

Молекулярная биология, 2022, T. 56, № 2, стр. 181-205
Прогерин и его роль в ускоренном и естественном старении
a Петербургский институт ядерной физики им. Б.П. Константинова, Курчатовский научный центр
188300 Гатчина, Россия
b Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Россия
* E-mail: m_mosev@mail.ru
Поступила в редакцию 18.02.2021
После доработки 21.06.2021
Принята к публикации 21.06.2021
- EDN: QDNSBR
- DOI: 10.31857/S0026898422020124
Аннотация
Известные теории старения базируются на определенном метаболическом изъяне, негативно влияющем на жизнедеятельность клетки, будь то окислительный стресс, накопление дефектов в ДНК, исчерпание теломер, нарушения эпигенетических процессов. С позиций рассматриваемой в настоящем обзоре теории старения в основе всех этих нарушений лежит накопление на внутренней стороне ядерной оболочки белка прогерина – дефектного предшественника белка ядерного матрикса ламина А, сохранившего подлежащий удалению С-концевой остаток цистеина с присоединенной гидрофобной олигоизопреновой цепочкой. С помощью этой цепочки молекулы прогерина прикрепляются к внутренней мембране оболочки ядра, оттесняя примыкавшие к ней фибриллы ядерного матрикса и периферию хроматина. При этом изменяются морфология и форма ядра, нарушаются свойства его оболочки и вмонтированных в нее поровых комплексов. По мере накопления прогерина структурные искажения в ядре нарастают, вызывая все большие нарушения ядерно-цитоплазматического транспорта макромолекул, что ведет к нарушению клеточного метаболизма. Это приводит к нарастающей со временем гибели клеток и старению организма. Такой механизм старения приводит к развитию синдрома ускоренного старения (прогерии) Хатчинсона–Гилфорда (Hutchinson–Gilford progeria syndrome). Повышенная продукция прогерина при этой болезни связана с точечной мутацией c.1824C→T в экзоне 11 гена LMNA, кодирующего ламины А и С. Эта мутация стимулирует нестандартный сплайсинг первичного транскрипта при формировании мРНК предшественника ламина А, в результате чего образуется прогерин. Больные прогерией дети, получившие от одного из родителей указанную мутацию, быстро стареют и умирают в возрасте до 15 лет. Лечение прогерии направлено на предотвращение образования прогерина или уничтожение уже накопленного прогерина. В последнем случае перспективно применение рапамицина и его аналогов, а также других веществ, активирующих систему аутофагии, способную очищать клетку от прогерина. Обнаружение прогерина, хотя и в значительно меньшем количестве, у людей, не страдающих прогерией, указывает на участие этого белка и в “естественном” старении. Максимальный возраст, до которого может дожить человек, можно вычислить, если учитывать роль прогерина в сокращении теломер. Обнадеживающие предварительные результаты по очистке клеток от прогерина позволяют разработать оптимальную процедуру периодической очистки организма человека от прогерина, применение которой снизит темп старения.
ВВЕДЕНИЕ
Жить долго и счастливо – естественное стремление людей во все времена. Однако совсем недавно это стремление приблизилось к реальности благодаря двум обстоятельствам: научному прогрессу, позволяющему реально решать проблему долголетия, и нарастающей потребности общества в максимальной отдаче людей, достигших высшей квалификации в своем деле. В настоящее время создалась парадоксальная ситуация: Для того, чтобы достичь вершин знаний и опыта, необходимы 40–45 лет, и хорошо, если остается 15–20 “здоровых” лет, когда специалисты трудятся полноценно. Но именно их вклад определяет прогресс цивилизации, от которого в значительной степени зависит, можно ли избежать или преодолеть неизбежные в будущем глобальные катастрофы. Некоторые из них можно предсказать, опираясь на историю Земли, в которой были и глобальные потепления (до дневной температуры +70°С), и глобальные оледенения, когда Земля на миллионы лет превращалась в снежный шар, а также длившиеся сотни тысяч лет истечения лавы, столкновения Земли с крупными космическими телами и обнуления магнитного поля Земли. Эти катастрофы могут повториться в любой момент, а человечество к противостоянию им еще не готово. В пока еще далеком будущем добавятся необратимые явления, вызванные затвердением ядра и мантии Земли, инволюцией Солнца, а возможно, и столкновение галактик (подробно см. [1]). Для противостояния этим вызовам человечеству придется мобилизовать весь интеллектуальный потенциал, достойным вкладом в который станет оздоровление и значительное увеличение продолжительности жизни человека. Очевидным следствием станет увеличение длительности квалифицированного труда.
Современные теории старения, как правило, привязаны к определенным нарушениям метаболизма клетки, которые со временем становятся причиной ее гибели. К таким нарушениям относятся образование в митохондриях активных форм кислорода, вызывающих окислительный стресс [2–5], накопление нерепарированных повреждений ДНК [3, 6], исчерпание теломер [7, 8], нарушение эпигенетических модификаций [3].
Представленная в настоящем обзоре теория старения также основана на определенном нарушении метаболизма клетки, а именно, на нарушении транспорта макромолекул через оболочку клеточного ядра, обусловленного накоплением токсичного белка прогерина. Согласно представленной концепции, это нарушение влечет за собой уже упомянутые нарушения метаболизма, а накопление прогерина может оказаться базовой причиной старения клеток и всего организма.
Транспорт макромолекул из цитоплазмы в ядро и в обратном направлении осуществляется через поровые комплексы, вмонтированные в ядерную оболочку, образованную внутренней и внешней липидными мембранами.
Через поровые комплексы из цитоплазмы в ядро импортируются структурные белки и ферменты, а также рибосомные белки, которые входят в состав большой и малой субъединиц рибосомы, формирующихся в ядре. Эти крупные рибонуклеопротеиновые структуры через поровые комплексы переходят в цитоплазму, где из них собираются полные рибосомы. Из ядра в цитоплазму экспортируются также тРНК и мРНК, необходимые для синтеза белка на рибосомах.
Ядерно-цитоплазматический транспорт хорошо организован. Через ядерные поры в обоих направлениях курсируют транспортные молекулярные комплексы (экспортины и импортины) с грузом в одном направлении и “порожняком” обратно. При доставке груза из ядра в цитоплазму поступление энергии, необходимой при снятии груза, обеспечивает присутствующая в экспортине GTPаза (белок Ran), осуществляющая гидролиз ассоциированного с ним GTP до GDP с освобождением значительного количества энергии [9]. По возвращении в ядро GDP обменивается на GTP и транспортный цикл повторяется [10–12].
Белок с сигналом ядерной локализации связывается с импортином в цитоплазме и направляется в ядро, где к ним подключается RanGTP с последующим гидролизом GTP, и груз освобождается.
Чтобы выполнять свои функции, сложно устроенные ядерные поры должны сохранять форму и ориентацию, что возможно при определенной жесткости ядерной оболочки, в которую они вмонтированы.
Достаточную жесткость ядерной оболочке и округлую форму ядру придает плотное прилегание к оболочке периферийной области (ламины) фибриллярного ядерного матрикса [13], а также определенных участков хроматина. Вся эта конструкция фиксируется с помощью белков-скрепок.
Ранние исследования ядерного матрикса были проведены И.Б. Збарским и Г.П. Георгиевым [14]. Тогда же получили первые электронно-микроскопические снимки ядерного матрикса как гранулярно-фибриллярной структуры, заполняющей объем ядра [15].
В дальнейшем выявили фундаментальные функции ядерного матрикса как каркаса, организующего общее пространство, занимаемое хроматином в ядре, и его конформацию в форме закрепленных в матриксе петель [16]. В ядерном матриксе локализованы многокомпонентные ферментные комплексы, обеспечивающие выполнение таких функций, как репликация ДНК, репарация повреждений ДНК и транскрипция. Аппарат сплайсинга также закреплен в матриксе [17–20].
Очевидно, что размещение точек закрепления хроматина в ядерном матриксе строго детерминировано и скоординировано с локализацией указанных ферментных комплексов. Это возможно при условии высокоупорядоченной структуры самого ядерного матрикса.
Однако неповрежденный ядерный матрикс наблюдать не удается, поскольку для этого необходимо удалить из ядра все другие компоненты: растворимые белки и нуклеиновые кислоты. Кроме того, образцы (заключенные в смолу ультратонкие срезы), применяемые в электронно-микроскопических исследованиях, подвергаются жестким процедурам обезвоживания, обезжиривания и др. На таких срезах ядерный матрикс выглядит как малоупорядоченная сеть фибрилл, заполняющая весь объем ядра (рис. 1). На периферии плотность сети повышена. Этот периферийный слой ядерного матрикса, подстилающий оболочку ядра, именуется ламиной.
Фибриллы, по-видимому, могут содержать РНК, так как РНКаза способна разрушить эту структуру [17, 20, 22, 23]. Однако в фибриллах ядерного матрикса преобладают белки, за которыми закрепилось название ламины [24]. Всего насчитывают четыре ламина: А, В1, В2 и С, молекулярные массы которых составляют 60–70 кДа. Молекулы ламинов имеют преимущественно фибриллярную структуру, в электронном микроскопе они выглядят, как палочки. Функционируют ламины, как правило, в форме димеров [25, 26].
Ламины А и С кодируются одним геном LMNA, состоящим из 12 экзонов. Все они входят в состав мРНК преламина А. В результате альтернативного сплайсинга образуется мРНК ламина С, содержащая первые 10 экзонов [27]. Ламины В1 и В2 близки по составу, но кодируются отдельными генами.
Ламины образуют два типа фибрилл, формирующих ядерный матрикс. В состав фибрилл одного типа входят ламины А и С, фибриллы другого типа содержат ламины В1 и В2 [13].
В норме периферийная область ядерного матрикса (ламина) не только плотно прилегает к ядерной оболочке (подстилает ее), но во многих точках прикреплена к ней с помощью белков, интегрированных во внутреннюю мембрану ядерной оболочки. Фибриллы, сформированные ламинами А и С, связываются с интегрированными белками, которые имеют также выход в цитоплазму, где взаимодействуют с цитоплазматическими промежуточными фибриллами [28, 29]. Фибриллы, состоящие из ламинов типа В, связаны со встроенными во внутреннюю мембрану ядерной оболочки молекулами рецептора ламина В (lamin B receptor, LBR). С-концевые гидрофобные домены LBR интегрированы во внутреннюю мембрану ядерной оболочки без выхода в цитоплазму, тогда как N-концевой гидрофильный участок LBR (более 200 аминокислотных остатков) выступает в нуклеоплазму, где связывается с фибриллами, образованными ламинами В, а также с хроматином, который оказывается “подтянутым” к ядерной оболочке [30–34].
Предваряя дальнейшее обсуждение, необходимо отметить, что состав и структура ядерного матрикса не стандартны, они могут существенно различаться в клетках разного типа и на разных стадиях развития. Так, в активно делящихся эмбриональных стволовых клетках ламины А/С (точнее фибриллы, сформированные этими ламинами) практически отсутствуют. Они появляются, когда клетки вступают в дифференцировку [35, 36] и играют в этом процессе ключевую роль, направляя его в нужное русло [37]. Наглядно роль ламинов А/С в процессе дифференцировки показана на примере мезенхимальных стволовых клеток [38]. Эти мультипотентные клетки были выделены из костного мозга молодых мужчин и выращены in vitro. Путем трансфекции в эти клетки вносили либо активно экспрессируемый ген LMNA (повышение синтеза ламинов А/С и их содержания в ядерном матриксе), либо малые интерферирующие РНК, подавляющие экспрессию гена LMNA (снижение содержания ламинов А/С в ядерном матриксе).
Оказалось, что в первом случае мезенхимальные стволовые клетки дифференцируются преимущественно в костные клетки (остеобласты), а во втором – в жировые клетки (адипоциты). Механизм воздействия ламинов А/С на дифференцировку окончательно не установлен [37], но предложена его версия [38], основанная на том факте, что при повышении в ядерном матриксе содержания ламинов А/С возрастает содержание в ядре β-катенина – внутриклеточного компонента сигнальной системы Wnt/β-катенин, способствующей дифференцировке [39]. Секретируемый компонент этой системы – белок Wnt – воздействует на рецепторы подлежащих дифференцировке стволовых клеток. По сигналу, переданному рецептором в цитоплазму, находящийся там β-катенин, ассоциированный с определенными факторами транскрипции, транспортируется в ядро, где под действием экзогенных транскрипционных факторов происходит активация генов, направляющих дифференцировку в русло образования остеобластов. В этой схеме много белых пятен. В частности, не ясна роль ламинов А/С в поступлении β‑катенина в ядро. Учитывая это, можно предположить, что в данном случае имеет значение обогащение периферической области ядерного матрикса фибриллами, сформированными из ламинов А/С, которые через линкерные белки, интегрированные в ядерную оболочку, связаны с цитоплазматическими промежуточными фибриллами, служащими транспортными артериями. Каким-то образом эта система ядерных и цитоплазматических фибрилл способствует проникновению в ядро β-катенина и сопровождающих его факторов транскрипции.
С возрастом содержание ламинов А/С в ядерном матриксе клеток костного мозга снижается [40]. Соответственно, меньшее количество β-катенина и ассоциированных с ним факторов транскрипции поступает в ядро, вследствие чего путь адипоцитарной дифференцировки стволовых клеток становится все более вероятным. Именно этим объясняется развитие у пожилых людей остеопороза (снижение прочности и объема костной ткани, замещаемой жировой тканью), приводящего к повышению хрупкости костей, тяжелым переломам при безобидных в молодости падениях.
Таким образом, очевидна ключевая роль ядерного матрикса, сформированного из ламинов, в клеточном метаболизме. Поэтому разрушение этой сложной конструкции не пройдет бесследно для жизнедеятельности клетки и может привести к ее гибели. С мутациями в гене LMNA, кодирующем ламины А и С, связаны более 10 серьезных заболеваний, получивших общее название ламинопатии [41, 42]. Мы рассмотрим одно из таких заболеваний, имеющее прямое отношение к теме этой статьи.
СИНДРОМ УСКОРЕННОГО СТАРЕНИЯ (ПРОГЕРИЯ)
Ускоренное старение, или синдром прогерии Хатчинсона–Гилфорда (Hutchinson–Gilford progeria syndrome, HGPS) –редкое (один случай на 4 млн новорожденных), но тяжелое заболевание, при котором признаки старения проявляются через несколько месяцев после рождения. Больные прогерией живут не более 15 лет. За это время они проходят все периоды жизни человека (рис. 2). Смерть наступает, как правило, из-за отказа сердечно-сосудистой системы [43–45].

Причиной прогерии является точечная мутация в гене LMNA (экзон 11; с.1824C> T), возникшая в линии половых клеток одного из родителей. Эта мутация не передается по наследству из-за того, что ее носители старятся и умирают в детском возрасте.
Указанная мутация стимулирует (делает узнаваемым для сплайсинга) скрытый (криптический) сайт сплайсинга в экзоне 11 первичного транскрипта гена LMNA.
мРНК ламина С содержит 10 первых экзонов и кодирует ламин С, а мРНК ламина А состоит из 12 экзонов. В норме эта мРНК кодирует предшественник ламина А (преламин А), который проходит несколько этапов процессинга, прежде чем превратиться в зрелый ламин А. Один из этапов образования зрелого ламина А – осуществляемое фарнезилтрансферазой присоединение к остатку цистеина вблизи С-конца преламина А гидрофобной молекулы, состоящей из трех звеньев изопрена. Этот процесс (изопренилирование, фарнезилирование) характерен для белков, которым предстоит функционировать в контакте с липидными структурами клетки. В норме на заключительном этапе процессинга протеиназа Zmpste24 “отрубает” С-концевую область (15 аминокислотных остатков) с фарнезилированным остатком цистеина, образуя зрелый ламин А (рис. 3а).
Рис. 3.
Схемы нормального процессинга преламина А с образованием зрелого ламина А (а) и аберрантного процессинга укороченного (мутантного) преламина А с образованием прогерина (б).
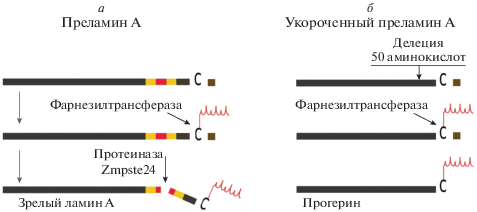
В сплайсинге первичного транскрипта гена LMNA с мутацией с.1824C>T участвует не обычно используемый, а криптический сайт в экзоне 11, вследствие чего этот экзон становится короче на 150 нуклеотидных звеньев. Соответственно, в мутантном преламине А отсутствует участок из 50 аминокислотных остатков вблизи C-конца [46, 47]. Но именно в этом участке расположен сайт, по которому в норме протеиназа Zmpste24 отрезает С-концевую область, содержащую фарнезилированныый цистеин. Поэтому из мутантного преламина А не образуется зрелый ламин А, а сохраняется фарнезилированный С-концевой остаток цистеина (рис. 3б). Эта молекула получила название прогерин [48, 49].
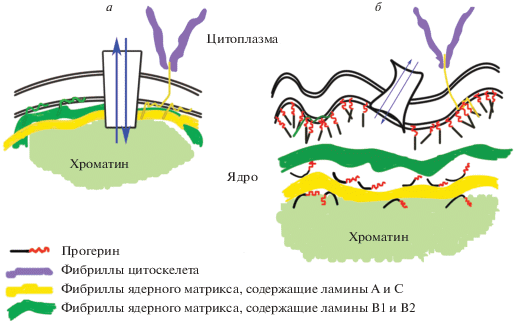
Прогерин, в отличие от зрелого ламина А, не способен участвовать в формировании фибрилл. Поступившие в ядро молекулы прогерина, используя присутствующую в них гидрофобную олигоизопреновую цепочку как якорь, закрепляются на внутренней мембране ядерной оболочки. При этом основная часть палочкообразной молекулы прогерина находится вне мембраны. Связываясь с мембраной, молекулы прогерина оттесняют периферийные фибриллы ядерного матрикса (ламину) и край хроматина, нарушая их контакт с ядерной оболочкой (рис. 4). Из-за этого нарушается вся система внутренней организации клеточного ядра [50–56].
Рис. 4.
Фрагменты ядерной оболочки. а – Не поврежденная ядерная оболочка, которую подстилают изнутри фибриллы ядерного матрикса и периферия хроматина. б – Ядерная оболочка ослаблена вмешательством прогерина, оттеснившего от нее фибриллы ядерного матрикса и периферию хроматина.
Недавно обнаружен также негативный эффект взаимодействия прогерина с ламином А [57]. Можно предположить, что “прилипание” прогерина к фибриллам, сформированным ламинами А и С (рис. 4), способствует их удалению от ядерной оболочки. Показано, что прием ингибитора такого взаимодействия (прогеринина) существенно увеличивал продолжительность жизни мышей, моделирующих прогерию [57].
Ядерная оболочка, лишившись опоры, теряет прочность, а само ядро утрачивает форму, сморщивается, материал в нем перераспределяется. Эти нарушения наблюдали многие авторы, используя обычную и иммунофлуоресцентную микроскопию (рис. 4а) [46, 58–62]. При этом нарушается конструкция и ориентация вмонтированных в ядерную оболочку поровых комплексов, проходимость которых снижается.
Именно нарушение ядерно-цитоплазматиоческого транспорта влияет и на другие метаболические процессы, от которых зависит состояние клетки. Следует заметить, что мы не касаемся случаев, когда транспорт через ядерную оболочку полностью заблокирован. Такая ситуация возможна при достаточном накоплении прогерина, но это означает скорую гибель клетки. Рассмотрена промежуточная ситуация, когда транспорт замедлен, пропорции искажены, но процесс не остановлен.
Наиболее существенно может тормозиться транспорт крупных частиц, таких как субъединицы рибосом и мРНК, что приводит к снижению синтеза белков до уровня, не способного обеспечить своевременную замену вышедших из строя белков на вновь образованные. Вследствие нарушения синтеза белков и их транспорта в ядро при прогерии существенно изменяется состав ядерных белков, что подтверждено прямым определением и сравнением с составом в нормальных клетках [63]. Эти же причины лежат в основе нарушения системы белков, участвующих в ядерно-цитоплазматическом транспорте (белки поровых комплексов, транспортные молекулы и энергоносители), что делает этот транспорт еще более дефектным.
Снижение синтеза белков при прогерии отражается на всех процессах, происходящих в цитоплазме, но наиболее ярко это проявляется в нарушении работы митохондрий, где происходит синтез АТР. В качестве побочных продуктов при этом образуются активные формы кислорода, пероксиды, свободные радикалы. В митохондриях функционируют антиоксидантные системы, нейтрализующие эти активные радикалы.
В норме антиоксидантные системы справляются с этой задачей, и количество активных форм кислорода, избежавших нейтрализации, незначительно. Соответственно, системы утилизации поврежденных клеточных структур – протеасомы и аутофагия – успевают освобождать клетки от “хлама”, благодаря чему их жизнедеятельность не нарушается.
Однако при прогерии приток антиоксидантных систем в митохондрии ослаблен. Вследствие этого в клетках накапливаются активные формы кислорода, которые окисляют белки, липиды, нуклеиновые кислоты (окислительный стресс) [64–67], и системы утилизации не справляются с возросшей нагрузкой. Ситуация усугубляется тем, что сами эти системы подвергаются атакам активных форм кислорода, а скорость их обновления, как и других клеточных структур, снижена. Скопление поврежденных макромолекул ускоряет гибель клеток.
Прогерин, как уже сказано, оттесняет хроматин от точек закрепления на ядерной оболочке. Это нарушает конформацию периферии хроматина. Стандартно периферия хроматина находится в конденсированном состоянии (в конформации гетерохроматина) [68]. В этих условиях ДНК надежно защищена от внешних воздействий. При прогерии эта компактная конформация нарушается. К изменению архитектуры хроматина приводит также нарушение притока из цитоплазмы гистон-метилтрансферазы [69, 70]. В результате ДНК оказывается более доступной для деградирующих воздействий.
Значительную долю ДНК на периферии хроматина составляют теломеры, располагающиеся на концах хромосом. Теломеры, образованные множественными повторами, состоящими из шести нуклеотидов TTAGGG, сокращаются с концов на несколько звеньев при каждом акте репликации хромосомной ДНК, предотвращая деградацию кодирующих генов [6, 71].
Сокращения теломер при репликации ДНК линейных хромосом неизбежны. Их можно считать плановыми, и клетки организма к ним приспособлены. Так, вступившие в дифференцировку эмбриональные клетки имеют теломеры, размер которых достаточен для прохождения числа клеточных циклов, позволяющих достичь состояния терминально дифференцированных (специализированных) клеток.
В области теломер происходят не только плановые (репликативные) сокращения концов хромосом, но, как и в других областях хромосом, возникают случайные повреждения ДНК, в том числе, двухцепочечные разрывы. В нормальных условиях системы репарации, присутствующие в клеточном ядре, ликвидируют эти повреждения, поддерживая жизнеспособность клетки. Однако при прогерии ситуация многократно ухудшается.
Вследствие нарушения компактной конформации периферии хроматина учащаются повреждения теломерной ДНК как активными формами кислорода (окислительный стресс), так и нуклеазами [72].
Среди возможных воздействий на ставшие доступными теломеры отметим транскрипцию [7, 73]. Транскрипты берут начало в субтеломерной области хромосомной ДНК и распространяются в область теломер при отсутствии определенной точки терминации. Поэтому транскрипты теломер представляют собой набор гетерогенных по размеру длинных некодирующих РНК (lncRNA). Присутствие этих РНК подавляет активность теломеразы [74, 75]. К тому же процесс транскрипции теломер активирует экзонуклеазу, деградирующую теломеры [76]. Ситуация усугубляется как нарушением ядерно-цитоплазматического транспорта, вызванным сокращением поступления белков из цитоплазмы, в том числе, входящих в состав теломеразы, так и усиленным воздействием на теломерную ДНК активных форм кислорода.
В результате всех этих “внеплановых” воздействий происходит ускоренное сокращение теломер и кодирующие гены оказываются близко к концу хомосомы. Это приводит к преждевременной остановке клеточного цикла и возможной гибели клетки [77, 78].
При прогерии очевидной базовой причиной событий, ведущих к гибели клеток и старению организма, является накопление в клеточных ядрах прогерина [65]. Следовательно, противостоять следует именно накоплению прогерина. Существуют два очевидных подхода к решению этой проблемы: воспрепятствовать образованию прогерина, либо очистить клетки от уже образованного прогерина. В исследованиях с использованием модельных животных (мышей) и культур клеток больных прогерией уже получены определенные результаты. Эффективными показали себя ингибиторы фарнезилтрансферазы, уже применявшиеся ранее в онкологии с целью инактивации онкобелка Ras, активируемого при фарнезилировании. На модельных животных, культурах фибробластов больных прогерией, а также на культуре клеток HeLa, несущих конструкции, продуцирующие прогерин, показано, что подавление активности фарнезилтрансферазы улучшает состояние клеток. В частности, применение лонафарниба (lonafarnib) и золедроната (zoledronate) как самостоятельно, так и в комбинации с другими агентами, например, с правастатином (pravastatin), позволяет ядру принять правильную форму и нормализует клеточный метаболизм [49, 65, 79–88]. Получены данные, свидетельстующие, что аналогичным действием обладают некоторые аминопиридины [89].
Лонафарниб опробован также на группе детей, больных прогерией. Согласно предварительным результатам, систематический прием этого ингибитора фарнезилтрансферазы в течение 2 лет снизило смертность больных прогерией детей более чем в 3 раза по сравнению с больными прогерией детьми того же возраста, не принимавшими этот препарат [90].
Однако следует иметь в виду, что фарнезилирование – это модификация, необходимая для функционирования ряда белков, участвующих в клеточном метаболизме. К ним относится и преламин А, фарнезилирование которого входит в схему процессинга, в результате которого образуется ламин А. Поэтому использование такого подхода может быть сопряжено с неприемлемыми побочными эффектами. Возможно, по этим причинам предпринятые несколько лет назад клинические испытания с применением ингибиторов фарнезилирования еще не завершены [62, 91].
Разрабатываются также подходы к лечению прогерии, направленные на предотвращение образования несущего делецию преламина А, а следовательно, и прогерина. В первую очередь, такой подход может быть осуществлен на уровне мРНК.
С этой целью применяют синтетические антисмысловые олигонуклеотиды, которые содержат те же азотистые основания, что и природные нуклеиновые кислоты, но в остальные элементы внесены изменения, защищающие олигонуклеотид от действия нуклеаз, но не препятствуют их комплементарному связыванию с природными нуклеиновыми кислотами. Обычно к олигонуклеотиду прикрепляют молекулу-проводника, облегчающую проникновение олигонуклеотида в клетку. В организм препарат вводят путем внутривенной инъекции. О результате такого эксперимента судят по состоянию легкодоступных для исследования фибробластов кожи пациентов или экспериментальных животных. Этот прием можно применить к мРНК преламина А с делецией. Олигонуклеотид должен быть комплементарен специфичному для этой мРНК участку на стыке концов делетированной области. “Прилипший” олигонуклеотид не позволит осуществить синтез белка.
На практике образование прогерина предотвращалось на уровне сплайсинга. В этом случае задача антисмыслового олигонуклеотида состояла в том, чтобы перекрыть измененный сайт сплайсинга экзона 11 в первичном транскрипте мутантного гена LMNA, что будет препятствовать образованию “неправильного” разрыва при сплайсинге.
Это воздействие антисмыслового олигонуклеотида существенно улучшало состояние клеток: содержание прогерина в них снижалось. При этом ядро принимало правильную форму, восстанавливались модификациии гистонов в гетерохроматине, возобновлялось нормальное функционирование генов и т.д. [92, 93]. Такой подход считается перспективным и в настоящее время [94–96]. Очевидную проблему представляет необходимость внедрения антисмысловой конструкции, если не во все, то в большинство клеток организма.
Для уменьшения продукции прогерина у носителей соответствующей мутации в гене LMNA предложен подход, основанный на снижении активности гена LMNA при взаимодействии с его промотором транс-изомера ретиноевой кислоты (производное витамина А). Совместное применение транс-ретиноевой кислоты и рапамицина показало высокую эффективность восстановлении нормального фенотипа фибробластов, полученных от больных прогерией [97].
Установлено, что витамин D улучшает морфологию ядер в культурах клеток, взятых у больных прогерией. При этом количество прогерина существенно уменьшается, по-видимому, вследствие снижения активности гена LMNA [98].
Все чаще рассматривают возможность применения генной терапии при прогерии [62, 99–101]. Речь идет о редактировании мутантного гена LMNA с целью коррекции описанной точечной мутации. За основу применяемой с этой целью конструкции принята одна из бактериальных систем, предназначенных для разрушения проникшей в клетку чужой ДНК, главным образом, фаговой. В геноме бактерий присутствует домен, содержащий сегменты, гомологичные участкам возможных “вражеских” ДНК. Транскрипты этих сегментов в комплексе с нуклеазой выявляют проникшую в клетку гомологичную ДНК, связываются с нею, предоставляя возможность нуклеазе приступить к работе – деструкции чужеродной (фаговой) ДНК [102, 103]. В клетках эукариот подобных систем нет, но если их встроить в эукариотические клетки, то они могут в них функционировать.
В интересующем нас случае конструкция CRISPR/Cas9, заимствованная у бактерий Streptococcus pyogenes, несет гидовую РНК (guiding RNA; gRNA), содержащую сегмент, гомологичный целевому (содержащему мутацию) участку в экзоне 11 гена LMNA. Эта РНК должна связаться с гомологичным участком ДНК и предоставить эндонуклеазе Cas9 возможность произвести в нем двухцепочечные разрывы. Эти разрывы могут быть ликвидированы системами репарации клетки с образованием делеций, что приведет к сдвигу рамки в транскрипте, изменяющем состав последующих кодонов. Поэтому белковые продукты обработанных таким способом мутантных генов LMNA будут иметь на С-конце совсем иные аминокислотные остатки и фарнезилирование не произойдет. Скорее всего, эти белки подвергнутся гидролизу. Указанные манипуляции не затрагивают ламин С, ограниченный первыми 10-ю экзонами, поэтому он образуется в нормальных количествах.
Полученные результаты умеренно позитивны. После внесения указанной конструкции в клетки (фибробласты), полученные от мышей, моделирующих прогерию, и от больных прогерией, клеточное ядро приобретает нормальную форму, что предполагает восстановление метаболизма. Инъекции той же конструкции мышам, моделирующим прогерию, увеличили продолжительность их жизни на 25% [101]. Методически близкое и по полученным результатам исследование проведено параллельно другой группой [99].
Следующим этапом предотвращения образования прогерина при прогерии должно стать истинное редактирование несущего мутацию гена LMNA, т.е. возвращение его к дикому типу. Предполагалось, что в описанную выше конструкцию внесут немутантную ДНК, которая в процессе репарации двухцепочечных разрывов заместит гомологичный мутантный вариант [104].
На практике реализован вариант, не требующий внесения разрывов в ДНК [105]. С этой целью в приведенную выше конструкцию вместо эндонуклеазы Cas9 внесен фермент, способный дезаминировать аденин в составе ДНК [106]. Образующийся при этом гипоксантин (соответствующее звено в ДНК – инозин) узнается при репликации как гуанин. В результате восстанавливается немутантная пара G-C. Согласно [105], в культуре фибробластов, принадлежавших больным прогерией, такое редактирование охватывало до 91% клеток. Морфология ядер в этих клетках нормализовалась. Введение того же препарата мышам, моделирующим прогерию, вдвое увеличивало срок их жизни. Очевидно, что эффективность лечения прогерии будет зависеть от числа клеток в органах и тканях, в которых осуществлено искомое редактирование. Проблемой могут оказаться терминально дифференцированные специализированные клетки органов и тканей, прекратившие репликацию хромосомной ДНК, ибо без репликации ДНК возврат к дикому типу не может быть завершен.
Описанные выше подходы направлены на предотвращение образования прогерина, но ни один из них не выполняет эту задачу полностью. Поэтому актуальной остается проблема уничтожения уже образовавшегося и накапливающегося в клетках прогерина. Оказалось, что эта задача выполнима. Такой вывод следовал из результатов, полученных с применением антибиотика рапамицина и его аналогов (эверолимуса и других), которые воздействуют на метаболизм клеток млекопитающих. Эти антибиотики используются при трансплантации органов в качестве иммунодепрессантов. Показано также действие рапамицина при нейродегенеративных заболеваниях [107, 108].
Для обсуждения проблемы старения важно отметить способность рапамицина вступать в контакт с серин-треониновой киназой mTOR (mechanistic Target Of Rapamycin) [109–112]. mTOR является ключевым элементом инициируемого инсулином “каскада старения”, контролируя многие процессы жизнедеятельности клетки [51, 113]. В частности, mTOR подавляет аутофагию [113, 114]. В этой системе очистки клетки от вредящих ей структур, в том числе белков, ферменты, осуществляющие деструкцию, заключены в липидные пузырьки (липосомы), куда доставляются подлежащие уничтожению структуры [115–120]. Важным открытием стал тот факт, что к таким “вредным” структурам аутофагия может причислить и прогерин. Подавляя активность киназы mTOR, рапамицин активирует аутофагию, чем способствует очистке клеток от прогерина [60, 121].
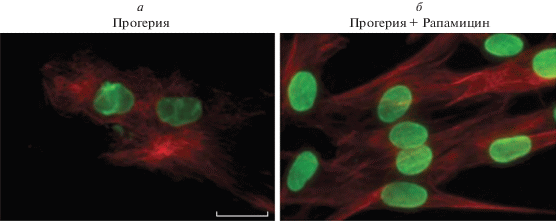
В присутствии рапамицина в культуре фибробластов кожи больного прогерией наблюдается визуальная нормализация клеточного ядра (рис. 5) и, по крайней мере частично, восстанавливается ядерно-цитоплазматический транспорт макромолекул, что проявляется в нормализации эпигенетических процессов (модификации ДНК и гистонов) [60, 122]. Эта инициированная активацией аутофагии цепочка восстановительных процессов приводит к оздоровлению организма и отдалению его старения [123–125].
Рис. 5.
Иммунофлуоресцентная микроскопия ядер в культуре фибробластов, взятых у больного прогерией. а – Контрольная культура (без применения лекарственного средства). б – В культуру в течение 14 дней вносили рапамицин. Красный цвет – антитела к тубулину. Зеленый цвет – антитела к ядерным белкам ламинам А/С. Масштабная линия – 10 мкм [60]. Перепечатано из [60] с разрешения издательства.
Создается впечатление, требующее, однако, дополнительных доказательств, что аутофагия, как система очистки, начинает узнавать прогерин как своего “клиента” только после ее активации рапамицином, хотя и без того данная система эффективна в отношении других “испорченных” структур. Возможно, под действием рапамицина активируются некие дополнительные элементы системы аутофагии, благодаря чему прогерин распознается как структура, подлежащая уничтожению.
Сходным с рапамицином действием обладает антиоксидант сульфорафан (sulforaphane). Это содержащее серу органическое соединение растительного происхождения в наибольшем количестве присутствует у крестоцветных, особенно у брокколи. Под влиянием сульфорафана в фибробластах, полученных от больных прогерией, повышалась эффективность аутофагии и происходила очистка этих клеток от прогерина. Хорошего эффекта удалось добиться при последовательном (раздельном) применении сульфорафана и лонафариба – ингибитора фарнезилтрансферазы [63, 126].
Очисткой клеток от поврежденных белков занимаются также протеасомы – органеллы, содержащие наборы протеолитических ферментов [127, 128].
Блокирование протеасомного пути с помощью пептидного ингибитора протеасом MG132 не ухудшило очистку от прогерина фибробластов, взятых у больных прогерией. Следовательно, протеасомы не участвуют в утилизации прогерина. Более того, оказалось, что подавление протеасомного пути очистки клеток приводит к активации аутофагии. Показано также, что внесенный в клетки MG132 препятствует действию факторов сплайсинга SRSF1 и SRSF-5 (Serine/arginine-rich splicing factor), которые содействуют использованию “скрытого” сайта при сплайсинге первичного транскрипта гена LMNA с образованием впоследствии прогерина. Таким образом, введение MG132 оказывает двойной эффект: усиливает очистку клеток от прогерина и снижает его синтез. В результате, как и при применении рапамицина, наблюдается нормализация формы и морфологии клеточного ядра [129, 130].
В настоящее время предложено значительное число веществ и схем их применения при прогерии [62, 87, 130]. На клеточных культурах и моделирующих прогерию мышах испытывают как отдельные вещества, так и их комбинации, направленные на максимальное снижение содержания прогерина в клетке, нормализации таким образом структуры ядра и транспорта через его оболочку. Клинические испытания должны окончательно выявить наиболее эффективные и безопасные из них.
ЕСТЕСТВЕННОЕ СТАРЕНИЕ И ПРОГЕРИН
Принципиально важным стало обнаружение прогерина в клетках здоровых людей, причем с возрастом прогерин накапливается [44, 49, 131–134]. Это означает, что существует возможность (с малой, но конечной вероятностью) ошибочного использования “скрытого” сайта сплайсинга в экзоне 11 гена LMNA дикого типа, вследствие чего образуется мРНК не преламина А, а предшественника прогерина. У людей в возрасте около 80 лет количество прогерина составляет примерно 1% от содержания нормального (зрелого) ламина А, что, казалось бы, немного, но локализация прогерина вблизи ядерной оболочки может оказаться причиной нарушения ее контакта с фибриллами ядерного матрикса и с хроматином со всеми последствиями, наблюдаемыми при прогерии. Разница в том, что в этом случае прогерин накапливается медленнее, а нарушения клеточного метаболизма нарастают не столь резко. Такая трактовка делает накопление прогерина важной, если не определяющей, причиной старения и в отсутствие прогерии.
Существенно также, что в клетках сохраняется некоторое количество преламина А, избежавшего воздействия протеиназы Zmpste24 и поэтому сохранившего С-концевой остаток цистеина, несущий гидрофобную цепочку (рис. 3). По своим свойствам, в том числе по способности связываться с внутренней мембраной оболочки ядра и, следовательно, по токсичности преламин А практически не отличается от прогерина. Мыши, у которых нарушена функция протеиназы Zmpste24 и потому не может быть завершен процессинг преламина А, имеют такие же признаки раннего старения, как и мыши, моделирующие прогерию [135–137].
Обнаружение прогерина у людей без прогерии позволяет предположить, что и в этом случае, уже касающемся каждого человека, все нарушения метаболизма, ведущие к старению, имеют то же происхождение, что и при прогерии – накопление в клеточных ядрах прогерина и фарнезилированного преламина А. Вызванные этим структурные изменения в ядре и нарушения ядерно-цитоплазматического транспорта приводят к нарушениям клеточного метаболизма, приводящим к гибели клеток. Это указывает также на отсутствие принципиальных различий между естественным старением и прогерией. Разный темп старения определяется различиями в скорости накопления прогерина в клеточных ядрах.
Приведем ряд доводов, подтверждающих эту гипотезу.
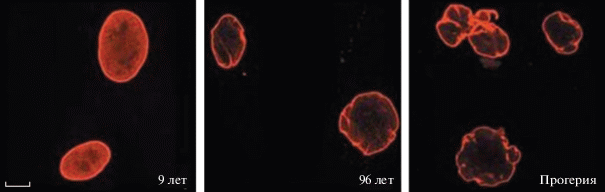
(а) У “естественно” состарившихся людей клеточные ядра имеют такие же нарушения формы и морфологии, как и у больных прогерией “состарившихся” детей [44] (рис. 6б).
Рис. 6.
Нарушение формы и морфологии клеточного ядра при “естественном” старении. а – Ребенок, 9 лет; б – престарелый человек, 96 лет; в – ребенок, больной прогерией. Иммунофлуоресцентная микроскопия. Использованы антитела к ламину А. Масштабная линия – 10 мкм. Перепечатано из [44] с разрешения издательства.
(б) Потенциал рапамицина как средства, продлевающего жизнь, впервые был показан на мышах без прогерии (животным начинали давать рапамицин после 300 дней жизни) [138]. Как и при прогерии, рапамицин блокирует в “каскаде старения” киназу mTOR, благодаря чему возрастает активность аутофагии и улучшается очистка клеток от прогерина. Это позволяет ядру поддерживать транспорт через его оболочку. Вследствие нормализации метаболизма клетки дольше сохраняют стабильность, что на уровне организма проявляется в продлении жизни.
В разное время титул “эликсир долголетия” присуждали и другим веществам, а именно, полифенолу ресвератролу [139–142], полиамину спермидину [143, 144], метформину [118, 145]. Примечательно, что все они активируют аутофагию, а следовательно, и очистку клеток от прогерина. Голодание, приводящее к увеличению длительности жизни, также активирует аутофагию [146, 147], как и регулярные физические упражнения [148]. Естественно, сказанное нельзя считать прямым доказательством главной роли удаления прогерина, так как аутофагия очищает клетку и от других молекул и надмолекулярных структур, например, от ставших непригодными митохондрий. Однако очистка клеток от прогерина может в значительной степени предотвратить образование других токсичных форм.
Важно отметить, что существуют вещества, не подавляющие киназу mTOR, а, наоборот, повышающие ее активность. К таким веществам относятся некоторые аминокислоты, в первую очередь, метионин. В опытах на животных установлено, что добавка к пище метионина вредна для здоровья и сокращает жизнь. Отсюда и изменение отношения к белковой диете – от положительного к весьма настороженному [149]. Негативное действие метионина следует объяснить снижением активности аутофагии под действием комплекса, включающего активированную киназу mTOR. Ослабление аутофагии означает накопление в клетках веществ, подлежавших утилизации, в том числе прогерина, нарушает метаболизм клеток и ведет к их гибели.
Известно позитивное воздействие на организм аминокислоты глицина. Одно из свойств глицина – способность стимулировать фермент, деметилирующий метионин [149]. Поэтому прием глицина может в значительной степени компенсировать вредные последствия белков. Кстати, в растительных белках метионина меньше, чем в животных.
(в) Можно задать вопрос, почему прогерин не накапливается в поколениях, а в каждом новом организме все начинается с чистого листа. Дело в том, что у эмбриональных клеток, способных, благодаря действию теломеразы [150], к бесконечному делению, ген LMNA, кодирующий ламины А и С, не активен. Следовательно, фарнезилированным предшественникам ламина А взяться неоткуда. Ген LMNA активируется только тогда, когда эмбриональные стволовые клетки превращаются в зрелые стволовые клетки, в которых отсутствует теломеразная активность, и за ограниченное число делений они должны стать дифференцированными по типу окружающих их специализированных клеток данного органа [35, 36, 151]. Половые клетки относятся к эмбриональным. В них ген LMNA не активен и фарнезилированные предшественники ламина А не накапливаются. Соответственно, нет их в зиготе и в ранних эмбрионах вплоть до начала формирования органов, связанных с образованием зрелых стволовых и дифференцированных клеток. Можно сказать, что эволюция умело провела корабль жизни между Сциллой и Харибдой, управляя геном LMNA так, что существование человечества не ограничивается во времени и числе поколений, а у отдельных личностей срок жизни ограничен, в первую очередь, вследствие накопления прогерина в клетках их органов и тканей.
(г) При прогерии происходит быстрое физическое старение, но интеллект не затрагивается [62, 100, 152–154]. То же наблюдается у людей, не страдающих прогерией. Если сравнить людей в возрасте 17–25 и 60 лет, то очевидно различие их физических, но не интеллектуальных возможностей. Даже очень пожилые физически немощные люди могут обладать “светлым умом”.
Объяснить эту особенность интеллекта позволили данные, засвидетельствовавшие практическое отсутствие ламина А в нервных клетках (нейроны, глия), при том что ламин С присутствует в нормальном количестве [155]. Присутствие ламина С означает, что ген LMNA функционирует, как и система сплайсинга.
В исследованиях, проведенных на мышах, в том числе моделирующих прогерию [152, 155], установлен следующий механизм этого явления.
В некодирующей области мРНК преламина А и прогерина содержится участок, образующий комплекс со специфичной для нервных клеток микроРНК miR-9. Этот комплекс блокирует взаимодействие мРНК с рибосомой, вследствие чего преламин А и прогерин не образуются. Логично предположить, что благодаря их отсутствию нервные клетки нормально функционируют значительно дольше других клеток. Многие нейроны сохраняются в течение всей жизни организма [156]. Это обеспечивает целостность нейронных сетей, что может оказаться важным условием длительного сохранения памяти.
Нервные клетки можно получить нетрадиционно путем направленной дифференцировки мультипотентных клеток, происходящих из соматических клеток (фибробластов) больных прогерией. Интересно, что в них, как и в нервных клетках, активируется синтез miR-9, блокирующей синтез преламина А и прогерина [153].
Казалось бы, все ясно: нервные клетки сохраняют нормальный метаболизм благодаря тому, что в них не накапливаются фарнезилированные белки-предшественники ламина А. Однако дальнейшие эксперименты ситуацию осложнили.
Значение отсутствия прогерина для длительного сохранения нормального метаболизма нервных клеток изучали на линии мышей (HG-C), которые продуцировали мРНК прогерина, лишенную участка, с которым связывается miR‑9, благодаря чему в нейронах, как в других клетках, синтезируется прогерин [157]. Ожидаемый эффект токсичности прогерина проявился в нейронах периферической нервной системы, но не в центральной нервной системе, нервные клетки которой сохранили нормальный метаболизм.
Полученные результаты, из которых следовало, что нервные клетки центральной нервной системы (но не периферической) имеют дополнительный механизм защиты, направленный на нейтрализацию фарнезилированных предшественников ламина А в нервной клетке, удивили самих авторов. Ими отмечено также, что у мышей, моделирующих прогерию, помимо центральной нервной системы есть другие ткани (печень, почки), в клетках которых присутствие прогерина не препятствует их функционированию [157]. Очевидно, что клетки этих тканей, в отличие от фибробластов, мышечных клеток и ряда других, обладают свойствами, препятствующими проявлениям токсичности фарнезилированными предшественниками ламина А. Идентификация этих свойств позволит найти новые подходы к лечению как преждевременного, так и естественного старения. Пока же предполагаемая принципиальная близость процессов ускоренного и естественного старения позволяет использовать результаты, уже полученные при лечении больных прогерией, для увеличения продолжительности жизни всех людей. Эта перспектива привлекла к решению задачи излечения от такой редкой болезни, как прогерия, многих исследователей.
Так как редкие ошибки сплайсинга, приводящие к образованию прогерина, устранить вряд ли возможно, то при естественном старении следует применять подходы, направленные на снижение его содержания в клетках. К таким подходам относится активация аутофагии, способной утилизировать прогерин, а также фарнезилированный преламин А, случайно избежавший действия протеиназы Zmpste24 (рис. 3 и текст). В этом направлении достигнуты значительные успехи. Активирующие аутофагию рапамицин и его аналоги, а также другие аналогично действующие “эликсиры долголетия” можно рассматривать как лекарства первого поколения от старения. Следует ожидать, что вскоре появятся конкретные рекомендации по применению этих веществ (возможно, в комбинациях) для периодической очистки организма от токсичных фарнезилированных предшественников ламина А. Лечение должно приводить к реальному омоложению (снижению биологического возраста).
Является ли прогерин основным или даже единственным фактором, запускающим механизмы естественного старения, как это имеет место при прогерии? Ответить на этот вопрос помогут данные о присутствии прогерина у животных, срок жизни которых разнится от 2–3 лет у мышей до более 200 лет у китов. Особый интерес представляют такие эксперименты на животных, темп старения которых близок к таковому у детей, больных прогерией (кошки, собаки и др.). Возможно, такие исследования уже ведутся.
ПОНЯТИЕ МАКСИМАЛЬНОГО ВОЗРАСТА
Нельзя пройти мимо того обстоятельства, что многие исследователи придерживаются концепции существования механизма, ставящего определенный предел продолжительности жизни [158–160]. Согласно этой концепции, каждый вид имеет свою максимальную (предельную) длительность жизни. Приближенно ее оценивают по достоверно установленному возрасту известных долгожителей. Этот возраст у людей составляет 122 года. Согласно этому критерию, максимальная продолжительность жизни кошек и собак может составить 25 лет, лошади – около 40 лет, гренландского кита – свыше 200 лет.
В качестве аргумента в пользу существования фиксированного максимального возраста приводят следующее рассуждение. По сравнению с 19‑м веком благодаря, главным образом, успехам медицины средняя продолжительность жизни человека почти удвоилась. Значительно увеличилось число людей, живущих 90 и более лет, однако максимальный возраст (около 120 лет) не изменился.
Оказалось, что у людей старше 100 лет теломеры на концах хромосом соматических клетках практически исчерпаны (объектом исследования были лейкоциты) [160–165]. Эти люди близки к достижению возраста, когда клеточные циклы в клетках, еще находящихся в процессе дифференцировки, будут заблокированы из-за достижения теломерами предельно малых размеров. Тогда органы перестанут пополняться зрелыми специализированными клетками, что и приведет к гибели организма.
Следует задаться вопросом, почему длина теломер вновь образованных зрелых (специализированных) клеток, например, лейкоцитов, зависит от возраста организма, в котором они сформировались, т.е. чем значительнее возраст, тем короче теломеры во вновь образованных зрелых клетках.
Пополнение органов специализированными клетками и в молодости, и в старости происходит в результате дифференцировки клеток из популяции ранних предшественников, поддерживаемой в течение всей жизни благодаря теломеразной активности. Клетки из этой популяции, вступив в процесс дифференцировки, дают начало одному или, как это имеет место при кроветворении или образовании костной и жировой тканей, нескольким типам специализированных клеток. В процессе дифференцировки теломераза не активна, и укорочение теломер неизбежно. Сам процесс дифференцировки в каждом конкретном случае (например, при созревании лейкоцитов) стандартен и, казалось бы, не должен иметь возрастных отличий. Наблюдаемое у зрелых лейкоцитов снижение размеров теломер с возрастом организма может означать, что этот процесс инициирован еще в популяции клеток-предшественников. В этих плюрипотентных (но не эмбриональных) клетках сохраняется теломеразная активность, но возможна также активация гена LMNA, кодирующего ламины А/С. Соответственно, в них будут накапливаться прогерин и фарнезилированный преламин А. В числе вызываемых нарушений отметим деконденсацию периферии хроматина. Вследствие этого локализованные там теломеры становятся доступными для активных форм кислорода и ферментов, что приводит к добавочному (не репликативному) укорочению теломер, которое поначалу компенсируется теломеразой. Когда теломераза в клетках-предшественниках перестает справляться с возрастающей нагрузкой, начинается возрастное укорочение теломер. Поэтому у вступающих в дифференцировку клеток теломеры изначально тем короче, чем больше возраст организма.
Пока длина теломер достаточна для того, чтобы процесс дифференцировки с образованием прекративших деление специализированных клеток был завершен, сокращение их теломер не должно влиять на старение. Однако, если в клетках, еще находящихся в процессе дифференцировки, теломеры сокращены до критического размера, то они не смогут достичь зрелости из-за блокирования клеточных циклов. Органы, включая кровь, перестанут пополняться специализированными клетками. Возможно, именно этим определяется максимальный срок жизни.
У индивидов с природно длинными теломерами [159] и, добавим, с эффективной природной аутофагией, существенно снижающей уровень прогерина, этот срок приближен к максимальной продолжительности жизни человека. Подтверждающие результаты получены на животных [166, 167]11.
Приобретенные к настоящему времени опыт и знания дают основания предпринять действия, которые позволят увеличить длительность жизни, в том числе, максимальный ее срок. Очевидными мишенями такой терапии считаются прогерин (цель – максимально снизить содержание прогерина в клетках организма) и теломеры (цель – не допустить их укорочение до предела, блокирующего клеточный цикл).
Выше описаны исследования, которые показали, что рапамицин и его аналоги, ресвератрол, метформин, спермидин, блокируя киназу mTOR, способствуют активации аутофагии, утилизирующей прогерин. В опытах на животных показано, что прием этих препаратов оздоравливает организм и продлевает жизнь [125, 168–173].
Само снижение содержания прогерина, достигнутое с помощью указанных препаратов, должно снизить темп сокращения теломер благодаря восстановлению компактной структуры хроматина. Однако наиболее эффективно прямое наращивание теломер, в том числе в соматических клетках, путем активации в них теломеразы. Повышение теломеразной активности достигнуто в мышиной модели с использованием методов генной терапии. Модифицированные животные отличаются большей продолжительностью жизни [174, 175]. Такое же воздействие оказывает ряд продуктов растительного происхождения [176–179].
К наиболее известным активаторам теломеразы относится астрагал перепончатый (Astragalus membranaceus, AM). Традиционная китайская медицина рекомендует употреблять это растение с пищей для оздоровления и увеличения продолжительности жизни. Современные исследования показали, что полисахариды, флавониды, сапонины астрагала являются эффективными активаторами теломеразы. Экстракт сушеных корней AM (TA-65, Telomerase Activator-65) [174, 181–184] вызывал оздоровление возрастных мышей и крыс, получавших этот препарат с пищей [176, 185, 186]. При этом, что важно, не увеличивалась частота перерождений клеток в опухолевые [176]. Показано также, что у дрозофил, принимавших AM с пищей, возрастала продолжительность жизни [187].
Здесь следует сделать важное замечание. Мы отмечали одно свойство экстрактов AM – способность активировать теломеразу. Однако эти экстракты мультифункциональны. В число их функций входит активация аутофагии [188, 189] по тому же механизму (подавление киназы mTOR), который используют рапамицин и другие отмеченные выше активаторы аутофагии. Это означает, что прием экстрактов AM способствует утилизации прогерина, благодаря чему, в частности, нормализуется структура периферийного хроматина и замедляется повреждение (укорочение) теломер. Таким образом, сохраняя теломеры, экстракты AM действуют двояко: способствуют защите теломерной ДНК от повреждающих ее агентов и активируют наращивающую ее теломеразу.
TA-65 и ряд подобных препаратов испытываются на группах пациентов, болезни которых связывают со старением. Пока проведены кратковременные исследования (в течение нескольких месяцев). Во всех описанных случаях наблюдали оздоровление пациентов, принимавших экстракты AM [181, 190–192].
Для определения воздействия экстрактов AM, как и других препаратов, на продолжительность жизни эксперимент должен быть длительным и начинаться тогда, когда у испытуемых еще не доминируют необратимые возрастные изменения. Если такие исследования с 40–50-летними людьми начать сейчас, то результаты проявятся через 30–50 лет.
Уже предложены прописи лекарственных комплексов, предназначенных для оздоровления и продления жизни [164, 193–196]. Однако необходимо учитывать и возможные побочные эффекты присутствующих в этих прописях веществ. Так, рапамицин обладает свойствами иммунодепрессанта, а метформин снижает уровень глюкозы в крови.
Когда будет установлен оптимальный состав комплекса средств от старения, включая выбор дозы, и значительная часть населения станет его регулярно принимать, следует ожидать увеличения не только средней, но и максимальной длительности жизни.
Говоря об оптимальном составе лекарства, следует добавлять “на данном этапе исследований”. Оптимизация будет продолжаться по мере обнаружения новых эффективных веществ. Многое зависит от исследований на молекулярном и клеточном уровнях. В этих исследованиях еще много белых пятен. Так, экспериментально показана ключевая роль прогерина в старении при прогерии. Весьма вероятно, что прогерин участвует и в естественном старении людей, но прямыми экспериментами это еще не доказано. Поэтому исследования на культурах клеток очень пожилых людей, в частности, опыты по возвращению ядрам (рис. 6б) нормальной формы, как это делалось на клетках больных прогерией (рис. 5), представляют большой интерес. Морфология клеточных ядер, оценка содержания в них прогерина, длина теломер в лимфоцитах и фибробластах могут служить маркерами, характеризующими эффективность примененного лекарства от старения.
Можно задать вопрос, почему под жестким прессом эволюции сохраняется столь сложная процедура процессинга предшественников ламина А: присоединение к преламину А гидрофобной цепочки (фарнезилирование) и последующее ее удаление (рис. 3а). Напрашивается ответ, что смысл этой процедуры состоит в том, чтобы малая, но конечная доля сохранившихся фарнезилированных молекул преламина А и прогерин локализовались в ядрах соматических клеток. Их накопление вызовет описанные выше нарушения структуры ядра и клеточного метаболизма (рис. 4), являющиеся причиной старения клеток и всего организма. Этим обеспечивается смена поколений. При этом сохранение популяции обеспечивают половые и эмбриональные стволовые клетки, в которых ген LMNA не активен, а, следовательно, предшественники ламина А, инициирующие старение, не образуются.
Говоря о старении, необходимо иметь в виду, что основные факторы, влияющие на старение, и сами механизмы старения могут существенно отличаться у разных видов животных. У человека, соматические клетки которого лишены теломеразной активности, существенную роль в старении может играть достигшее предела укорочение теломер у пролиферирующих клеток. Прием активаторов теломеразы продлевает существование этих клеток и оздоровляет организм. Однако у мелких грызунов, в том числе мышей и крыс, соматические клетки обладают теломеразной активностью, благодаря чему в культуре клеток число их делений не лимитировано [197]. Но живут они всего 3–5 лет. Главной причиной старения этих животных может быть несовершенная система репарации ДНК, о чем свидетельствует высокий уровень мутагенеза и злокачественных перерождений клеток. Лишение большинства соматических клеток способности к пролиферации открыло путь к появлению крупных животных с большим количеством клеток. Отмечено, что соматические клетки не пролиферируют уже у крупного грызуна бобра [197]. По мере увеличения размеров тела животных происходило совершенствование систем репарации ДНК, обусловленное необходимостью снижения вероятности злокачественного перерождения в расчете на одну клетку. Иначе многие особи не доживали бы до репродуктивного возраста. Особенно в этом преуспели слоны и киты. В геноме слонов обнаружены не менее 20 копий гена TP53, который кодирует белок, участвующий в контроле клеточного цикла. Геном человека содержит только один такой ген [198]. Крупные киты живут почти в 3 раза дольше человека, у них в 100 раз больше клеток, и они не подвержены онкологическим заболеваниям. Простой расчет показывает, что система репарации ДНК у китов должна быть в 300 раз эффективнее, чем у человека.
Особый интерес вызывают голые землекопы (naked mole-rats) – уже приобретшие известность мелкие грызуны, обитающие в Западной Африке. В лабораториях они появились около 40 лет тому назад. “Ветераны” не только живы, но способны производить потомство и не обнаруживают признаков старения ([199], для обзора см. [1]). Ближайшие родственники голого землекопа – морские свинки, мыши и крысы – живут 2–5 лет и умирают с явными признаками старения. У голых землекопов, содержащихся в лабораториях и зоопарках, выявлено лишь несколько случаев онкологических заболеваний [200]. Обращает внимание обнаружение во внеклеточном матриксе голых землекопов сверхвысокомолекулярной гиалуроновой кислоты (6.5 млн Да), которая, как полагают, эффективно защищает клетки от стрессовых состояний [201].
У голых землекопов обнаружена высокая активность аутофагии, осуществляющей утилизацию прогерина [202]. К сожалению, данные о накоплении прогерина у этих животных-долгожителей отсутствуют. Представляется, что главные открытия в расшифровке механизмов их здорового долголетия еще впереди [203].
Дальнейшие исследования на разных видах животных позволят также уточнить, насколько универсальна вовлеченность прогерина в процессы старения.
Об интересе к рассмотренным в настоящем обзоре подходам к объяснению причин старения и к созданию на базе уже полученных результатов лекарств, отдаляющих старение, свидетельствует быстрый рост числа публикаций, намечающих, в частности, направления дальнейших исследований [204–211].
Список литературы
Мосевицкий М.И. (2018) Распространенность жизни и уникальность разума. Санкт Петербург: СпецЛит.
van der Pol A., van Gilst W.H., Voors A.A., van der Meer P. (2019) Treating oxidative stress in heart failure: past, present and future. Eur. J. Heart Fail. 21, 425–435. https://doi.org/10.1002/ejhf.1320
Прошкина Е.Н., Соловьёвa И.А., Шапошниковa М.В., Москалевa А.A. (2020) Ключевые молекулярные механизмы старения, биомаркеры и потенциальные интервенции. Молекуляр. биология. 54, 883–921.
Romano A.D., Serviddio G., de Matthaeis A., Bellanti F., Vendemiale G. (2010) Oxidative stress and aging. J. Nephrol. 23(Suppl 15), S29–536.
Skulachev V.P., Shilovsky G.A., Putyatina T.S., Po-pov N.A., Markov A.V., Skulachev M.V., Sadovnichii V.A. (2020) Perspectives of Homo sapiens lifespan extension: focus on external or internal resources? Aging (Albany NY). 12, 5566–5584. https://doi.org/10.18632/aging.102981
Best B.P. (2009). Nuclear DNA damage as a direct cause of aging. Rejuvenation Res. 12, 199–208. https://doi.org/10.1089/rej.2009.0847
Olovnikov A.M. (1973) A theory of merginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 41, 181–190. https://doi.org/10.1016/0022-5193(73)90198-7
Михельсон В.М., Гамалея И.А. (2013) Теломерная теория старения (обзор). Saarbrücken: Palmarium Acad. Publ. 101.
Snow C.J., Dar A., Dutta A., Kehlenbach R.H., Paschal B.M. (2013) Defective nuclear import of TPR in progeria reflects the ran sensitivity of large cargo transport. J. Cell Biol. 201, 541–557. https://doi.org/10.1083/jcb.201212117
Fasci D., van Ingen H., Scheltema R.A., Heck A.J.R. (2018) Histone interaction landscapes visualized by crosslinking mass spectrometry in intact cell nuclei. Mol. Cell. Proteomics. 17, 2018–2033. https://doi.org/10.1074/mcp.RA118.000924
Dworak N., Makosa D., Chatterjee M., Jividen K., Yang C.S., Snow C., Simke W.C., Johnson I.G., Kelley J.B., Paschal B.M. (2019) A nuclear lamina-chromatin-Ran GTPase axis modulates nuclear import and DNA damage signaling. Aging Cell. 18, e12851. https://doi.org/10.1111/acel
Güttler T., Görlich D. (2011) Ran-dependent nuclear export mediators: a structural perspective. EMBO J. 30, 3457–3474. https://doi.org/10.1038/emboj.2011.287
Goldberg M.W., Huttenlauch I., Hutchison C.J., Stick R. (2008) Filaments made from A- and B-type lamins differ in structure and organization. J. Cell Sci. 121, 215–225.https://doi.org/10.1242/jcs.022020
Zbarsky I.B., Georgiev G.P. (1959) Cytological characteristics of protein and nucleoprotein fractions of cell nuclei. Biochim. Biophys. Acta. 32(1), 301–302. https://doi.org/10.1016/0006-3002(59)90600-6
Георгиев Г.П., Ченцов Ю.С. (1963) Об ультраструктуре ядра. Основные структурные элементы клеточных ядер и их нуклеопротеидный состав. Биофизика. 8, 50 –57.
Earnshaw W.C., Laemmli U.K. (1983) Architecture of metaphase chromosomes and chromosome scaffolds. J. Cell Biol. 96, 84–93. https://doi.org/10.1083/jcb.96.1.84
Smith H.C., Puvion E., Buchholtz L.A., Berezney R. (1984) Spatial distribution of DNA loop attachment and replicational sites in the nuclear matrix. J. Cell Biol. 99, 1794–1802. https://doi.org/10.1083/jcb.99.5.1794
Mortillaro M.J., Blencowe B.J., Wei X., Nakayasu H., Du L., Warren S.L., Sharp P.A., Berezney R. (1996) A hyperphosphorylated form of the large subunit of RNA polymerase II is associated with splicing complexes and the nuclear matrix. Proc. Natl. Acad. Sci. USA. 93, 8253–8257. https://doi.org/10.1073/pnas.93.16.8253
Wei X., Somanathan S., Samarabandu J., Berezney R. (1999) Three-dimensional visualization of transcription sites and their association with splicing factor-rich nuclear speckles. J. Cell Biol. 146, 543–558.
Razin S.V., Iarovaia O.V., Y.S. Vassetzky Y.S. (2014) A requiem to the nuclear matrix: from a controversial concept to 3D organization of the nucleus. Chromosoma. 123, 217–224. https://doi.org/10.1007/s00412-014-0459-8
Вальтер С.Н., Качурин А.Л., Попов Ю.В., Мосевицкий М.И. (1984) Наблюдение в тонких срезах печени внутриядерного каркаса, образованного сетью структурированных фибрилл. Докл. АН СССР. 279, 1249–1251.
Adolph K.W. (1980) Organization of chromosomes in HeLa cells: isolation of histone-depleted nuclei and nuclear scaffolds. J. Cell Sci. 42, 291–304. PMID: 7400238
Fey E.G., Krochmalnic G., Penman S. (1986) The nonchromatin substructures of the nucleus: the ribonucleoprotein (RNP)-containing and RNP-depleted matrices analyzed by sequential fractionation and resinless section electron microscopy. J. Cell Biol. 102, 1654–1665. https://doi.org/10.1083/jcb.102.5.1654
Gerace L., Blobel G. (1980) The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell. 19(1), 277–287. doi: https://doi.org/10.1016/0092-8674(80)90409-2
Turgay Y., Eibauer M., Goldman A.E., Shimi T., Khayat M., Ben-Harush K., Dubrovsky-Gaupp A., Sapra K.T., Goldman R.D., Medalia O. (2017) The molecular architecture of lamins in somatic cells. Nature. 543, 261–264. https://doi.org/10.1038/nature21382
Ahn J., Jo I., Kang S.M., Hong S., Kim S., Jeong S., Kim Y.H., Park B.J., Ha N.C. (2019). Structural basis for lamin assembly at the molecular level. Nat. Commun. 10(1), 3757. https://doi.org/10.1038/s41467-019-11684-x
Lin F., Worman H.J. (1993) Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 268, 16321–16326.
Stroud M.J., Banerjee I., Veevers J., Chen J. (2014) Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ. Res. 114, 538–548. https://doi.org/10.1161/CIRCRESAHA.114.301236
Stroud M.J. (2018) Linker of nucleoskeleton and cytoskeleton complex proteins in cardiomyopathy. Biophys. Rev. 10, 1033–1051. https://doi.org/10.1007/s12551-018-0431-6
Worman H.J., Yuan J., Blobel G., Georgatos S.D. (1988) A lamin B receptor in the nuclear envelope. Proc. Natl. Acad. Sci. USA. 85, 8531–8534. https://doi.org/10.1073/pnas.85.22.8531
Smith S., Blobel G. (1993) The first membrane spanning region of the lamin B receptor is efficient for sorting to the inner nuclear membrane. J. Cell Biol. 120, 631–637.
Olins A.L., Rhodes G., Welch D.B., Zwerger M., Olins D.E. (2010) Lamin B receptor: multi-tasking at the nuclear envelope. Nucleus. 1, 53–70. https://doi.org/10.4161/nucl.1.1.10515
Liokatis S., Edlich C., Soupsana K., Giannios I., Panagiotidou P., Tripsianes K., Sattler M., Georgatos S.D., Politou A.S. (2012) Solution structure and molecular interactions of lamin B receptor tudor domain. J. Biol. Chem. 287, 1032–1042. https://doi.org/10.1074/jbc.M111.281303
Nikolakaki E., Mylonis I., Giannakouros T. (2017) Lamin B receptor: interplay between structure, function and localization. Cells. 6, 28. https://doi.org/10.3390/cells6030028
Constantinescu D., Gray H.L., Sammak P.J., Schatten G.P., Csoka A.B. (2006) Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells. 24, 177–185. https://doi.org/10.1634/stemcells.2004-0159
Gruenbaum Y., Foisner R. (2015) Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 84, 131–164. https://doi.org/10.1146/annurev-biochem-060614-034115
Zhang H., Petrie M.V., He Y., Peace J.M., Chiolo I.E., Aparicio O.M. (2019) Dynamic relocalization of replication origins by Fkh1 requires execution of DDK function and Cdc45 loading at origins. Elife. 8, pii: e45512. https://doi.org/10.7554/eLife.45512
Bermeo S., Vidal C., Zhou H., Duque G. 2015. Lamin A/C acts as an essential factor in mesenchymal stem cell differentiation through the regulation of the dynamics of the Wnt/β-catenin pathway. J. Cell Biochem. 116, 2344–2353.
Davidson K.C., Adams A.M., Goodson J.M., McDonald C.E., Potter J.C., Berndt J.D., Biechele T.L., Taylor R.J., Moon R.T. (2012) Wnt/β-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc. Natl. Acad. Sci. USA. 109, 4485–4490.
Duque G., Rivas D. (2006) Age-related changes in lamin A/C expression in the osteoarticular system: laminopathies as a potential new aging mechanism. Mech. Ageing Dev. 127, 378–383. https://doi.org/10.1016/j.mad.2005.12.007
Forleo C., Carmosino M., Resta N., Rampazzo A., Valecce R., Sorrentino S., Iacoviello M., Pisani F., Procino G., Gerbino A., Scardapane A., Simone C., Calore M., Torretta S., Svelto M., Favale S. (2015) Clinical and functional characterization of a novel mutation in lamin A/C gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS One. 10(4), e0121723. https://doi.org/10.1371/journal.pone.0121723
Crasto S., My I., Pasquale E.D. (2020) The broad spectrum of LMNA cardiac diseases: from olecular mechanisms to clinical phenotype. Front. Physiol. 11, 761. https://doi.org/10.3389/fphys.2020.00761
Pollex R.L., Hegele R.A. (2004) Hutchinson–Gilford progeria syndrome. Clin. Genet. 66, 375–381. https://doi.org/10.1111/j.1399-0004.2004.00315.x
Scaffidi P., Misteli T. (2006) Lamin A-dependent nuclear defects in human aging. Science. 312(5776), 1059–1063. https://doi.org/10.1126/science.1127168
Merideth M.A., Gordon L.B., Clauss S., Sachdev V., Smith A.C., Perry M.B., Brewer C.C., Zalewski C., Kim H.J., Solomon B., Brooks B.P., Gerber L.H., Turner M.L., Domingo D.L., Hart T.C., Graf J., Reynolds J.C., Gropman A., Yanovski J.A., Gerhard-Herman M., Collins F.S., Nabel E.G., Cannon R.O. 3rd, Gahl W.A., Introne W.J. (2008) Phenotype and course of Hutchinson–Gilford progeria syndrome. N. Engl. J. Med. 358, 592–604. https://doi.org/10.1056/NEJMoa0706898
Coutinho H.D.M., Falcão-Silva V.S., Gregório Fernandes Gonçalves G.F., da Nóbrega R.B. (2009) Molecular ageing in progeroid syndromes: Hutchinson–Gilford progeria syndrome as a model. Immun. Ageing. 20, 6–14. https://doi.org/10.1186/1742-4933-6-4
Eriksson M., Brown W.T., Gordon L.B., Glynn M.W., Singer J., Scott L., Erdos M.R., Robbins C.M., Moses T.Y., Berglund P., Dutra A., Pak E., Durkin S., Csoka A.B., Boehnke M., Glover T.W., Collins F.S. (2003) Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature. 423(6937), 293–298. https://doi.org/10.1038/nature01629
De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C.L., Munnich A., Le Merrer M., Lévy N. (2003). Lamin A truncation in Hutchinson–Gilford progeria. Science. 300(5628), 2055. https://doi.org/10.1126/science.1084125
Capell B.C., Erdos M.R., Madigan J.P., Fiordalisi J.J., Varga R., Conneely K.N., Gordon L.B., Der C.J., Cox A.D., Collins F.S. (2005) Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 102, 12879–12884. https://doi.org/10.1073/pnas.0506001102
Glynn M.W., Glover T.W. (2005) Incomplete processing of mutant lamin A in Hutchinson–Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum. Mol. Genet. 14, 2959–2969. https://doi.org/10.1093/hmg/ddi326
Cenni V., Capanni C., Mattioli E., Schena E., Squarzoni S., Bacalini M.G., Garagnani P., Salvioli S., Franceschi C., Lattanzi G. (2020) Lamin A involvement in ageing processes. Ageing Res. Rev. 62, 101073. https://doi.org/10.1016/j.arr.2020.101073
Chojnowski A., Ong P.F., Wong E.S., Lim J.S., Mutalif R.A., Navasankari R., Dutta B., Yang H., Liow Y.Y., Sze S.K., Boudier T., Wright G.D., Colman A., Burke B., Stewart C.L., Dreesen O. (2015) Progerin reduces LAP2α-telomere association in Hutchinson–Gilford progeria. Elife. 4, e07759. https://doi.org/10.7554/eLife.07759
Chojnowski A., Ong P.F., Wong E.S., Lim J.S., Mutalif R.A., Navasankari R., Dutta B., Yang H., Liow Y.Y., Sze S.K., Boudier T., Wright G.D, Colman A., Burke B., Stewart C.L., Dreesen O. (2020) Heterochromatin loss as a determinant of progerin-induced DNA damage in Hutchinson–Gilford progeria. Aging Cell. 19, e13108. https://doi.org/10.1111/acel.13108
Romero-Bueno R., de la Cruz Ruiz P., Artal-Sanz M., Askjaer P., Dobrzynska A. (2019) Nuclear organization in stress and aging. Cells. 8, 664. https://doi.org/10.3390/cells8070664
Martins F., Sousa J., Pereira C.D., da Cruz e Silva O.A.B., Rebelo S. (2020) Nuclear envelope dysfunction and its contribution to the aging process. Aging Cell. 19, e13143. https://doi.org/10.1111/acel.13143
Arii J., Maeda F., Maruzuru Y., Koyanagi N., Kato A., Mori Y., Kawaguchi Y. (2020) ESCRT-III controls nuclear envelope deformation induced by progerin. Sci. Rep. 10, 18877. https://doi.org/10.1038/s41598-020-75852-6
Kang S.M., Yoon M.H., Ahn J., Kim J.E., Kim S.Y., Kang S.Y., Joo J., Park S., Cho J.H., Woo T.G., Oh A.Y., Chung K.J., An S.Y., Hwang T.S., Lee S.Y., Kim J.S., Ha N.C., Song G.Y., Park B.J. (2021) Progerinin, an optimized progerin-lamin A binding inhibitor, ameliorates premature senescence phenotypes of Hutchinson–Gilford progeria syndrome. Commun. Biol. 4, 5. https://doi.org/10.1038/s42003-020-01540-w
Goldman R.D., Shumaker D.K., Erdos M.R., Eriksson M., Goldman A.E., Gordon L.B., Gruenbaum Y., Khuon S., Mendez M., Varga R., Collins F.S. (2004) accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 101, 8963–8968. https://doi.org/10.1073/pnas.0402943101
Dahl K.N., Scaffidi P., Islam M.F., Yodh A.G., Wilson K.L., Misteli T. (2006) Distinct structural and mechanical properties of the nuclear lamina in Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 103, 10271–10276.
Cao K., Graziotto J.J., Blair C.D., Mazzulli J.R., Erdos M.R., Krainc D., Collins F.S. (2011) Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson–Gilford progeria syndrome. Cells. Sci. Transl. Med. 3, 89ra58. https://doi.org/10.1126/scitranslmed.3002346
Noda A., Mishima S., Hirai Y., Hamasaki K., Landes R.D., Mitani H., Haga K., Kiyono T., Nakamura N., Kodama Y. (2015) Progerin, the protein responsible for the Hutchinson–Gilford progeria syndrome, increases the unrepaired DNA damages following exposure to ionizing radiation. Genes Environ. 37, 13. https://doi.org/10.1186/s41021-015-0018-4
Saxena S., Kumar S. (2020) pharmacotherapy to gene editing: potential therapeutic approaches for Hutchinson–Gilford progeria syndrome. Geroscience. 42, 467–494. https://doi.org/10.1007/s11357-020-00167-3
Gabriel D., Roedl D., Gordon L.B., Djabali K. (2015) Sulforaphane enhances progerin clearance in Hutchinson–Gilford progeria fibroblasts. Aging Cell. 14, 78–91. https://doi.org/10.1111/acel.12300
Rivera-Torres J., Acín-Perez R., Cabezas-Sánchez P., Osorio F.G., Gonzalez-Gómez C., Megias D., Cá-mara C., López-Otín C., Enríquez J.A., Luque-García J.L., Andrés V. (2013) identification of mitochondrial dysfunction in Hutchinson–Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J. Proteomics. 91, 466–477.
Bidault G., Garcia M., Capeau J., Morichon R., Vigouroux C., Béréziat V. (2020). Progerin expression induces inflammation, oxidative stress and senescence in human coronary endothelial cells. Cells. 9(5), 1201. https://doi.org/10.3390/cells9051201
Chen W.M., Chiang J.C., Lin Y.C., Lin Y.N., Chuang P.Y., Chang Y.C., Chen C.C., Wu K.Y., Hsieh J.C., Chen S.K., Huang W.P., Chen B.P.C., Lee H. (2020) Lysophosphatidic acid receptor LPA3 prevents oxidative stress and cellular senescence in Hutchinson–Gilford progeria syndrome. Aging Cell. 19, e13064. https://doi.org/10.1111/acel.13064
Mao X., Bharti P., Thaivalappil A., Cao K. (2020) peroxisomal abnormalities and catalase deficiency in Hutchinson–Gilford progeria syndrome. Aging (Albany NY). 12, 5195–5208. https://doi.org/10.18632/aging.102941
Bandaria J.N., Qin P., Berk V., Chu S., Yildiz A. (2016) Shelterin protects chromosome ends by compacting telomeric chromatin. Cell. 164, 735–746. https://doi.org/10.1016/j.cell.2016.01.036
Prokocimer M., Barkan R., Gruenbaum Y. (2013) Hutchinson–Gilford progeria syndrome through the lens of transcription. Aging Cell. 12, 533–543. https://doi.org/10.1111/acel.12070
Arancio W., Pizzolanti G., Genovese S.I., Pitrone M., Giordano C. (2014) Epigenetic involvement in Hutchinson–Gilford progeria syndrome: a mini-review. Gerontology. 60, 197–203. https://doi.org/10.1159/000357206
Bär C., Blasco M.A. (2016). Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Res. 5, F1000 Faculty Rev-89. https://doi.org/10.12688/f1000research.7020.1
Gavia-García G., Rosado-Pérez J., Arista-Ugalde T.L., Aguiñiga-Sánchez I., Santiago-Osorio E., Mendoza-Núñez V.M. (2021) Telomere length and oxidative stress and its relation with metabolic syndrome components in the aging. Biology (Basel). 10(4), 253. https://doi.org/10.3390/biology10040253
Schoeftner S., Blasco M.A. (2008) Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell. Biol. 10, 228–236. https://doi.org/10.1038/ncb1685
Redon S., Reichenbach P., Lingner J. (2010) The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucl. Acids Res. 38, 5797–5806. https://doi.org/10.1093/nar/gkq296
Jiang X., Wang L., Xie S., Chen Y., Song S., Lu Y., Lu D. (2020) Long noncoding RNA MEG3 blocks telomerase activity in human liver cancer stem cells epigenetically. Stem Cell Res. Ther. 11(1), 518. https://doi.org/10.1186/s13287-020-02036-4
Pfeiffer V., Lingner J. (2012) TERRA promotes telomere shortening through exonuclease 1-mediated resection of chromosome ends. PLoS Genet. 8(6), e1002747. https://doi.org/10.1371/journal.pgen.1002747
Huang S., Risques R.A., Martin G.M., Rabinovitch P.S., Oshima J. (2008) Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Exp. Cell. Res. 314(1), 82–91. https://doi.org/10.1016/j.yexcr.2007.08.004
Aguado J., Sola-Carvajal A., Cancila V., Revêchon G., Ong P.F., Jones-Weinert C.W., Wallén Arzt E., Lattanzi G., Dreesen O., Tripodo C., Rossiello F., Eriksson M., d’Adda di Fagagna F. (2019) Inhibition of DNA damage response at telomeres improves the detrimental phenotypes of Hutchinson–Gilford progeria syndrome. Nat. Commun. 10(1), 4990. https://doi.org/10.1038/s41467-019-13018-3
Mallampalli M.P., Huyer G., Bendale P., Gelb M.H., Michaelis S. (2005) inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 102, 14416–14421. https://doi.org/10.1073/pnas.0503712102
Toth J.I., Yang S.H., Qiao X., Beigneux A.P., Gelb M.H., Moulson C.L., Miner J.H., Young S.G., Fong L.G. (2005) Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl. Acad. Sci. USA. 102, 12873–12878. https://doi.org/10.1073/pnas.0505767102
Yang S.H., Bergo M.O., Toth JI., Qiao X., Hu Y., Sandoval S., Meta M., Bendale P, Gelb M.H., Young S.G., Fong L.G. (2005) blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson–Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA. 102, 10291–10296. https://doi.org/10.1073/pnas.0504641102
Yang S.H., Meta M., Qiao X., Frost D., Bauch J., Coffinier C., Majumdar S., Bergo M.O., Young S.G., Fong L.G. (2006) A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson–Gilford progeria syndrome mutation. J. Clin. Invest. 116, 2115–2121. https://doi.org/10.1172/JCI28968
Fong L.G., Frost D., Meta M., Qiao X., Yang S.H., Coffinier C., Young S.G. (2006) A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 311(5767), 1621–1623. https://doi.org/10.1126/science.1124875
Wang Y., Panteleyev A.A., Owens D.M., Djabali K., Stewart C.L., Worman H.J. (2008) Epidermal expression of the truncated prelamin A causing Hutchinson–Gilford progeria syndrome: effects on keratinocytes, hair and skin. Hum. Mol. Genet. 17, 2357–2369. https://doi.org/10.1093/hmg/ddn136
Wang Y., Ostlund C., Worman H.J. (2010) Blocking protein farnesylation improves nuclear shape abnormalities in keratinocytes of mice expressing the prelamin A variant in Hutchinson–Gilford progeria syndrome. Nucleus. 1, 432–439. https://doi.org/10.4161/nucl.1.5.12972
Cubria M.B., Suarez S., Masoudi A., Oftadeh R, Kamalapathy P., DuBose A., Erdos M.R., Cabral W.A., Karim L., Collins F.S., Snyder B.D., Nazarian A. (2020). Evaluation of musculoskeletal phenotype of the G608G progeria mouse model with lonafarnib, pravastatin, and zoledronic acid as treatment groups Proc. Natl. Acad. Sci. USA. 117, 12029–12040. https://doi.org/10.1073/pnas.1906713117
Lai W.F., Wong W.T. (2020) Progress and trends in the development of therapies for Hutchinson–Gilford progeria syndrome. Aging Cell. 19(7), e13175. https://doi.org/10.1111/acel.13175
Dhillon S. (2021) Lonafarnib: first approval. Drugs. 81, 283–289. https://doi.org/10.1007/s40265-020-01464-z
Blondel S., Egesipe A.L., Picardi P., Jaskowiak A.L., Notarnicola M., Ragot J., Tournois J., Le Corf A., Brinon B., Poydenot P., Georges P., Navarro C., Pitrez P.R., Ferreira L., Bollot G., Bauvais C., Laustriat D., Mejat A., De Sandre-Giovannoli A., Levy N., Bifulco M., Peschanski M., Nissan X. (2016) Drug screening on Hutchinson–Gilford progeria pluripotent stem cells reveals aminopyrimidines as new modulators of farnesylation. Cell. Death Dis. 7(2), 2105. https://doi.org/10.1038/cddis.2015.374
Gordon L.B., Shappell H., Massaro J., D’Agostino R.B. Sr., Brazier J., Campbell S.E., Kleinman M.E., Kieran M.W. (2018) association of lonafarnib treatment vs no treatment with mortality rate in patients with Hutchinson–Gilford progeria syndrome. JAMA. 319, 1687–1695. https://doi.org/10.1001/jama.2018.3264
Young S.G., Yang S.H., Davies B.S., Jung H.J., Fong L.G. (2013) Targeting protein prenylation in progeria. Sci. Transl. Med. 5(171), 171ps3. https://doi.org/10.1126/scitranslmed.3005229
Scaffidi P., Misteli T. (2005). Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 11, 440–445. https://doi.org/10.1038/nm1204
Osorio F.G., Navarro C.L., Cadiñanos J., López-Mejía I.C., Quirós P.M., Bartoli C., Rivera J., Tazi J., Guzmán G., Varela I., Depetris D., de Carlos F., Cobo J., Andrés V., De Sandre-Giovannoli A., Freije J.M., Lévy N., López-Otín C. (2011) Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 3(106), 106ra107. https://doi.org/10.1126/scitranslmed.3002847
Erdos M.R., Cabral W.A., Tavarez U.L., Cao K., Gvozdenovic-Jeremic J., Narisu N., Zerfas P.M., Crumley S., Boku Y., Hanson G., Mourich D.V., Kole R., Eckhaus M.A., Gordon L.B., Collins F.S. (2021) A targeted antisense therapeutic approach for Hutchinson–Gilford progeria syndrome. Nat. Med. 27, 536–545. https://doi.org/10.1038/s41591-021-01274-0
Puttaraju M., Jackson M., Klein S., Shilo A., Bennett C.F., Gordon L., Rigo F., Misteli T. (2021) Systematic screening identifies therapeutic antisense oligonucleotides for Hutchinson–Gilford progeria syndrome. Nat. Med. 27, 526–535. https://doi.org/10.1038/s41591-021-01262-4
Revêchon G., Whisenant D., Eriksson M. (2021) Splice-inhibition therapy targets progeria. Nat. Med. 27, 377–379. https://doi.org/10.1038/s41591-021-01267-z
Pellegrini C., Columbaro M., Capanni C., D’Apice M.R., Cavallo C., Murdocca M., Lattanzi G., Squarzoni S. (2015) All-trans retinoic acid and rapamycin normalize Hutchinson–Gilford progeria fibroblast phenotype. Oncotarget. 6, 2914–2928. https://doi.org/10.18632/oncotarget.4939
Kreienkamp R., Croke M., Neumann M.A., Bedia-Diaz G., Graziano S., Dusso A., Dorsett D., Carlberg C., Gonzalo S. (2016) Vitamin D receptor signaling improves Hutchinson–Gilford progeria syndrome cellular phenotypes. Oncotarget. 7(21), 30018–30031. https://doi.org/10.18632/oncotarget.9065
Beyret E., Liao H.K., Yamamoto M., Hernandez-Benitez R., Fu Y., Erikson G., Reddy P., Izpisua Belmonte J. (2019) Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson–Gilford progeria syndrome. Nat. Med. 25, 419–422. https://doi.org/10.1038/s41591-019-0343-4
Piekarowicz K., Machowska M.,Volha Dzianisava V., Rzepecki R. (2019) Hutchinson–Gilford progeria syndrome–current status and prospects for gene therapy treatment. Cells. 8(2), 88. https://doi.org/10.3390/cells8020088
Santiago-Fernández O., Osorio F.G., Quesada V., Rodríguez F., Basso S., Maeso D., Rolas L, Barkaway A., Nourshargh S., Folgueras A.R., Freije J.M.P., López-Otín C. (2019) Development of a CRISPR/Cas9-based therapy for Hutchinson–Gilford progeria syndrome. Nat. Med. 25, 423–426. https://doi.org/10.1038/s41591-018-0338-6
Marraffini L.A., Sontheimer E.J. (2010) CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 11, 181–190. https://doi.org/10.1038/nrg2749
Wiedenheft B. (2013). In defense of phage: viral suppressors of CRISPR-mediated adaptive immunity in bacteria. RNA Biol. 10, 886–890. https://doi.org/10.4161/rna.23591
Wu S.-S., Li Q.-C., Yin C.-Q., Xue W., Song C.-Q. (2020) Advances in CRISPR/Cas-based gene therapy in human genetic diseases. Theranostics. 10, 4374–4382. https://doi.org/10.7150/thno.43360
Koblan L.W., Erdos M.R., Wilson C., Cabral W.A., Levy J.M., Xiong Z.M., Tavarez U.L., Davison L.M., Gete Y.G., Mao X., Newby G.A., Doherty S.P., Narisu N., Sheng Q., Krilow C., Lin C.Y., Gordon L.B., Cao K., Collins F.S., Brown J.D., Liu D.R. (2021) In vivo base editing rescues Hutchinson–Gilford progeria syndrome in mice. Nature. 589(7843), 608–614. https://doi.org/10.1038/s41586-020-03086-7
Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. (2017) Programmable base editing of A-T to G-C in genomic DNA without DNA cleavage. Nature. 551(7681), 464–471. https://doi.org/10.1038/nature24644
Graziotto J.J., Cao K., Collins F.S., Krainc D. (2012) Rapamycin activates autophagy in Hutchinson–Gilford progeria syndrome: implications for normal aging and age-dependent neurodegenerative disorders. Autophagy. 8, 147–151. https://doi.org/10.4161/auto.8.1.18331
Ehninger D., Neff F, Xie K. (2014) Longevity, aging and rapamycin. Cell. Mol. Life Sci. 71, 4325–4346. https://doi.org/10.1007/s00018-014-1677-1
Mendelsohn A.R., Larrick J.W. (2011) Rapamycin as an antiaging therapeutic?: targeting mammalian target of rapamycin to treat Hutchinson–Gilford progeria and neurodegenerative diseases. Rejuvenation Res. 14, 437–441. https://doi.org/10.1089/rej.2011.1238
Ramos F.J., Chen S.C., Garelick M.G, Dai D.F., Liao C.Y., Schreiber K.H., MacKay V.L., An E.H., Strong R., Ladiges W.C., Rabinovitch P.S., Kaeberlein M., Kennedy B.K. (2012) Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 4(144), 144ra103. https://doi.org/10.1126/scitranslmed.3003802
Yang H., Rudge D.G., Koos J.D., Vaidialingam B., Yang H.J., Pavletich N.P. (2013) mTOR kinase structure, mechanism and regulation. Nature. 497, 217–223. https://doi.org/10.1038/nature12122
Clements C.S., Bikkul M.U., Ofosu W., Eskiw C., Tree D., Makarov E., Kill I.R., Bridger J.M. (2019) Presence and distribution of progerin in HGPS cells is ameliorated by drugs that impact on the mevalonate and mTOR pathways. Biogerontology. 20, 337–358. https://doi.org/10.1007/s10522-019-09807-4
Papadopoli D., Boulay K., Kazak L., Pollak M., Mallette F.A., Topisirovic I., Hulea L. (2019) mTOR as a central regulator of lifespan and aging. F1000Res. 8, F1000 Faculty Rev-998. https://doi.org/10.12688/f1000research.17196.1
Saxton R.A., Sabatini D.M. (2017) mTOR signaling in growth, metabolism, and disease. Cell. 169, 361–371.
Huang J.U., Klionsky D.J. (2007) Autophagy and human disease. Cell Cycle. 6, 1837–1849. https://doi.org/10.4161/cc.6.15.4511
Kim Y.Ch., Guan K.-L. (2015) mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest. 125, 25–32. https://doi.org/10.1172/JCI73939
Sotthibundhu A. (2016) Rapamycin regulates autophagy and cell adhesion in induced pluripotent stem cells. Stem Cell Res. Ther. 7, 166. https://doi.org/10.1186/s13287-016-0425-x
Almendáriz-Palacios C., Gillespie Z.E., Janzen M., Martinez V., Bridger J.M., Harkness T.A.A., Mousseau D.D., Eskiw C.H. (2020) The nuclear lamina: protein accumulation and disease. Biomedicines. 8(7), 188. https://doi.org/10.3390/biomedicines8070188
Saegusa C., Hosoya M., Nishiyama T., Saeki T., Fujimoto C., Okano H., Fujioka M., Ogawa K. (2020) Low-dose rapamycin-induced autophagy in cochlear outer sulcus cells. Laryngoscope Investig. Otolaryngol. 5, 520–528. https://doi.org/10.1002/lio2.392
Mizushima N., Levine B.N. (2020) Autophagy in human diseases. N. Engl. J. Med. 383(16), 1564–1576. https://doi.org/10.1056/NEJMra2022774
Lu X., Djabali K. (2018) Autophagic removal of farnesylated carboxy-terminal lamin peptides. Cells. 7(4), 33. https://doi.org/10.3390/cells7040033
Cenni V., Capanni C., Columbaro M., Ortolani M., D’Apice M.R., Novelli G., Fini M., Marmiroli S., Scarano E., Maraldi N.M., Squarzoni S., Prencipe S., Lattanzi G. (2011) Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur. J. Histochem. 55(4), e36. https://doi.org/10.4081/ejh.2011.e36
Strong R., Miller R.A., Bogue M., Fernandez E., Javors M.A., Libert S., Marinez P.A., Murphy M.P., Musi N., Nelson J.F., Petrascheck M., Reifsnyder P., Richardson A., Salmon A.B., Macchiarini F., Harrison D.E. (2020) Rapamycin-mediated mouse lifespan extension: late-life dosage regimes with sex-specific effects. Aging Cell. 19(11), e13269. https://doi.org/10.1111/acel.13269. 68
Zhang Y., Zhang J., Wang S. (2021) The role of rapamycin in healthspan extension via the delay of organ aging. Ageing Res Rev. 70, 101376. https://doi.org/10.1016/j.arr.2021.101376
Garay R.P. (2021) Investigational drugs and nutrients for human longevity. Recent clinical trials registered in ClinicalTrials.gov and clinicaltrialsregister.eu. Expert Opin. Investig. Drugs. 30, 749–758. https://doi.org/10.1080/13543784.2021.1939306
Peters J.M., Franke W.W., Kleinschmidt J.A. (1994) Distinct 19S and 20S subcomplexes of the 26S proteasome and their distribution in the nucleus and the cytoplasm. J. Biol. Chem. 269, 7709–7718.
Сорокин А.В., Ким Е.Р., Овчинников Л.П. (2009) Протеасомная система деградации и процессинга белков. Усп. Биол. Хим. 49, 3–76.
Harhouri K., Navarro C., Depetris D., Mattei M.G., Nissan X., Cau P., De Sandre-Giovannoli A., Lévy N. (2017) MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol. Med. 9, 1294–1313. https://doi.org/10.15252/emmm.201607315
Harhouri K., Frankel D., Bartoli C., Roll P., De Sandre-Giovannoli A., Lévy N. (2018) An overview of treatment strategies for Hutchinson–Gilford progeria syndrome. Ucleus. 9, 246–257. https://doi.org/10.1080/19491034.2018.1460045
McClintock D., Ratner D., Lokuge M., Owens D.M., Gordon L.B., Collins F.S., Djabali K. (2007) The mutant form of lamin A that causes Hutchinson–Gilford progeria is a biomarker of cellular aging in human skin. PLoS One. 2, e1269. https://doi.org/10.1371/journal.pone.0001269
Rodriguez S., Coppedè F., Sagelius H., Eriksson M. (2009) Increased expression of the Hutchinson–Gilford progeria syndrome truncated lamin a transcript during cell aging. Eur. J. Hum. Genet. 17, 928–937. https://doi.org/10.1038/ejhg.2008.270
Ashapkin V.V., Kutueva L.I., Kurchashova S.Y., Kireev I.I. (2019) Are there common mechanisms between the Hutchinson–Gilford progeria syndrome and natural aging? Front. Genet. 10, 455. https://doi.org/10.3389/fgene.2019.00455
Kreienkamp R., Gonzalo S. (2020) Metabolic dysfunction in Hutchinson–Gilford progeria syndrome. Cells. 9, 395. https://doi.org/10.3390/cells9020395
Osorio F.G., Varela I., Lara E., Puente X.S., Espada J., Santoro R., Freije J.M., Fraga M.F., López-Otín C. (2010) Nuclear envelope alterations generate an aging-like epigenetic pattern in mice deficient in Zmpste24 metalloprotease. Aging Cell. 9, 947–957. https://doi.org/10.1111/j.1474-9726.2010.00621.x
Worman H.J., Michaelis S. (2018). Permanently farnesylated prelamin A, progeria, and atherosclerosis. Circulation. 138, 283–286. https://doi.org/10.1161/CIRCULATIONAHA.118.034480
Kawakami Y., Hambright W.S., Takayama K., Mu X., Lu A., Cummins J.H., Matsumoto T., Yurube T., Kuroda R., Kurosaka M., Fu F.H., Robbins P.D., Niedernhofer L.J., Huard J. (2019) Rapamycin rescues age-related changes in muscle-derived stem/progenitor cells from progeroid mice. Mol. Ther. Methods Clin. Dev. 14, 64–76. https://doi.org/10.1016/j.omtm.2019.05.011
Harrison D.E., Strong R., Sharp Z.D., Nelson J.F., Astle C.M., Flurkey K., Nadon N.L., Wilkinson J.E., Frenkel K., Carter C.S., Pahor M., Javors M.A., Fernandez E., Miller R.A. (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 460(7253), 392–395. https://doi.org/10.1038/nature08221
Stacchiotti A., Corsetti G. (2020) Natural compounds and autophagy: allies against neurodegeneration. Front Cell Dev. Biol. 8, 555409. https://doi.org/10.3389/fcell.2020.555409
Yessenkyzy A., Saliev T., Zhanaliyeva M., Masoud A.R., Umbayev B., Sergazy S., Krivykh E., Gulyayev A., Nurgozhin T. (2020) Polyphenols as caloric-restriction mimetics and autophagy inducers in aging research. Nutrients. 12(5), 1344. https://doi.org/10.3390/nu12051344
García-Aguilar A., Palomino O., Benito M., Guillén C. (2021) Dietary polyphenols in metabolic and neurodegenerative diseases: molecular targets in autophagy and biological effects. Antioxidants (Basel). 10(2), 142. https://doi.org/10.3390/antiox10020142
Maduro A.T., Luís C., Soares R. (2021) Ageing, cellular senescence and the impact of diet: an overview. Porto. Biomed. J. 6(1), e120. https://doi.org/10.1097/j.pbj.0000000000000120
Pietrocola F., Lachkar S., Enot D.P., Niso-Santano M., Bravo-San Pedro J.M., Sica V., Izzo V., Maiuri M.C., Madeo F., Mariño G., Kroemer G. (2015) Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2, 509–516. https://doi.org/10.1038/cdd.2014.215
Eisenberg T., Abdellatif M., Schroeder S., Primessnig U., Stekovic S., Pendl T., Harger A., Schipke J., Zimmermann A., Schmidt A., Tong M., Ruckenstuhl C., Dammbrueck C., Gross A.S., Herbst V., Magnes C., Trausinger G., Narath S., Meinitzer A., Hu Z., Kirsch A., Eller K., Carmona-Gutierrez D., Büttner S., Pietrocola F., Knittelfelder O., Schrepfer E., Rockenfeller P., Simonini C., Rahn A., Horsch M., Moreth K., Beckers J., Fuchs H., Gailus-Durner V., Neff F., Janik D., Rathkolb B., Rozman J., de Angelis M.H., Moustafa T., Haemmerle G., Mayr M., Willeit P., von Frieling-Salewsky M., Pieske B., Scorrano L., Pieber T., Pechlaner R., Willeit J., Sigrist S.J., Linke W.A., Mühlfeld C., Sadoshima J., Dengjel J., Kiechl S., Kroemer G., Sedej S., Madeo F. (2016) Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438. https://doi.org/10.1038/nm.4222
Finley J. (2018) Cellular stress and AMPK activation as a common mechanism of action linking the effects of metformin and diverse compounds that alleviate accelerated aging defects in Hutchinson–Gilford progeria syndrome. Med. Hypotheses. 118, 151–162. https://doi.org/10.1016/j.mehy.2018.06.029
Mariño G., Pietrocola F., Madeo F., Kroemer G. (2014) Caloric restriction mimetics: natural/physiological pharmacological autophagy inducers. Autophagy. 10, 1879–1882. https://doi.org/10.4161/auto.36413
Escobar K.A., Cole N.H., Mermier C.M., VanDusseldorp A.T. (2019) Autophagy and aging: maintaining the proteome through exercise and caloric restriction. Aging Cell. 18, e12876. https://doi.org/10.1111/acel.12876
Martin-Rincon M., Morales-Alamo D., Calbet J.A.L. (2018) Exercise-mediated modulation of autophagy in skeletal muscle. Scand. J. Med. Sci. Sports. 28, 772–781. https://doi.org/10.1111/sms.12945
Park S.S., Seo Y.K., Kwon K.-S. (2019) Sarcopenia targeting with autophagy mechanism by exercise. BMB Rep. 52, 64–69. https://doi.org/10.5483/BMBRep.2019.52.1.292
Babygirija R., Lamming D.W. (2021) The regulation of healthspan and lifespan by dietary amino acids. Transl. Med. Aging. 5, 17–30. https://doi.org/10.1016/j.tma.2021.05.001
Kim J.S., Choi H.W., Choi S., Do J.T. (2011) Reprogrammed pluripotent stem cells from somatic cells. Int. J. Stem Cells. 4(1), 1–8. https://doi.org/10.15283/ijsc.2011.4.1.1
Jung H.-J, Tu Y., Yang S.H., Tatar A., Nobumori C., Wu D., Young S.G., Fong L.G. (2014) New LMNA knock-in mice provide a molecular mechanism for the “segmental aging” in hutchinson-gilford progeria syndrome. Hum. Mol. Genet. 23, 1506–1515.
Nissan X., Blondel S., Navarro C., Maury Y., Denis C., Girard M., Martinat C., De Sandre-Giovannoli A., Levy N., Peschanski M. (2012) Unique preservation of neural cells in Hutchinson–Gilford progeria syndrome is due to the expression of the neural-specific miR-9 microRNA. Cell Rep. 2, 1–9. https://doi.org/10.1016/j.celrep.2012.05.015
Baek J.H., Schmidt E., Viceconte N., Strandgren C., Pernold K., Richard T.J., Van Leeuwen F.W., Dantuma N.P., Damberg P., Hultenby K., Ulfhake B., Mugnaini E., Rozell B., Eriksson M. (2015). Expression of progerin in aging mouse brains reveals structural nuclear abnormalities without detectible significant alterations in gene expression, hippocampal stem cells or behavior. Hum. Mol. Genet. 24, 1305–1321. https://doi.org/10.1093/hmg/ddu541
Jung H.-J., Coffinier C., Choe Y., Beigneux A.P., Davies B.S., Yang S.H., Barnes R.H. 2nd, Hong J., Sun T., Pleasure S.J., Young S.G., Fong L.G. (2012) Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA. 109, E423–E431. https://doi.org/10.1073/pnas.1111780109
Schlachetzki J.C.M., Toda T., Mertens J. (2020) When function follows form: nuclear compartment structure and the epigenetic landscape of the aging neuron. Exp. Gerontol. 133, 110876. https://doi.org/10.1016/j.exger.2020.110876
Yang S.H., Procaccia S., Jung H.J., Nobumori C., Tatar A., Tu Y., Bayguinov Y.R., Hwang S.J., Tran D., Ward S.M., Fong L.G., Young S.G. (2015) Mice that express farnesylated versions of prelamin a in neurons develop achalasia. Hum. Mol. Genet. 24, 2826–2840. https://doi.org/10.1093/hmg/ddv043
Dong X., Milholland B., Vijg J. (2016) Evidence for a limit to human lifespan. Nature. 538, 257–259. https://doi.org/10.1038/nature19793
Steenstrup T., Kark J.D., Verhulst S., Thinggaard M., Hjelmborg J.V.B., Dalgård C., Kyvik K.O., Christiansen L., Mangino M., Spector T.D., Petersen I., Kimura M., Benetos A., Labat C., Sinnreich R., Hwang S.J., Levy D., Hunt S.C., Fitzpatrick A.L., Chen W., Berenson G.S., Barbieri M., Paolisso G., Gadalla S.M, Savage S.A., Christensen K., Yashin A.I., Arbeev K.G., Aviv A. (2017) Telomeres and the natural lifespan limit in humans. Aging (Albany NY). 9, 1130–1142. https://doi.org/10.18632/aging.101216
Tricola G.M., Simons M.J.P., Atema E., Boughton R.K., Brown J.L., Dearborn D.C., Divoky G., Eimes J.A., Huntington C.E., Kitaysky A.S., Juola F.A., Lank D.B., Litwa H.P., Mulder E.G.A., Nisbet I.C.T., Okanoya K., Safran R.J., Schoech S.J., Schreiber E.A., Thompson P.M., Verhulst S., Wheelwright N.T., Winkler D.W., Young R., Vleck C.M., Haussmann M.F. (2018) The rate of telomere loss is related to maximum lifespan in birds. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 373(1741), 20160445. https://doi.org/10.1098/rstb.2016.0445
Cawthon R.M., Smith K.R., O’Brien E., Sivat-chenko A., Kerber R.A. (2003) Association between telomere length in blood and mortality in people aged 60 years or older. Lancet. 361, 393–395. https://doi.org/10.1016/S0140-6736(03)12384-7
Aubert G., Lansdorp P.M. (2008) Telomeres and aging. Physiol. Rev. 88, 557–579. https://doi.org/10.1152/physrev.00026.2007
Celtikci. B., Erkmen G.K., Dikmen Z.G. (2020) Regulation and effect of telomerase and telomeric length in stem cells. Curr. Stem Cell Res. Ther. 16, 809–823. https://doi.org/10.2174/1574888X15666200422104423
Ros M., Carrascosa J.M. (2020) Current nutritional and pharmacological anti-aging interventions. Biochim. Biophys. Acta – Mol. Basis Dis. 1866(3), 165612. https://doi.org/10.1016/j.bbadis.2019.165612
Vaiserman A, Krasnienkov D. (2021) Telomere length as a marker of biological age: state-of-the-art. Front. Genet. Open Issues. Future Perspectives. 21, 630186. https://doi.org/10.3389/fgene.2020.630186
Muñoz-Lorente M.A., Cano-Martin A.C., Blasco M.A. (2019) Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 10(1), 4723. https://doi.org/10.1038/s41467-019-12664-x
Froy H., Underwood S.L., Dorrens J., Seeker L.A., Watt K., Wilbourn R.V., Pilkington J.G., Harrington L., Pemberton J.M., Nussey D.H. (2021) Heritable variation in telomere length predicts mortality in Soay sheep. Proc. Natl. Acad. Sci. USA. 118, e2020563118. https://doi.org/10.1073/pnas.2020563118
Wilkinson J.E., Burmeister L., Brooks S.V., Chan C.C., Friedline S., Harrison D.E., Hejtmancik J.F., Nadon N., Strong R., Wood L.K., Woodward M.A., Miller R.A. (2012) Rapamycin slows aging in mice. Aging Cell. 11, 675–682. https://doi.org/10.1111/j.1474-9726.2012.00832.x
Li Y.R., Li S., Lin C.C. (2018) Effect of resveratrol and pterostilbene on aging and longevity. Biofactors.44, 69–82. https://doi.org/10.1002/biof.1400
Weichhart T. (2018) mTOR as regulator of lifespan, aging, and cellular senescence: a mini-review. Geronto-logy. 64, 127–134. https://doi.org/10.1159/000484629
Blagosklonny M.V. (2019) Rapamycin for longevity: opinion article. Aging (Albany NY). 11, 8048–8067. https://doi.org/10.18632/aging.102355
Glossmann H.H., Lutz O.M.D. (2019) Metformin and aging: Gerontology. 65, 581–590. https://doi.org/10.1159/000502257
Bjedov I., Rallis C. (2020) The target of rapamycin signalling pathway in ageing and lifespan regulation. Genes (Basel). 11, 1043. https://doi.org/10.3390/genes11091043
Bernardes de Jesus B., Vera E., Schneeberger K., Tejera A.M., Ayuso E, Bosch F., Blasco M.A. (2012) Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 4, 691–704. https://doi.org/10.1002/emmm.201200245
Boccardi V., Herbig U. (2012) Telomerase gene therapy: a novel approach to combat aging. EMBO Mol. Med. 4, 685–687. https://doi.org/10.1002/emmm.201200246
Bernardes de Jesus B., Schneeberger K., Vera E., Tejera A., Harley C.B., Blasco M.A. (2011) The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell. 10, 604–621. https://doi.org/10.1111/j.1474-9726.2011.00700.x
Salvador L., Singaravelu G., Harley C.B., Flom P., Suram A., Raffaele J.M. (2016) A natural product telomerase activator lengthens telomeres in humans: a randomized, double blind, and placebo controlled study. Rejuvenation Res. 19, 478–484. https://doi.org/10.1089/rej.2015.1793
Tsoukalas D., Fragkiadaki P., Docea A.O., Alegakis A.K., Sarandi E., Thanasoula M., Spandidos D.A., Tsatsakis A., Razgonova M.P., Calina D. (2019) Discovery of potent telomerase activators: unfolding new therapeutic and anti-aging perspectives. Mol. Med. Rep. 20, 3701–3708. https://doi.org/10.3892/mmr.2019.10614
Prieto-Oliveira P. (2021) Telomerase activation in the treatment of aging or degenerative diseases: a systematic review. Mol. Cell. Biochem. 476(2), 599–607. https://doi.org/10.1007/s11010-020-03929-x
Whittemore K., Vera E, Martínez-Nevado E, Sanpera C, Blasco MA. (2019) Telomere shortening rate predicts species life span. Proc. Natl. Acad. Sci. USA. 116, 15122–15127. https://doi.org/10.1073/pnas.1902452116
Fernandez M.L., Thomas M.S., Lemos B.S., DiMarco D.M., Missimer A., Melough M., Chun O.K., Murillo A.G., Alyousef H.M., Medina-Vera I. (2018) TA-65, a telomerase activator improves cardiovascular markers in patients with metabolic syndrome. Curr. Pharm. Des. 24, 1905–1911. https://doi.org/10.2174/1381612824666180316114832
Ait-Ghezala G., Hassan S., Tweed M., Paris D., Crynen G., Zakirova Z., Crynen S., Crawford F. (2016) Identification of telomerase-activating blends from naturally occurring compounds. Altern. Ther. Health Med. 22, 6–14. PMID: .27433836
Березуцкий М.А., Дурнова Н.А., Власова Я.А. (2019) Экспериментальные и клинические исследования механизмов антивозрастных эффектов химических соединений Astragalus membranaceus (обзор литературы). Усп. геронтол. 32, 702–710.
Sharma R., Martins N. (2020) Telomeres, DNA damage and ageing: potential leads from ayurvedic rasayana (anti-ageing) drugs. J. Clin. Med. 9(8), 2544. https://doi.org/10.3390/jcm9082544
Alshinnawy A.S., El-Sayed W.M., Taha A.M., Sayed A.A., Salem A.M. (2020) Astragalus membranaceus and Punica granatum alleviate infertility and kidney dysfunction induced by aging in male rats. Turk. J. Biol. 44, 166–175. https://doi.org/10.3906/biy-2001-5
Bernardes de Jesus B., Blasco M.A. (2013) Telomerase at the intersection of cancer and aging. Trends Genet. 29, 513–520. https://doi.org/10.1016/j.tig.2013.06.007
Yang F., Xiu M., Yang S., Li X., Tuo W., Su Y., He J., Liu Y.(2021) extension of drosophila lifespan by astragalus polysaccharide through a mechanism dependent on antioxidant and insulin/IGF-1 signaling. Evid. Based Complement. Alternat. Med. 2021, 6686748. https://doi.org/10.1155/2021/6686748.9999
Shan H., Zheng X., Li M. (2019) The effects of astragalus membranaceus active extracts on autophagy-related diseases. Int. J. Mol. Sci. 20(8), 1904. https://doi.org/10.3390/ijms20081904
Zhang X., Liang T., Yang W., Zhang L., Wu S., Yan C., Li Q. (2020) Astragalus membranaceus injection suppresses production of interleukin-6 by activating autophagy through the AMPK-mTOR pathway in lipopolysaccharide-stimulated macrophages. Oxid. Med. Cell. Longev. 2020, 1364147. https://doi.org/10.1155/2020/1364147
Harley C.B., Liu W., Flom PL., Raffaele J.M. (2013) A natural product telomerase activator as part of a health maintenance program: metabolic and cardiovascular response. Rejuvenation Res. 16, 386–395. https://doi.org/10.1089/rej.2013.1430
Liu P., Zhao H., Luo Y. (2017) Anti-aging implications of Astragalus membranaceus (Huangqi): a well-known chinese tonic. Aging Dis. 8, 868–886. https://doi.org/10.14336/AD.2017.0816
Maier R., Bawamia B., Bennaceur K., Dunn S., Marsay L., Amoah R., Kasim A., Filby A., Austin D., Hancock H., Spyridopoulos I. (2020) Telomerase activation to reverse immunosenescence in elderly patients with acute coronary syndrome: protocol for a randomized pilot trial. JMIR Res. Protoc. 9, e19456. https://doi.org/10.2196/1945632965237
Егоров Е.Е. (2020) Здоровое старение: антиоксиданты, разобщители и/или теломераза? Молекуляр. биология. 54, 355–361.
Pignatti C., D’Adamo S., Stefanelli C., Flaigni F., Cetrullo S. (2020). Nutrients and pathways that regulate health span and life span. Geriatrics (Basel). 5(4), 95. https://doi.org/10.3390/geriatrics5040095
Ukraintseva S., Arbeev K., Duan M., Akushevich I., Kulminski A., Stallard E., Yashin A. (2021) Decline in biological resilience as key manifestation of aging: potential mechanisms and role in health and longevity. Mech. Ageing Dev. 194, 111418. https://doi.org/10.1016/j.mad.2020.111418
Yu M., Zhang H., Wang B., Zhang Y., Zheng X., Shao B., Zhuge Q., Jin K. (2021) Key signaling pathways in aging and potential interventions for healthy aging. Cells. 10(3), 660. https://doi.org/10.3390/cells10030660
Gorbunova V., Seluanov A. (2009) Coevolution of telomerase activity and body mass in mammals: from mice to beavers. Mech. Ageing Dev. 130(1–2), 3–9. https://doi.org/10.1016/j.mad.2008.02.008
Abegglen L.M., Caulin A.F., Chan A., Lee K., Robinson R., Campbell M.S., Kiso W.K., Schmitt D.L., Waddell P.J., Bhaskara S., Jensen S.T., Maley C.C., Schiffman J.D. (2015) Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA. 314, 1850–1860. https://doi.org/10.1001/jama.2015.13134
Ruby J.G., Smith M., Rochelle Buffenstein R. (2018) Naked mole-rat mortality rates defy gompertzian laws by not increasing with age. Elife. 7, e31157. https://doi.org/10.7554/eLife.31157
Seluanov A., Gladyshev V.N., Vijg J., Gorbunova V. (2018) Mechanisms of cancer resistance in long-lived mammals. Nat. Rev. Cancer. 18(7), 433–441. https://doi.org/10.1038/s41568-018-0004-9
Takasugi M., Firsanov D., Tombline G., Ning H., Ablaeva J., Seluanov A., Gorbunova V. (2020) Naked mole-rat very-high-molecular-mass hyaluronan exhibits superior cytoprotective properties. Nat. Commun. 11, 2376. https://doi.org/10.1038/s41467-020-16050-w
Zhao S., Lin L., Kan G., Xu C., Tang Q., Yu C., Cui S. (2014). High autophagy in the naked mole rat may play a significant role in maintaining good health. Cell. Physiol. Biochemistry. 33(2), 321–332. https://doi.org/10.1159/000356672
Brassard J.A. Fekete N., Garnier A., Hoesli C.A. (2016) Hutchinson–Gilford progeria syndrome as a model for vascular aging. Biogerontology. 17, 129–145. https://doi.org/10.1007/s10522-015-9602-z
Smith E.S.J., Park T.J., Holmes M.M., Buffenstein R. (2021) Some exciting future directions for work on naked mole-rats. Adv. Exp. Med. Biol. 1319, 409–420. https://doi.org/10.1007/978-3-030-65943-1_17
Macicior J., Marcos-Ramiro B., Ortega-Gutiérrez S. (2021) Small-molecule therapeutic perspectives for the treatment of progeria. Int. J. Mol Sci. 22(13), 7190. https://doi.org/10.3390/ijms22137190
Cabral W.A., Tavarez U.L., Beeram I., Yeritsyan D., Boku Y.D., Eckhaus M.A., Nazarian A., Erdos M.R., Collins F.S. (2021) Genetic reduction of mTOR extends lifespan in a mouse model of Hutchinson–Gilford progeria syndrome. Aging Cell. 20(9), e13457. https://doi.org/10.1111/acel.13457
Kychygina A., Dall’Osto M., Allen J.A.M., Cadoret J.C., Piras V., Pickett H.A., Crabbe L. (2021) Progerin impairs 3D genome organization and induces fragile telomeres by limiting the dNTP pools. Sci. Rep. 11(1), 13195. https://doi.org/10.1038/s41598-021-92631-z
Coppedè F. (2021) Mutations involved in premature-ageing syndromes. Appl. Clin. Genet. 14, 279–295. https://doi.org/10.2147/TACG.S273525
Yu M., Zhang H., Wang B., Zhang Y., Zheng X., Shao B., Zhuge Q., Jin K. (2021) Key signaling pathways in aging and potential interventions for healthy aging. Cells. 10(3), 660. https://doi.org/10.3390/cells10030660
Cabral W.A., Tavarez U.L., Beeram I., Yeritsyan D., Boku Y.D., Eckhaus M.A., Nazarian A., Erdos M.R., Collins F.S. (2021) Genetic reduction of mTOR extends lifespan in a mouse model of Hutchinson–Gilford progeria syndrome. Aging Cell. 20(9), e13457. https://doi.org/10.1111/acel.13457
Chen N.Y., Kim P.H., Fong L.G., Young S.G. (2020) Nuclear membrane ruptures, cell death, and tissue damage in the setting of nuclear lamin deficiencies. Progress and trends. Nucleus. 11, 237–249. https://doi.org/10.1080/19491034.2020.1815410
Dreesen O. (2020) Towards delineating the chain of events that cause premature senescence in the accelerated aging syndrome Hutchinson–Gilford progeria (HGPS). Biochem. Soc. Trans. 48, 981–991. https://doi.org/10.1042/BST20190882
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология