дегроном, достаточно дегрона из 23 а.к. [82]. В качестве домена LOV использовали домен LOV2 фототропина A. thaliana (AtLOV2). Изучение кинетики деградации белка RFP-AtLOV2-ODC(23) показало, что после 2 ч облучения синим светом (λ = 470 нм) в клетках остается не более 10% белка, а время полудеградации составляет 38 мин. После помещения клеток в темноту белок RFP-AtLOV2-ODC(23) появлялся через 1 ч. Такой же подход с использованием AtLOV2-ODC-домена впоследствии применяли для деградации других белков [84, 86].
Предложен и другой модуль [83], обеспечивающий деградацию белка при облучении синим светом, – домен LOV2 из фототропина 1 Avena sativa (AsLOV2), к С-концу которого присоединен пептид RRRG, вызывающий протеасомную деградацию белков в клетках млекопитающих [89]. Этот модуль назван доменом B-LID (Blue-Light Inducible Degradation). Домен B-LID использовали и в комбинированном подходе, который позволяет при облучении клеток синим светом (λ = = 460 нм) одновременно подавлять активность промотора гена, кодирующего целевой белок, и вызывать протеасомную деградацию этого белка [85]. Созданная система, названная Blue-OFF, состояла из репрессора KRAB-EL222 и модуля B‑LID, обеспечивающего деградацию белка. Оба компонента содержали светочувствительные домены LOV. Облучение способствовало димеризации и связыванию фоточувствительного транскрипционного фактора EL222 со специфической последовательностью ДНК; в результате транскрипция с промотора прекращалась под действием репрессорного домена KRAB (Krüppel associated box), входящего в состав модуля KRAB-EL222. Эффективность системы Blue-OFF исследована с использованием люциферазы светлячка (firefly luciferase, FLuc) в качестве белка-мишени. Работу регулятора транскрипции KRAB-EL222 анализировали, используя ген люциферазы под контролем промотора SV40, за которым располагалась последовательность (C120)5, связывающая KRAB-EL222. К С‑концу люциферазы с целью контроля эффективности фотоиндуцируемой деградации присоединен модуль B-LID. После облучения клеток HEK-293T, экспрессирующих люциферазу FLuc-B-LID, светом с длиной волны 460 нм в течение 8 ч уровень люциферазы снижался до 10% от исходного при наличии обоих модулей (KRAB-EL222 и B-LID) и только на 50% в присутствии одного из них. Таким образом показана эффективность работы системы Blue-OFF. Следует добавить, что уровень FLuc-B-LID, сниженный в результате облучения, восстанавливался, когда клетки помещали на 12 ч в темноту. Аналогичные результаты получены и на других клеточных линиях (HeLa, CHO-K1, NIH/3T3 и COS-7).
Понятно, что оптогенетический подход к контролируемой деградации белков имеет и свои преимущества, и свои недостатки по сравнению с методами, использующими химические индукторы. То, что индукция деградации белка, содержащего домен LOV, происходит под действием света и не требует добавления к клеткам химического агента, который потенциально может влиять на их метаболизм, является несомненным преимуществом этого подхода. Однако использование именно синего света ограничивает применение оптогенетического подхода в основном клеточными культурами. Применение этого подхода in vivo достаточно проблематично, поскольку синий свет не способен проникать через кожные покровы и, более того, вызывает окислительный стресс [90]. Дополнительно отметим, что все разработанные к настоящему времени варианты оптогенетического подхода используют убиквитиннезависимую деградацию белков-мишеней.
ЗАКЛЮЧЕНИЕ
Разработанные в последние годы методы воздействия на геном или мРНК позволяют определить роль тех или иных клеточных компонентов и, в первую очередь, белков, в различных клеточных процессах. Однако в некоторых случаях более информативным или удобным представляется воздействие непосредственно на экспрессированный белок, причем в идеале это воздействие должно быть контролируемым во времени и обратимым. В данном обзоре мы рассмотрели основные подходы к контролируемой деградации белков в клетке и постарались оценить их достоинства и недостатки. Некоторые из этих подходов (dTAG, HaloTag) показали свою эффективность не только в культурах клеток, но и на животных моделях. Несомненно, перспективен подход с использованием молекул PROTAC, который позволил вплотную подойти к созданию нового типа противоопухолевых препаратов. Два препарата PROTAC проходят в настоящее время клинические испытания. Мы, однако, не претендуем на охват всех существующих систем контролируемой деградации белков, поскольку эта область активно развивается и постоянно появляются новые системы, позволяющие включать–выключать деградацию исследуемых белков.
Работа поддержана Российским научным фондом (грант № 17-14-01107-П).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов
Список литературы
Glickman M.H., Ciechanover A. (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82, 373–428.
Wang X., Herr R.A., Chua W.-J., Lybarger L., Wiertz E.J.H.J., Hansen T.H. (2007) Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell Biol. 177, 613–624.
Tokarev A.A., Munguia J., Guatelli J.C. (2011) Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J. Virol. 85, 51–63.
Thrower J.S., Hoffman L., Rechsteiner M., Pickart C.M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102.
Kravtsova-Ivantsiv Y., Ciechanover. A. (2012) Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 125, 539—548.
Boutet S. C., Disatnik M. H., Chan L. S., lori K., Lando T.A. (2007) Regulation of Pax3 by proteasomal degradation of monoubiquitinated protein in skeletal muscle progenitors. Cell. 130, 349–362.
Kravtsova-Ivantsiv Y., Cohen S., Ciechanover A. (2009) Modification by single ubiquitin moieties rather than polyubiquitination is sufficient for proteasomal processing of the p105 NF-kappaB precursor. Mol. Cell. 33, 496–504.
Komander D. (2009) The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 37, 937–953.
Ikeda F., Dikic I. (2008) Atypical ubiquitin chains: new molecular signals. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 9, 536–542
Бунеева О.А., Медведев А.Е. (2016) Роль атипичного убиквитинирования в клеточной регуляции. Биомед. химия. 62, 496–509.
Meyer H.J., Rape M. (2014) Enhanced protein degradation by branched ubiquitin chains. Cell. 157, 910–921.
Ohtake F., Tsuchiya H., Saeki Y., Tanaka K. (2018) K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc. Natl. Acad. Sci. USA. 115, E1401–E1408.
Yau R.G., Doerner K., Castellanos E.R., Haakonsen D.L., Werner A., Wang N., Yang X.W., Martinez-Martin N., Matsumoto M.L., Dixit V.M., Rape M. (2017) Assembly and function of heterotypic ubiquitin chains in cell-cycle and protein quality control. Cell. 171, 918–933.
Pickart C.M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533.
Fasanaro P., Capogrossi M.C., Martelli F. (2010) Regulation of the endothelial cell cycle by the ubiquitin-proteasome system. Cardiovasc. Res. 85, 272–280.
Craig K.L., Tyers M. (1999) The F-box: a new motif for ubiquitin dependent proteolysis in cell cycle regulation and signal transduction. Prog. Biophys. Mol. Biol. 72, 299–328.
Pearce M.J., Mintseris J., Ferreyra J., Gygi S.P., Darwin K.H. (2008) Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science. 322, 1104–1107.
Sheard L.B., Tan X., Mao H., Withers J., Ben-Nissan G., Hinds T.R., Kobayashi Y., Hsu F., Sharon M., Brow-se J., He S.Y., Rizo J., Howe G.A., Zheng N. (2010) Jasmonate perception by inositol-phosphate-potentiated COI1-JAZ co-receptor. Nature. 468, 400–405.
Tan X., Calderon-Villalobos L.I.A., Sharon M., Zheng C., Robinson C.V., Estelle M., Zheng N. (2007) Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature. 446, 640–645.
Kepinski S., Leyser O. (2005) The Arabidopsis F-box protein TIR1 is an auxin receptor. Nature. 435, 446–451.
Dharmasiril N., Dharmasiril S., Estelle M. (2005) The F-box protein TIR1 is an auxin receptor. Nature. 435, 441–445.
Thines B., Katsir L., Melotto M., Niu Y., Mandaokar A., Liu G., Nomura K., He S.Y., Howe G.A., Browse J. (2007) JAZ repressor proteins are targets of the SCF(COI1) complex during jasmonate signalling. Nature. 448, 661–665.
Staswick P.E. (2008) JAZing up jasmonate signaling. Trends Plant Sci. 13, 66–71.
Teale W.D., Paponov I.A., Palme K. (2006) Auxin in action: signalling, transport and the control of plant growth and development. Nat. Rev. Mol. Cell. Biol. 7, 847–859.
Gray W.M., Kepinski S., Rouse D., Leyser O., Estelle M. (2001) Auxin regulates SCF(TIR1)-dependent degradation of AUX/IAA proteins. Nature. 414, 271–276.
Ulmasov T., Hagen G., Guilfoyle T.J. (1997) ARF1, a transcription factor that binds to auxin response elements. Science. 276, 1865–1868.
Ulmasov T., Murfett J., Hagen G., Guilfoyle T.J. (1997) Aux/IAA proteins repress expression of reporter genes containing natural and highly active synthetic auxin response elements. Plant Cell. 9, 1963–1971.
Ulmasov T., Hagen G., Guilfoyle T.J. (1999) Dimerization and DNA binding of auxin response factors. Plant J. 19, 309–319.
Nishimura K., Fukagawa T., Takisawa H., Kakimoto T., Kanemaki M. (2009) An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods. 6, 917–923.
Kanke M., Nishimura K., Kanemaki M., Kakimoto T., Takahashi T.S., Nakagawa T., Masukata H. (2011) Auxin-inducible protein depletion system in fission yeast. BMC Cell Biol. 12, 1–16.
Kreidenweiss A., Hopkins A.V., Mordmüller B. (2013) 2A and the auxin-based degron system facilitate control of protein levels in Plasmodium falciparum. PLoS One. 8, 1–6.
Brown K.M., Long S., Sibley D.L. (2018) Conditional knockdown of proteins using auxin-inducible degron (AID) fusions in Toxoplasma gondii. Bio Protoc. 8, e2728.
Trost M., Blattner A.C., Lehner C.F. (2016) Regulated protein depletion by the auxin-inducible degradation system in Drosophila melanogaster. Fly. 10, 35–46.
Zhang L., Ward J.D., Cheng Z., Dernburg A.F. (2015) The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development. 142, 4374–4384.
Holland A.J., Fachinetti D., Han J.S., Cleveland D.W. (2012) Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl. Acad. Sci. USA. 109, E3350–E3357.
Morawska M., Ulrich H.D. (2013) An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast. 30, 341–351.
Kubota T. (2013) The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell. 50, 1–8.
Brosh R., Hrynyk I., Shen J., Waghray A., Zheng N., Lemischka I.R. (2016) A dual molecular analogue tuner for dissecting protein function in mammalian cells. Nat. Commun. 7, 1–13.
Zotova A., Pichugin A., Atemasova A., Knyazhans-kaya E., Lopatukhina E., Mitkin N., Holmuhamedov E., Gottikh M., Kuprash D., Filatov A., Mazurov D. (2019) Isolation of gene-edited cells via knock-in of short glycophosphatidylinositol-anchored epitope tags. Sci. Rep. 9, 1–10.
Fell V.L., Schild-Poulter C. (2015) The Ku heterodimer: function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 763, 15–29.
Sathyan K.M., McKenna B.D., Anderson W.D., Duarte F.M., Core L., Guertin M.J. (2019) An improved auxin-inducible degron system preserves native protein levels and enables rapid and specific protein depletion. Genes Dev. 33, 1441–1455.
Barrass J.D., Mendoza-Ochoa G.I., Maudlin I.E., Sani E., Beggs J.D. (2019) Tuning degradation to achieve specific and efficient protein depletion. J. Vis. Exp. 149, 1–18.
Li S., Prasanna X., Salo V.T., Vattulainen I., Ikonen E. (2019) An efficient auxin-inducible degron system with low basal degradation in human cells. Nat. Meth. 16, 866–869.
Tanaka S., Miyazawa-Onami M., Iida T., Araki H. (2015) iAID: an improved auxin- inducible degron system for the construction of a ‘tight’ conditional mutant in the budding yeast Saccharomyces cerevisiae. Yeast. 32, 567–581.
Belli G., Gari E., Piedrafita L., Aldea M., Herrero E. (1998) An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucl. Acids Res. 26, 942–947.
Natsume T., Kiyomitsu T., Saga Y., Kanemaki M.T. (2016) Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donor. Cell Repts. 15, 210–218.
Turner J.G., Ellis C., Devoto A. (2002) The jasmonate signal pathway. Plant Cell. 14, S153–S164.
Shikata M., Matsuda Y., Ando K., Nishii A., Takemura M., Yokota F., Kohchi T. (2004) Characterization of Arabidopsis ZIM, a member of a novel plant specific GATA factor gene family. J. Exp. Bot. 55, 631–639.
Melotto M., Mecey C., Niu Y., Chung H.S., Katsir L., Yao J., Zeng W., Thines B., Staswick P., Browse J., Howe G.A., He S.Y. (2008) A critical role of two positively charged amino acids in the Jas motif of Arabidopsis JAZ proteins in mediating coronatine- and jasmonoyl isoleucine-dependent interaction with the COI1 F-box protein. Plant J. 55, 979–988.
Feys B., Benedetti C.E., Penfold C.N., Turner J.G. (1994) Arabidopsis mutants selected for resistance to the phytotoxin coronatine are male sterile, insensitive to methyl jasmonate, and resistant to a bacterial pathogen. Plant Cell. 6, 751–759.
Sheard, L.B., Tan X., Mao H., Withers J., Ben-Nissan G., Hinds T.R., Kobayashi Y., Hsu F., Sharon M., Browse J., He S.Y., Rizo J., Howe G.A., Zheng N. (2010) Jasmonate perception by inositol-phosphate-potentiated COI1-JAZ co-receptor. Nature. 468, 400–405.
Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A., Scott T.G., Vittori S., Perry J.A., Qi J., Winter G.E., Wong K., Gray N.S., Bradner J.E. (2018) The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441.
Bosu D.R., Kipreos E.T. (2008) Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell Div. 3, 7.
Los G.V., Encell L.P., McDougall M.G., Hartzell D.D., Karassina N., Zimprich C., Wood M.G., Learish R., Friedman Ohana R., Urh M., Simpson D., Mendez J., Zimmerman K., Otto P., Vidugiris G., Zhu J., Darzins A., Klaubert D.H., Bulleit R.F., Wood K.V. (2008) HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382.
Neklesa T.K., Tae H.S., Schneekloth A.R., Stulberg M.J., Corson T.W., Sundberg T.B., Raina K., Holley S.A., Crews M.C. (2011) Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 7, 538–543.
Lins L., Brasseur R. (1995) The hydrophobic effect in protein folding. FASEB J. 9, 535–540.
Kubota H. (2009) Quality control against misfolded proteins in the cytosol: a network for cell survival. J. Biochem. 146, 609–616.
Tomoshige S., Naito M., Hashimoto Y., Ishikawa M. (2015) Degradation of HaloTag-fused nuclear proteins using bestatin-HaloTag ligand hybrid molecules. Org. Biomol. Chem. 13, 9746–9750.
Buckley D.L., Raina K., Darricarrere N., Hines J., Gustafson J.L., Smith I.E., Miah A.H., Harling J.D., Crews C.M. (2015) HaloPROTACS: use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chem. Biol. 10, 1831–1837.
Tovell H., Testa A., Maniaci C., Zhou H., Prescott A.R., Macartney T., Ciulli A., Alessi D.R. (2019) Rapid and reversible knockdown of endogenously tagged endosomal proteins via an optimized HaloPROTAC degrader. ACS Chem. Biol. 14, 882–892.
Pettersson M., Crews C.M. (2019) Proteolysis TArgeting chimeras (PROTACs) – past, present and future. Drug Discov. Today Technol. 31, 15–27.
Sakamoto K.M., Kim K.B., Kumagai A., Mercurio F., Crews C.M., Deshaies R.J. (2001) Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA. 98, 8554–8559.
Schneekloth A.R., Pucheault M., Tae H.S., Crews C.M. (2008) Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg. Med. Chem. Lett. 18, 5904–5908.
Buckley D.L., Molle I.V., Careiss P.C., Tae H.S., Michel J., Noblin D.J., Jorgensen W.L., Ciulli A., Crews C.M. (2012) Targeting the von Hippel–Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J. Am. Chem. Soc. 134, 4465–4468.
Itoh Y., Ishikawa M., Naito M., Hashimoto Y. (2010) Protein knockdown using methyl bestatin–ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid binding proteins. J. Am. Chem. Soc. 132, 5820–5826.
Gandhi A.K., Kang J., Havens C.G., Conklin T., Ning Y., Wu L., Ito T., Ando H., Waldman M.F., Thakurta A., Klippel A., Handa H., Daniel T.O., Schafer P.H., Chopra R. (2014) Immunomodulatory agents lenalidomide and pomalidomide costimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4CRBN. Br. J. Haematol. 164, 811–821.
Gadd M.S., Testa A., Lucas X., Chan K-H., Chen W., Lamont D.J., Zengerle M., Ciulli A. (2017) Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13, 514–521.
Chessum N.E.A., Sharp S.Y., Caldwell J.J., Pasqua A.E., Wilding B., Colombano G., Collins I., Ozer B., Richards M., Rowlands M., Stubbs M., Burke R., McAndrew P.C., Clarke P.A., Workman P., Cheeseman M.D., Jones K. (2018) Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766). J. Med. Chem. 61, 918–933.
Nowak R.P., DeAngelo S.L., Buckley D., He Z., Donovan K.A., An J., Safaee N., Jedrychowski M.P., Ponthier C.M., Ishoey M., Zhang T., Mancias J.D., Gray N.S., Bradner J.E., Fischer E.S. (2018) Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 14, 706–714.
Hines J., Gough J.D., Corson T.W., Crews C.M. (2013) Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl. Acad. Sci. USA. 110, 8942–8947.
Khan S., He Y., Zhang X., Yuan Y., Pu S., Kong Q., Zheng G., Zhou D. (2020) PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene. 39, 4909–4924.
Li X., Song Y. (2020) Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 13: 50.
Zeng S., Huang W., Zheng X., Cheng L., Zhang Z., Wang J., Shen Z. (2021) Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: recent progress and future challenges. Eur. J. Med. Chem. 210, 112981.
Kregel S., Wang C., Han X., Xiao L., Fernandez-Salas E., Bawa P., McCollum B.L., Wilder-Romans K., Apel I.J., Cao X., Speers C., Wang S., Chinnaiyan A.M. (2020) Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia. 22, 111–119.
Chung H.K., Jacobs C.L., Huo Y., Yang J., Krumm S.A., Plemper R.K., Tsien R.Y., Lin M.Z. (2015) Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 11, 713–720.
Brass V., Berke J.M., Montserret R., Blum H.E., Penin F., Moradpour D. (2008) Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc. Natl. Acad. Sci. USA. 105, 14545–14550.
McPhee F., Sheaffer A.K., Friborg J., Hernandez D., Falk P., Zhai G., Levine S., Chaniewski S., Yu F., Barry D., Chen C., Lee M.S., Mosure K., Sun L., Sinz M., Meanwell N.A., Colonno R.J., Knipe J., Scola P. (2012) Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob. Agents Chemother. 56, 5387–5396.
Fay E.J., Aron S.L., Stone I.A., Waring B.M., Plemper R.K., Langlois R.A. (2018) engineered small-molecule control of influenza a virus replication. J. Virol. 93, e01677–18.
Rago F., DiMare M.T., Elliott G., Ruddy D.A., Sovath S., Kerr G., Bhang H.C., Jagani Z. (2019) Degron mediated BRM/SMARCA2 depletion uncovers novel combination partners for treatment of BRG1/SMARCA4-mutant cancers. Biochem. Biophys. Res. Commun. 508, 109–116.
Zhu W., Zhang B., Li M., Mo F., Mi T., Wu Y., Teng Z., Zhou Q., Li W., Hu B. (2019) Precisely controlling endogenous protein dosage in hPSCs and derivatives to model FOXG1 syndrome. Nat. Commun. 10, 928.
Han X., Qian X., Bernstein J.G., Zhou H., Franzesi G.T., Stern P., Bronson R.T., Graybiel A.M., Desimone R., Boyden E.S. (2009) Millisecond-timescale optical control of neural dynamics in the nonhuman primate brain. Neuron. 62, 191–198.
Renicke C., Schuster D., Usherenko S., Essen L., Taxis C. (2013) A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem. Biol. 20, 619−626.
Bonger K.M., Rakhit R., Payumo A.Y., Chen J.K., Wandless T.J. (2014) A general method for regulating protein stability with light. ACS Chem. Biol. 9, 111–115.
Taxis C. (2017) Development of a synthetic switch to control protein stability in eukaryotic cells with light. Meth. Mol. Biol. 1596, 241−255.
Baaske J., Gonschorek P., Engesser R., Dominguez-Monedero A., Raute K., Fischbach P., Müller K., Cachat E., Schamel W.W.A., Minguet S., Davies J.D., Timmer J., Weber W., Zurbriggen M.D. (2018) Dual-controlled optogenetic system for the rapid down-regulation of protein levels in mammalian cells. Sci. Rep. 8, 15024.
Hasenjäger S., Trauth J., Hepp S., Goenrich J., Essen L., Taxis C. (2019) Optogenetic downregulation of protein levels with an ultrasensitive switch. ACS Synth. Biol. 8, 1026−1036.
Harper S.M., Neil L.C., Gardner K.H. (2003). Structural basis of a phototropin light switch. Science. 301, 1541–1544.
Jariel-Encontre I., Bossis G., Piechaczyk M. (2008). Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta. 1786, 153–177.
Bonger K.M., Chen L.C., Liu C.W., Wandless T.J. (2011) Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 7, 531–537.
Nakashima Y., Ohta S., Wolf A.M. (2017) Blue light-induced oxidative stress in live skin. Free Radic. Biol. Med. 108, 300–310.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология