

Молекулярная биология, 2022, T. 56, № 1, стр. 168-178
Конструирование in silico новой мультиэпитопной кандидатной пептидной вакцины против Q-лихорадки
S. Jabarzadeh a, A. Samiminemati a, M. Zeinoddini a, *
a Faculty of Chemistry and Chemical Engineering, Malek Ashtar University of Technology
Tehran, Iran
* E-mail: zeinoddini52@mut.ac.ir
Поступила в редакцию 06.12.2020
После доработки 07.04.2021
Принята к публикации 15.04.2021
- EDN: RVPJNU
- DOI: 10.31857/S0026898422010037
Аннотация
С появлением новых патогенов возникает необходимость в новых вакцинах. Эффективную вакцину отличает высокая иммуногенностью и безопасность, и этим критериям могут удовлетворять вакцины на основе эпитопов. Среди зоонозных инфекций особое место занимает Q-лихорадка, вызываемая бактерией Coxiella burnetii. Из-за многочисленных вспышек и пандемического потенциала это заболевание находится под пристальным вниманием эпидемиологов и клиницистов. Мы представляем синтетическую мультиэпитопную вакцину против Coxiella burnetii. Эта вакцина разработана с использованием иммуноинформатики. Изучены антигенные белки этого патогена и отобрано пять эпитопов Т-клеток. Антигенность, аллергенность и токсичность выбранных эпитопов оценивали с использованием серверов VaxiJen 2.0, AllerTOP и ToxinPred соответственно. Выбранные эпитопы объединили в пептидную последовательность с субъединицей В холерного токсина (CTXB) в качестве адъюванта. Сродство предложенной вакцины к молекулам главного комплекса гистосовместимости (MHC) классов I и II оценивали с использованием технологии молекулярного докинга. Полученный препарат характеризуется высокой иммуногенностью, стабильностью и периодом полувыведения, сопоставимым с используемым в программах вакцинации. Валидированную последовательность эпитопов можно рассматривать в качестве потенциальной вакцины для защиты от возбудителя Q-лихорадки.
Q-лихорадка – широко распространенное зоонозное заболевание, которое вызвано Coxiella burnetii, грамотрицательной облигатной внутриклеточной бактерией, открытой в 1935 году Ha-rold Cox и MacFarlane Burnet (см. обзоры [1, 2]). Эта бактерия принадлежит к семейству Coxiellaceae, способному к внутриклеточной внутривакуолярной персистенции как у беспозвоночных, так и у позвоночных хозяев.
C. burnetii хорошо переносит различные среды, в том числе кислые условия до pH примерно 4.5, высокие температуры до 62°C в течение 30 мин, УФ-облучение и давление до 300 000 кПа [3, 4]. Домашние животные, такие как крупный рогатый скот, овцы и козы, представляют собой основные источники C. burnetii. Эту инфекцию могут передавать человеку инфицированные насекомые, такие как клещи и комары, ей можно заразиться при прямом контакте с инфицированными животными, а также при употреблении их мяса, молока и других продуктов питания [3, 5]. Симптомы Q-лихорадки у людей изначально похожи на симптомы гриппа, но впоследствии они могут привести к вторичным хроническим состояниям, таким как гепатит, острый эндокардит, васкулит, лимфаденит и др. [6].
К настоящему времени этот бактериальный зооноз вызвал три крупные вспышки. В 1955 году первые случаи Q-лихорадки зарегистрировали в девяти африканских странах. В период с 2007 по 2010 гг. Нидерланды столкнулись с большой волной инфекций, вызванных ею. Самая крупная зоонозная вспышка Q-лихорадки произошла в Кайенне ‒ столице Французской Гвианы [6, 7].
Как упомянуто ранее, Q-лихорадка может передаваться людям или другим животным при прямом контакте или через инфицированные продукты. Однако о горизонтальной передаче этого заболевания (от человека человеку) сообщений нет [1]. Учитывая зоонозный потенциал Q-лихорадки на основании зарегистрированных крупных вспышек, встала задача предотвратить распространение этой инфекции, а значит приступить к разработке вакцин. До сих пор доступна только одна вакцина против Q-лихорадки, Q-VAX, ‒ в виде инактивированного формалином цельноклеточного препарата штамма Heinzerling C. burnetii фазы 1 [8].
Последние достижения в области геномного секвенирования улучшили наше понимание биологии микроорганизмов [9, 10] и привели к созданию баз данных по специфичным для организма белкам, нуклеотидным последовательностям, а также к доступности серверов для предсказания эпитопов [11, 12]. Предсказание эпитопов В- и Т‑клеток выполняют раздельно [13, 14].
В представленном исследовании, проведенном методом обратной вакцинологии, для разработки иммуноактивной пептидной/белковой вакцины против C. burnetii использовано несколько баз данных и серверов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Сбор данных по эпитопам. В этом разделе предложенные эпитопы исследовали в базе данных IEDB (https://www.iedb.org/) и выбирали некоторые поверхностные эпитопы. Отметим, что фильтры корректировали на основе использования вакцины против Coxiella burnetii (штамм RSA 493) со всеми линейными и структурными эпитопами. Результат этого поиска ‒ около 62 эпитопов и эпитопов поверхностных белков, которые отобрали из предложенных. Аминокислотная последовательность токсина Vibrio cholerae (субъединица B) получена из банка NCBI (https://www.ncbi.nlm.nih.gov/ protein/) с регистрационным номером AAV67882.1. Кроме того, из банка данных PDB (https:// www.rcsb.org/) была извлечена трехмерная структура молекул главного комплекса гистосовместимости (MHC) класса I и MHC класса II с идентификационными номерами 4UQ3 и 1DLH соответственно.
Множественное выравнивание последовательностей и выбор антигена. Полная последовательность целевого поверхностного белка получена из базы данных UniProt (https://www.uniprot.org/) в формате FASTA [15]. На следующем этапе с использованием базы NCBI BLAST (https://blast.ncbi. nlm.nih.gov/Blast.cgi) эту последовательность выравнивали с таковыми для белков человека и подтвердили, что между ними нет сходства.
Антигенность предсказанных эпитопов. Антигенность как В-клеточных, так и Т-клеточных эпитопов предсказана с использованием сервера VaxiJen 2.0 с точностью предсказания от 70 до 89% (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/) [16].
Прогнозирование аллергенности и токсичности. Аллергенность и токсичность эпитопов имеют решающее значение для вакцин на основе пептидов, поскольку некоторые предполагаемые эпитопы могут проявлять эти свойства и тем самым вызывать нежелательные перекрестные реакции иммунной системы. Выбранные в качестве эпитопов пептиды проверяли на аллергенность с помощью сервера AllerTOP (https://www.ddgpharmfac.net/AllerTOP/) [17]. Токсичность эпитопов анализировали с использованием базы данных (https://webs.iiitd.edu.in/raghava/toxinpred/algo.php).
Создание пептидов для химерного белка. На этом этапе эпитопы-кандидаты размещали в пептидной последовательности и соединяли вместе через жесткий линкер LysLys (KK), как показано на рис. 1. На следующем этапе аминокислотную последовательность адъюванта (AAV67882.1) ‒ субъединицы B токсина Vibrio cholera ‒ вводили в начало и конец полиэпитопной последовательности ‒ для повышения иммуногенности конструкта. Для присоединения адъювантных последовательностей к полиэпитопной использовали линкер PAPAP.
Рис. 1.
Последовательность разработанного иммуногена включает аминокислотные последовательности эпитопов Coxiella burnetii (штамм RSA 493), адъюванта и линкеров. Последовательности эпитопов показаны черным, адъюванта ‒ синим, линкеров KK (соединяют эпитопы) − зеленым, линкеров PAPAP (соединяют последовательности адъюванта с эпитопами) − красным цветом.

Физико-химические свойства и стабильность родственного белка. Для анализа химических и физических свойств полученного полипептида, таких как молекулярная масса, чистый заряд и период полураспада, его последовательность исследовали с использованием баз данных PepCalc (https://pepcalc.com) [18] и ProtParam (https:// web.expasy.org/cgi-bin/protparam/protparam). Стабильность белка рассмотрели и смоделировали с использованием программ IUpred 2.0 (https:// iupred2a.elte.hu/) [19], IsUnstruct (v2.02) (http:// bioinfo.protres.ru/IsUnstruct/) [20] и сервера FoldUnfold (http://bioinfo.protres.ru/ogu/) [21].
Предсказание вторичной и третичной структуры. Вторичная структура сконструированного пептида предсказана по методу GorIV с использованием базы данных PRABI (https://npsa-prabi.ibcp.fr/cgibin/ npsa_automat.pl?page=/NPSA/npsa_gor4.html). Трехмерная структура белка спрогнозирована с использованием сервера I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) [22]. Он предсказал трехмерную структуру белка с использованием моделирования de novo. I-TASSER ‒ это ранжированный подход к предсказанию структуры и функции белка, основанный на уровне подобия входных и шаблонных структур, доступных в банке данных PDB.
Уточнение модели и оценка качества. Уточнение предсказанной модели выполнено с использованием сервера 3Drefine (http://sysbio.rnet.missouri.edu/3Drefine/), чтобы уменьшить возможные структурные ошибки в предсказанной третичной структуре родственной белковой вакцины. Кроме того, структурное качество вакцины проверено на графике Рамачандрана с использованием сервера RAPAGE (https://servicesn.mbi.ucla.edu/SAVES/) и Z-балла сервера ProSA (https://prosa.services.came.sbg.ac.at/prosa.php) [23].
Молекулярный докинг. Процесс молекулярной стыковки выполняли для проверки аффинности связывания разработанной белковой последовательности с молекулами MHC класса I (HLA-A0201; PDB Acc. No. UQ3) и MHC класса II (HLA-DR; PDB Acc. No. 1DLH). Молекулярный докинг проводили с использованием онлайн-сервера ClusPro (https://cluspro.bu.edu/ login.php?redir=/queue.php) со сложным типом по умолчанию [24‒26]. На следующем этапе процесс повторяли с использованием программного обеспечения HEX 6.0. С этой целью выбирали следующие параметры: FFT Mode–3D fast life, диапазоны расстояний, скручивания, рецепторов и лигандов ‒ 40, 360, 180 и 180 соответственно, тип корреляции ‒ только форма, размер сетки ‒ 0.6.
Обратная трансляция пептида и проверка открытой рамки считывания (ORF). На заключительном этапе аминокислотную последовательность полученного полипептида обратно транслировали в нуклеотидную с использованием базы данных (https://www.bioinformatics.org/sms2/rev_trans.html). Открытую рамку считывания (ORF) связанной последовательности со значением по умолчанию для Escherichia coli исследовали в базе данных ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Выбор эпитопов и выравнивание последовательностей
В табл. 1 показаны эпитопы, выбранные на сервере IEDB. Последовательность пептидов (эпитопов) для вакцины C. burnetii сконструирована, отобрана и проанализирована в программе protein BLAST.
Таблица 1.
Эпитопы, выбранные для исследования
| UniProt (Acc. No.) | Белок | Эпитопы |
|---|---|---|
| H7C7D7 (CBU_1910) | Com1 | DIQSIVHHYLVNHPEVL GNVTLVEFFDY KYYAFHDALLS SEQITLQTAEKVGLNVA TPTFVIGNKALTKFGF |
| Q83DK8 (CBU_0718) | Гипотетический ассоциированный с мембраной белок | DDVAKLRGDLSSIIHKLTSFSKTEASM |
| Q83AL4 (CBU_1869) | Гипотетический экспортируемый белок | PITKKQLKTMSNYEVIAK IKLPRNRYRLVFTQQ GKHFDGIKVLKLSPQNTI |
| Q83F71 (CBU_0077) | Гипотетический трансмембранный белок | EVLTLLLNWVNYHE |
| Q83EL2 (CBU_0307) | Наружный белок мембраны | GVAYTYNRANAGLPTNK VPGYRNASSKRFVAP |
| Q83DT1 (OmpH) | OmpH | QELFVAQNKAMSDFM |
| Q83CG1 (CBU_1157) | Гипотетический экспортируемый белок | ISLLVFKNSHRVQLWAK RFDLSLMLNYPNSADRY |
Кроме того, чтобы подтвердить отсутствие сходства между последовательностями сконструированного полипептида и белков человека, полные последовательности из эпитопов-кандидатов, содержащихся в поверхностных белках C. burnetii, сопоставляли с белками человека. Результаты protein BLAST показаны в табл. 2.
Таблица 2.
Результаты исследования белков C. burnetii с помощью программы protein BLAST и предсказание антигенности
| Эпитоп | NCBI Blast | |||
|---|---|---|---|---|
| Минимальная идентичность, % Homo sapiens |
Максимальная идентичность, % Homo sapiens |
Минимальная идентичность, % Coxiella burnetii |
Максимальная идентичность, % Coxiella burnetii |
|
| H7C7D7 (CBU_1910) |
35.14 | 35.90 | 96.92 | 100.00 |
| Q83DK8 (CBU_0718) |
0 | 0 | 98.94 | 100.00 |
| Q83AL4 (CBU_1869) |
0 | 0 | 98.62 | 100.00 |
| Q83F71 (CBU_0077) |
0 | 0 | 99.24 | 100.00 |
| Q83EL2 (CBU_0307) |
0 | 0 | 35.11 | 100.00 |
| Q83DT1 (ompH) |
0 | 0 | 99.14 | 100.00 |
| Q83CG1 CBU_1157 |
0 | 0 | 99.14 | 100.00 |
Прогнозирование и выбор Т-клеточного эпитопа
Внутриклеточная природа заражения C. burnetii побудила нас ограничить выбор кандидатных пептидов только Т-клеточными эпитопами. Для предсказания эпитопов Т-клеток проводили трехэтапный скрининг иммуногенных (порог: 0.4) и аллергенных характеристик эпитопов (табл. 3). Все эпитопы по предсказанию были нетоксичными.
Таблица 3.
Прогнозируемые свойства выбранных Т-клеточных эпитопов
| Эпитоп | Вероятностный протективный аллерген (AllertTop) | Антиген (VaxiJen 2.0) |
|---|---|---|
| DIQSIVHHYLVNHPEVL | Аллерген | Антиген |
| GNVTLVEFFDY | Аллерген | Антиген |
| KYYAFHDALLS | Не аллерген | Не антиген |
| SEQITLQTAEKVGLNVA | Не аллерген | Антиген |
| TPTFVIGNKALTKFGF | Не аллерген | Антиген |
| DDVAKLRGDLSSIIHKLTSFSKTEASM | Не аллерген | Не антиген |
| PITKKQLKTMSNYEVIAK | Не аллерген | Не антиген |
| IKLPRNRYRLVFTQQ | Не аллерген | Не антиген |
| GKHFDGIKVLKLSPQNTI | Аллерген | Антиген |
| EVLTLLLNWVNYHE | Не аллерген | Не антиген |
| GVAYTYNRANAGLPTNK | Не аллерген | Не антиген |
| VPGYRNASSKRFVAP | Не аллерген | Антиген |
| ISLLVFKNSHRVQLWAK | Аллерген | Не антиген |
| RFDLSLMLNYPNSADRY | Не аллерген | Антиген |
| QELFVAQNKAMSDFM | Не аллерген | Не антиген |
Инженерия вакцин и физико-химические свойства
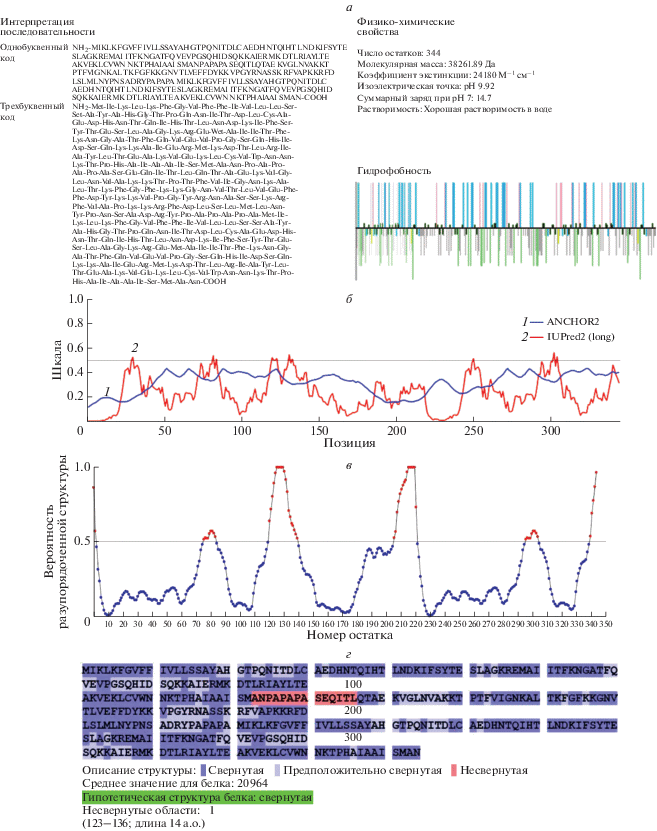
С помощью иммуноинформатического анализа отобрали пять эпитопов Т-клеток. Сконструированная вакцина-кандидат включала 344 аминокислоты, разделенные на следующие сегменты: CTxB в качестве адъюванта, Т-клеточные эпитопы и соответствующие линкеры. Физические и химические свойства конечной конструкции предсказаны с помощью сервера PepCalc. Результаты подтвердили, что полипептид с молекулярной массой около 38 261.89 Да – стабильный растворимый белок с pI 9.92 и расчетным суммарным зарядом около 14.7 (рис. 2а).
Рис. 2.
Свойства сконструированного полипептида. а ‒ Аминокислотная последовательность и физико-химические характеристики. Согласно результатам, получен полипептид с общим числом аминокислотных остатков (а.о.) 344, молекулярной массой 38261.89 Да, хорошо растворимый в воде. Кроме того, изоэлектрическая точка равна 9.92, а суммарный заряд при pH 7.0 ‒ около 14.7. б ‒ График стабильности белка, созданный с использованием сервера IUpred 2.0. Этот график подтверждает стабильность полученного полипептида, так как прогнозируемые нарушения в его структуре ниже порогового значения 0.5. в ‒ Результаты анализа по программе IsUnstruct и предсказание неупорядоченных участков на основе модели Изинга. г ‒ Предсказание по программе FoldUnfold в соответствии с аминокислотной последовательностью. Сервер FoldUnfold проверяет аминокислоту в последовательности. Свернутые и несвернутые области в последовательности показаны синим и красным цветом соответственно.
Стабильность белка
Стабильность родственного белка оценивали с использованием серверов IUpred 2.0, IsUnstruct (v2: 02) и FoldUnfold (масштаб: ожидаемое число контактов 8 Å, порог: 20.4, рамка усреднения: 11). Рассчитанная стабильность белка подтверждена, как показано соответственно на рис. 2б, 2в и 2г.
С использованием программы ProtParam для аминокислотного состава сконструированного полипептида предсказаны стабильность и период полужизни. Согласно результатам, показанным на рис. 3, структура белка стабильна, период его полужизни составляет примерно 30 ч в клетках млекопитающих, более 20 ч в дрожжах и более 10 ч в E. coli (рис 3).

Прогнозирование вторичной и третичной структуры
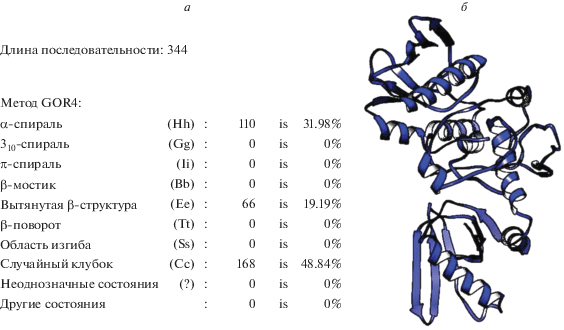
Прогноз вторичной и третичной структуры химерного полипептида проиллюстрирован на рис. 4. Как можно видеть, в α-спираль, удлиненную β-структуру и случайный клубок организовано соответственно 31.58, 19.30 и 49.12% аминокислотных остатков из общего числа 344. Кроме того, первичную 3D-модель предлагаемого иммуногена предсказали онлайн-сервером I-TASSER.
Уточнение модели и оценка качества
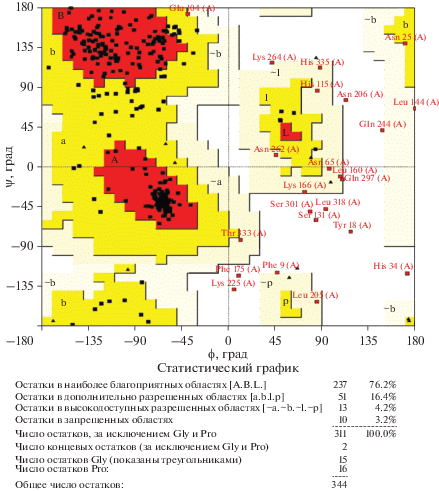
Процессы уточнения модели выполнены на сервере 3Drefine для выбранной модели полипептида. С этой целью анализировали всю структуру белка, включая элементы вторичной структуры, области петель и его боковые цепи. Пять факторов, включая оценку 3Drefine, GDT-TS, GDT-HA, RMSD, RWplus и MolProbity, ‒ основные параметры для процесса уточнения, которые отражают потенциальную энергию (3Drefine-оценочная функция и RWplus), оценку сходства (GDT-TS и GDT-HA), классификации и физическую реалистичность соответственно. Далее выбирали уточненную модель с соответствующими характеристиками для дальнейших оценок упомянутых факторов. Выявлено, что уточненная сервером 3Drefine модель имеет значения GDT-TS 1.0000, GDT-HA 0.9644, RMSD 0.375, MolProbity 3.395, оценку 3Drefine 22 028.8 и RWPlus ‒ 63 478.84. На следующем этапе геометрическое качество первичной и уточненной моделей проанализировали по графику Рамачандрана (рис. 5).
Молекулярный докинг
Результаты анализа молекулярного докинга подтвердили сродство химерных пептидов как MHC класса I, так и MHC класса II. Обнаружено, что химерный пептид проявлял большее сродство к MHC класса II с е-value, равным ‒920.88, тогда как для MHC класса I параметр составлял ‒772.51. В этом разделе альбумин использовали в качестве нейтрального белка. Кроме того, энергия связывания с MHC класса I и MHC класса II для сконструированного полипептида проанализирована с использованием онлайн-сервера Cluspro (https://cluspro.bu.edu/login.php). Результаты представлены в табл. 4 и табл. 5.
Таблица 4.
Результаты молекулярного докинга, проведенного с использованием сервера ClusPro, для предлагаемой последовательности потенциального иммуногена с MHC класса I
| Выбранная модель MHC I | Репрезентативность | Взвешенная Score-функция |
|---|---|---|
| 0 | Центр | ‒829.0 |
| Самая низкая энергия | ‒829.0 | |
| 1 | Центр | ‒805.6 |
| Самая низкая энергия | ‒805.6 | |
| 2 | Центр | ‒659.0 |
| Самая низкая энергия | ‒745.5 |
Таблица 5.
Результаты молекулярного докинга, проведенного с использованием сервера ClusPro, для предлагаемой последовательности иммуногена с MHC класса II
| Выбранная модель MHC II | Репрезентативность | Взвешенная Score-функция |
|---|---|---|
| 0 | Центр | ‒824.2 |
| Самая низкая энергия | ‒824.2 | |
| 1 | Центр | ‒631.3 |
| Самая низкая энергия | ‒708.2 | |
| 2 | Центр | ‒648.4 |
| Самая низкая энергия | ‒737.4 |
Обратная трансляция белка и дизайн конструктов
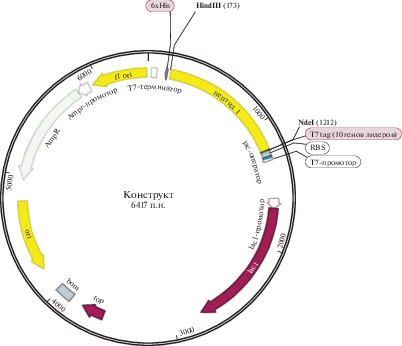
Чтобы сконструировать кассету для экспрессии в плазмидном векторе для синтеза целевого белка, необходимо осуществить обратный перевод аминокислотной последовательности в нуклеотидную. С этой целью конечную пептидную последовательность преобразовали в нуклеотидную с использованием инструментов биоинформатики. Поиск ORF последовательности проводили в программе ORFfinder. Полученную нуклеотидную последовательность использовали для моделирования клонирования в вектор PET21 с использованием автономного программного обеспечения SnapGene. Эти результаты показаны на рис. 6.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Недавно разработали несколько методов для индукции иммуногенности вакцин и снижения связанных с их применением рисков. В случае распространенного зоонозного заболевания, Q-лихорадки, доступна только одна коммерческая вакцина. Это инактивированная формалином бактерия C. burnetii [8]. В представленной работе описано создание полипептида, который может быть использован как иммуноген для вакцины против Q-лихорадки. Полипептид сконструирован на основе спрогнозированных функциональных и иммунодоминантных эпитопов C. burnetii. При предсказании эпитопов использовали несколько баз данных [27‒29]. В пептидных вакцинах на конец аминокислотной цепи можно ввести метку, например Arg-тег, кальмодулинсвязывающий пептид, целлюлозсвязывающий домен, DsbA, c-myc-тег, глутатион-S-трансферазу, FLAG-, HAT-тег, His-тег, мальтозсвязывающий белок, NusA, S-тег и др. [30]. Дальнейшую белковую инженерию можно применять для солюбилизации нерастворимого полипептида или стабилизации нестабильного [31]. После отбора эпитопов сконструированную аминокислотную последовательность обратно переводят в кодирующую ее нуклеиновую и полученный остов синтезируют и клонируют в подходящем хозяине для экспрессии целевого белка [32].
В последнее время опубликовано множество исследований по разработке вакцин на основе пептидов против инфекционных заболеваний. Например, Farhadi и др. [33] представили вакцину на основе пептидов, которая состоит из эпитопов В-клеток и линейных CD4+ Т-клеток, выбранных из белков внешней мембраны (Omps) Klebsiella pneumoniae. В другом исследовании Nosrati и др. [34] получили мультиэпитопную рекомбинантную вакцину из линейных В-клеточных и Т-клеточных связывающих эпитопов из гликопротеинов Gc и Gn вируса геморрагической лихорадки Крым–Конго. Полученный ими оптимизированный пептид состоял из 382 аминокислотных остатков, организованных в четыре домена, включая линейные В-клеточные эпитопы, Т-клеточные эпитопы и два адъюванта [34]. В 2020 году Aryanzad и соавт. [35] разработали и изготовили иммуногенный химерный белок против антигенов IpaD и IpaB из Shigella dysenteriae [35]. В 2021 году Sohali и др. [36] описали процедуру in silico для предсказания Т-клеточного эпитопа SARS-CoV-2 [36]. Jaydari и соавт. [37] сообщали о В- и Т‑клеточных эпитопах против C. burnetii. В этой вакцине эпитопы Com1 и OmpH в различном порядке были организованы в 3 группы: Т-клеточные, В-клеточные и общие Т- и В-клеточные. Авторы показали, что каркас, сконструированный из эпитопов В-клеток, проявляет наибольшую иммуногенность как в отношении антигенов Com1, так и OmpH.
Нами исследованы все Т-клеточные эпитопы семи антигенов C. burnetii, включая белки Com1 и OmpH (табл. 1). При разработке пептидных вакцин первостепенное значение имеет определение иммуногенных В-клеточных и Т-клеточных эпитопов. Т-клетки играют важную роль в иммунном ответе клетки на патогены. Бактерия C. burnetii ‒ облигатный внутриклеточный патоген, поэтому иммунные ответы на C. burnetii главным образом основаны на Т-клеточном иммунитете [38]. Из-за этого мы сосредоточились на Т-клеточных эпитопах с высоким уровнем иммуногенности и сродства к MHC класса I и II. Мы изучили все Т‑клеточные эпитопы семи антигенов C. burnetii, включая белки Com1 и OmpH (табл. 1). После проверки показателей иммуногенности, аллергенности, физико-химических характеристик и взаимодействия между вакциной-кандидатом и молекулами MHC была подтверждена пригодность вакцины-кандидата.
Иммуноинформатический подход ‒ рентабельный, безопасный и быстрый метод разработки вакцин. Эта работа была направлена на создание мультиэпитопной вакцины против зоонозной бактерии C. burnetii, вызывающей Q-лихорадку. В результате сконструирован и наработан мультиэпитопный иммуногенный полипептид высокой стабильности, пригодный для использования в качестве иммуногена в вакцине против Q-лихорадки.
Авторы благодарят исследовательский совет Malek-Ashtar University of Technology (MUT) за финансовую поддержку этого исследования.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
McQuiston J.H., Childs J.E., Thompson H.A. (2002) Q fever. J. Am. Vet. Med. Assoc. 221(6), 796–799. https://doi.org/10.2460/javma.2002.221.796
Marrier T.J. (2010) Q fever pneumonia. Infect. Dis. Clin. North Am. 24(1), 27–41. https://doi.org/10.1016/j.idc.2009.10.004
Angelakis E., Raoult D. (2010) Q fever. Vet. Microbiol. 140(3‒4), 297–309. https://doi.org/10.1016/j.vetmic.2009.07.016
Mori M., Mertens K., Cutler S.J., Santos A.S. (2017) Critical aspects for detection of Coxiella burnetii. Vector Borne Zoonotic Dis. 17(1), 33–41. https://doi.org/10.1089/vbz.2016.1958
Fenollar F., Fournier P.E., Raoult D. (2004) Molecular detection of Coxiella burnetii in the sera of patients with Q fever endocarditis or vascular infection. J. Clin. Microbiol. 42, 4919–4924. https://doi.org/10.1128/JCM.42.11.4919-24
Melenotte C., Protopopescu C., Million M., Edouard S., Carrieri M.P., Eldin C., Angelakis E., Djossou F., Bardin N., Fournier P.E., Mege J.L., Raoult D. (2018) Clinical features and complications of Coxiella burnetii infections from the French national reference center for Q fever. JAMA Netw. Open. 1(4), e181580. https://doi.org/10.1001/jamanetworkopen.2018.1580
Njeru J., Henning K., Pletz M.W., Heller R., Neubauer H. (2016) Q fever is an old and neglected zoonotic disease in Kenya: a systematic review. BMC Public Health. 16, 297. https://doi.org/10.1186/s12889-016-2929-9
Bond K.A., Franklin L.J., Sutton B., Firestone S.M. (2017) Q-Vax Q fever vaccine failures, Victoria, Australia 1994‒2013. Vaccine. 35(51), 7084–7087. https://doi.org/10.1016/j.vaccine.2017.10.088
Hornstra H.M., Priestley R.A., Georgia S.M., Kachur S., Birdsell D.N., Hilsabeck R., Gates L.T., Samuel J.E., Heinzen R.A., Kersh G.J., Keim P., Massung R.F., Pearson T. (2011) Rapid typing of Coxiella burnetii. PloS One. 6(11), e26201. https://doi.org/10.1371/journal.pone.0026201
Vigil A., Ortega R., Nakajima-Sasaki R., Pablo J., Molina D.M. (2010) Genome-wide profiling of humoral immune response to Coxiella burnetii infection by protein microarray. Proteomics. 10(12), 2259–2269. https://doi.org/10.1002/pmic.201000064
Florea L., Halldórsson B., Kohlbacher O., Schwartz R., Hoffman S., Istrail S. (2003) Epitope prediction algorithms for peptide-based vaccine design. Proc. IEEE Comput. Soc. Bioinform. Conf. 2, 17–26. https://doi.org/10.1109/CSB.2003.1227293
Zhang Q., Wang P., Kim Y., Haste-Andersen P., Beaver J., Bourne P.E., Bui H.H., Buus S., Frakild S., Greenbaum J., Lund O., Lundegaard C., Nielsen M., Ponomarenko J., Sette A., Zhu Z., Peters B. (2008) Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 36, 513–518. https://doi.org/10.1093/nar/gkn254
Saha S., Bhasin M., Raghava G.P.S. (2005) Bcipep: a database of B-cell epitopes. BMC Genom. 6, 79. https://doi.org/10.1186/1471-2164-6-79
Deavin A.J., Auton T.R., Greaney P.J. (1996) Statistical comparison of established T-cell epitope predictors against a large database of human and murine antigens. Mol. Immunol. 33(2), 145–155. https://doi.org/ 10.1016/0161-5890(95)00120-4
Consortium T. (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47(D1), D506–D515. https://doi.org/10.1093/nar/gky1049
Doytchinova I., Flower D. (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8(4), 1–7. https://doi.org/10.1186/1471-2105-8-4
Dimitrov I., Bangov I., Flower D., Doytchinova I. (2014) AllerTOP v.2 ‒ a server for in silico prediction of allergens. J. Mol. Model. 20(6), 2278–2284. https://doi.org/10.1007/s00894-014-2278-5
Lear S., Cobb S. (2016) Pep-Calc.com: a set of web utilities for the calculation of peptide and peptoid properties and automatic mass spectral peak assignment. J. Comput. Aided Mol. Des. 30(3), 271–277. https://doi.org/10.1007/s10822-016-9902-7
Dosztányi Z ., Csizmok V., Tompa P., Simon I. (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 21(16), 3433–3434. https://doi.org/10.1093/bioinformatics/bti541
Lobanov M.Y., Sokolovskiy I.V., Galzitskaya O.V. (2013) IsUnstruct : prediction of the residue status to be ordered or disordered in the protein chain by a method based on the Ising model. J. Biomol. Struct. Dyn. 31(10), 37–41. https://doi.org/10.1080/07391102.2012.718529
Galzitskaya O., Garbuzynskiy S. (2006) FoldUnfold: web server for the prediction of disordered regions in protein chain. Bioinformatics. 22(23), 2948–2949. https://doi.org/10.1093/bioinformatics/btl504
Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9(1), 40. https://doi.org/10.1186/1471-2105-9-40
Wiederstein M., Sippl M. (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35(Web Server issue), W407–W410. https://doi.org/10.1093/nar/gkm290
Vajda S., Yueh C., Beglov D., Bohnuud T., Mottarella S.E., Xia B., Hall D.R., Kozakov D. (2017) New additions to the ClusPro server motivated by CAPRI. Proteins. 85(3), 435–444. https://doi.org/10.1002/prot.25219
Kozakov D., Beglov D., Bohnood T., Mottarella S.E., Xia B., Hall R., Vajda S. (2013) How good is automated protein docking? Proteins. 81(12), 1–22. https://doi.org/10.1002/prot
Kozakov D., Hall D.R., Xia B., Porter K.A., PadhornyD., Yueh C., Beglov D., Vajda S. (2017) The ClusPro web server for protein – protein docking. Nat. Protoc. 12(2), 255–278. https://doi.org/10.1038/nprot.2016.169
Soria-Guerra R.E., Nieto-Gomez R., Govea-Alonso D.O., Rosales-Mendoza S. (2015) An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J. Biomed. Inform. 53, 405–414. https://doi.org/10.1016/j.jbi.2014.11.003
Wagstaff S.C., Laing G.D., Theakston R.D.G., Papaspyridis C., Harrison R.A. (2006) Bioinformatics and multiepitope DNA immunization to design rational snake antivenom. PLoS Med. 3(6), 832–844. https://doi.org/10.1371/journal.pmed.0030184
Patronov A., Doytchinova I. (2013) T-cell epitope vaccine design by immunoinformatics. Open Biol. 3(1), 120139. https://doi.org/10.1098/rsob.120139
Terpe K. (2003) Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 60(5), 523–533. https://doi.org/10.1007/s00253-002-1158-6
Kazlauskas R.J., Bornscheuer U.T. (2009) Finding better protein engineering strategies. Nat. Chem. Biol. 5(8), 526–529. https://doi.org/10.1038/nchembio0809-526
Ghorban Hosseini N., Tebianian M., Farhadi A., Hossein Khani A., Rahimi A., Mortazavi M., Hosseini S.Y., Taghizadeh M., Rezaei M., Mahdavi M. (2017) In silico analysis of L1/L2 sequences of human papillomaviruses: implication for universal vaccine design. Viral. Immunol. 30(3), 210–223. https://doi.org/10.1089/vim.2016.0142
Farhadi T., Karimia Z., Younes G., Nezafatb N., Hemmati S., Erfani N. (2015) Production of a novel multi-epitope vaccine based on outer membrane proteins of Klebsiella pneumoniae. Trends Pharm. Sci. 1, 167–172.
Nosrati M., Behbahani M., Mohabatkar H. (2019) Towards the first multi-epitope recombinant vaccine against Crimean–Congo hemorrhagic fever virus: a computer-aided vaccine design approach. J. Biomed. Informat. 93, 103160. https://doi.org/10.1016/j.jbi.2019.103160
Aryanzad S.A., Zeinoddini M., Haddadi A., Nazarian S., Sajedi R.H. (2020) In silico design of chimeric and immunogenic protein-containing IpaB and IpaD as a vaccine candidate against Shigella dysenteriae. Curr. Proteom. 17(4), 333–341. https://doi.org/10.2174/1570164617666190906145843
Sohail M.S., Ahmed S.F., Quadeer A.A., McKay M.R. (2021) In silico T cell epitope identification for SARS-CoV-2: progress and perspectives. Adv. Drug Del. Rev. 171, 29–47. https://doi.org/10.1016/j.addr.2021.01.007
Jaydari A., Forouharmehr A., Nazifi N. (2019) Determination of immunodominant scaffolds of Com1 and OmpH antigens of Coxiella burnetii. Microb Pathog. 126, 298–309. https://doi.org/10.1016/j.micpath.2018.11.012
Zhang G., Peng Y., Schoenlaub L., Elliott A., Mitchell W., Zhang Y. (2013) Formalin-inactivated Coxiella burnetii phase I vaccine-induced protection depends on B cells to produce protective IgM and IgG. Infect. Immun. 81(6), 2112–2122. https://doi.org/10.1128/IAI.00297-13
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология