

Молекулярная биология, 2022, T. 56, № 3, стр. 468-475
Полногеномное профилирование метилирования ДНК CD4+ T-лимфоцитов больных первично-прогрессирующим рассеянным склерозом свидетельствует о вовлеченности этого эпигенетического процесса в иммунопатогенез заболевания
И. С. Киселев a, b, *, О. Г. Кулакова a, b, Л. В. Данилова c, d, О. А. Батурина e, М. Р. Кабилов e, Е. В. Попова a, А. Н. Бойко a, f, О. О. Фаворова a, b
a Российский национальный исследовательский медицинский университет им. Н.И. Пирогова Министерства здравоохранения Российской Федерации
117997 Москва, Россия
b Национальный медицинский исследовательский центр кардиологии Министерства здравоохранения Российской Федерации
121552 Москва, Россия
c Институт общей генетики им. Н.И. Вавилова Российской академии наук
119991 Москва, Россия
d Johns Hopkins School of Medicine
21205 Baltimore, MD, USA
e Институт химической биологии и фундаментальной медицины Сибирского отделения Российской академии наук
630090 Новосибирск, Россия
f Федеральный центр мозга и нейротехнологий Федерального медико-биологического
агентства Российской Федерации
117997 Москва, Россия
* E-mail: kiselev.ivan.1991@gmail.com
Поступила в редакцию 18.10.2021
После доработки 18.10.2021
Принята к публикации 02.11.2021
- EDN: FAXIHU
- DOI: 10.31857/S0026898422030089
Аннотация
Рассеянный склероз (РС) – хроническое заболевание ЦНС, патогенез которого включает аутоиммунную и нейродегенеративную компоненты. В большинстве случаев у больных наблюдается ремиттирующая форма РС, однако у 10–15% пациентов развивается первично-прогрессирующий РС (ППРС), существенно отличающийся от ремиттирующего как по механизмам развития патологического процесса, так и по ряду демографических и клинических характеристик. Эти отличия могут объясняться особенностями эпигенетических механизмов регуляции генной экспрессии при ППРС, в том числе одного из них – метилирования ДНК. Особенности метилирования ДНК в различных клеточных популяциях при ППРС остаются практически не изученными. Цель настоящей работы состояла в выявлении дифференциально метилированных CpG-сайтов (ДМС) генома CD4+ Т-лимфоцитов, характеризующих ППРС. Полногеномный анализ метилирования ДНК CD4+ T-лимфоцитов проводили с использованием ДНК-микрочипов высокой плотности. Идентифицировано 108 ДМС, отличающих больных ППРС, никогда не принимавших иммуномодулирующие препараты, от здоровых индивидов. У больных гиперметилированными оказались 81% этих ДМС. Более половины всех ДМС находятся в области известных генов и входят в состав CpG-островков и соседних с ними областей, что говорит о высокой функциональной значимости этих ДМС при развитии ППРС. Анализ представленности генов, содержащих ДМС, в основных биологических процессах показал их участие в формировании иммунного ответа, процессинге и презентации антигена, развитии иммунной системы, регуляции адгезии клеток к внеклеточному матриксу. Результаты полногеномного анализа метилирования ДНК CD4+ T-лимфоцитов больных ППРС свидетельствуют о вовлеченности этой эпигенетической модификации в иммунопатогенез заболевания. Полученные данные могут помочь в углублении знаний об особенностях патогенеза этой тяжелой формы РС.
ВВЕДЕНИЕ
Рассеянный склероз (РС) – тяжелое хроническое заболевание ЦНС, сочетающее в своем патогенезе аутоиммунную и нейродегенеративную компоненты и сопровождающееся прогрессирующей неврологической дисфункцией [1]. Следствием неуклонного нарастания неврологического дефицита является необратимая инвалидизация пациентов в молодом трудоспособном возрасте, что обуславливает высокую социальную и экономическую значимость заболевания.
РС – это комплексное заболевание, которое развивается у лиц с генетической предрасположенностью под воздействием факторов внешней среды. При этом влияние внешних факторов на риск развития РС может быть в значительной мере опосредовано эпигенетическими механизмами регуляции экспрессии генов [2].
Течение РС обладает выраженной клинической гетерогенностью. В большинстве случаев у больных наблюдается ремиттирующая форма РС (РРС), характеризующаяся сменами периодов обострения и ремиссии [3]. Однако у 10–15% пациентов с самого начала болезни происходит непрерывное нарастание неврологического дефицита; такая тяжелая клинически обособленная форма РС получила название первично-прогрессирующего РС (ППРС) [4].
ППРС существенно отличается от РРС как по механизмам развития патологического процесса [5], так и по целому ряду демографических и клинических характеристик, таких как соотношение больных мужчин и женщин, средний возраст дебюта заболевания, клинические признаки дебюта, скорость прогрессирования и др. [4, 6]. Эти отличия могут объясняться особенностями эпигенетических механизмов регуляции генной экспрессии при ППРС.
Эпигенетические модификации генома не затрагивают последовательности ДНК, но при этом способны влиять на экспрессию генов в различных клетках и/или тканях [2]. Один из ключевых эпигенетических процессов – метилирование ДНК в положении С5 цитозинового основания в составе CpG-динуклеотидов. Метилирование CpG-сайтов в области промотора приводит к быстрому подавлению экспрессии генов с участием репрессорных белков [7]. Поиск специфических паттернов метилирования, характеризующих ППРС, может помочь в понимании особенностей развития заболевания.
Наибольшей информативностью при изучении метилирования ДНК обладает полногеномный анализ с использованием ДНК-микрочипов высокой плотности или высокопроизводительного секвенирования. Применение этих методов позволило выявить различия в паттернах метилирования ДНК из различных клеток и тканей больных РРС и здоровых добровольцев [8–12].
Первое исследование метилирования ДНК из мононуклеарных клеток периферической крови (МНК) больных ППРС выявило паттерн метилирования, отличающий эту форму заболевания и от больных РРС, и от здоровых доноров [8]. Однако МНК представляют собой гетерогенную клеточную популяцию, а, как показано ранее, паттерны метилирования ДНК в различных типах клеток могут значительно отличаться как в норме, так и при патологии [13]. Это приводит к необходимости изучения особенностей метилирования ДНК в отдельных субпопуляциях клеток крови при ППРС.
В настоящей работе мы изучили метилирование ДНК CD4+ T-лимфоцитов – одной из ключевых для патогенеза РС популяций лейкоцитов крови – у больных ППРС [14].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Характеристика групп. В исследование включены восемь больных ППРС (шесть женщин и двое мужчин) в возрасте от 28 до 58 лет. Диагноз устанавливали согласно критериям МакДональда в редакции 2014 года [15]. Средний балл по расширенной шкале инвалидизации (Expanded Disability Status Scale, EDSS) составил 4.29 ± 0.39. Больные, включенные в исследование, никогда не принимали иммуномодулирующих препаратов. В контрольную группу вошли восемь здоровых добровольцев (шесть женщин, двое мужчин) в возрасте от 28 до 50 лет без признаков неврологических заболеваний.
Все включенные в исследование индивиды были (согласно данным анкетирования) этническими русскими, все подписали информированное согласие на участие в эксперименте. Проведение исследования одобрено этическим комитетом ФГБОУ ВО РНИМУ им. Н.И. Пирогова МЗ РФ.
Выделение ДНК и полногеномный анализ профилей метилирования. Образцы периферической крови собирали в вакуумные пробирки с EDТА. МНК выделяли из крови путем центрифугирования в градиенте фиколл-гипака. Фракцию CD4+ Т-лимфоцитов выделяли из МНК с помощью микробус, конъюгированных с антителами к CD4-антигенам (“Miltenyi Biotec”, Германия). Геномную ДНК выделяли из CD4+ Т-лимфоцитов с помощью набора DNA Mini Kit (“Qiagen”, Германия) по методике производителя. Бисульфитную конверсию геномной ДНК проводили при помощи набора EZ DNA Methylation-Gold Kit (“Zymo Research”, США). Уровень метилирования ДНК оценивали с помощью биологических микрочипов высокой плотности Infinium HumanMethylation450 BeadChip на сканере чипов iScan (“Illumina”, США) в Центре коллективного пользования “Геномика” (ИХБФМ СО РАН).
Биоинформатический анализ. Анализ данных, в том числе первичная обработка данных и их нормирование, выполнены с использованием пакета methylumi [16] и стандартных средств среды R. Из анализа исключали пробы на ДНК-микрочипе, которые содержали однонуклеотидные полиморфизмы (SNP) на расстоянии ≤10 п.н. от исследуемого CpG-сайта; пробы, перекрывающиеся с повторяющимися последовательностями генома; и пробы, расположенные в половых хромосомах. Из рассмотрения также исключили пробы, у которых величина значимости обнаружения сигнала (detection p-value) была больше 0.05 хотя бы в одном из исследованных образцов. Для оценки уровней метилирования CpG-сайтов вычисляли показатель β, который определяется как отношение интенсивности сигнала метилирования от соответствующей пробы на ДНК-микрочипе к общей интенсивности сигнала (сумма интенсивностей метилированного и неметилированного сигналов) данной пробы. Значения β варьировали от 0 для неметилированных проб до 1 – в полностью метилированных.
Дифференциальное метилирование сайтов определяли с помощью пакета limma [17] среды R. CpG-сайт считали дифференциально метилированным (ДМС), если средние значения показателя бета этого сайта в сравниваемых группах различались более чем на 10% (|Δβ| > 0.1), а соответствующая величина p, определенная с помощью t-теста Стьюдента, была меньше 0.01. Локализацию конкретного CpG-сайта в CpG-островке определяли, используя аннотацию UCSC в hg19; ДМС, расположенные в 2 т.п.н. от CpG-островков, относили к соседним с ними областям (shore); локализованные в 2 т.п.н. от соседних областей – к отдаленным областям (shelf). Построение карт интенсивности сигналов ДМС осуществляли с использованием пакетов pheatmap и ggplot2 для среды R.
Статистический анализ перепредставленности известных биологических процессов (Biological Process) из базы данных Gene Onthology (GO) генами, содержащими ДМС, проводили с использованием web-инструмента GO Panther (http://www.pantherdb.org/ [18]). Отбирали значимо обогащенные процессы (p < 0.05), включающие не менее двух идентифицированных генов. В случае обогащения нескольких вложенных друг в друга процессов анализировали только обобщающий процесс более высокого уровня.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Чтобы оценить вовлечение эпигенетического механизма метилирования ДНК CD4+ Т-лимфоцитов в развитие ППРС, мы провели профилирование паттернов метилирования более 450 000 CpG-сайтов генома этой клеточной популяции в репрезентативных группах больных ППРС и здоровых индивидов. Сравнение уровней метилирования CpG-сайтов в этих группах позволило нам выявить 108 ДМС при ППРС. При иерархической кластеризации образцов ДНК на картах интенсивности сигналов ДМС (рис. 1) больные ППРС и здоровые индивиды группируются в обособленные кластеры.
Рис. 1.
Карта интенсивности сигналов ДМС генома CD4+ Т-лимфоцитов у больных ППРС и здоровых индивидов. Градиент серого от темного к светлому отражает повышение уровня метилирования ДНК в образцах. Дендрограмма сверху показывает иерархическую кластеризацию образцов: серый цвет – образцы больных ППРС, черный – здоровых индивидов. Слева представлена иерархическая кластеризация ДМС, где черным показана их локализация в CpG-островках (island), темно-серым – в соседних с ними областях (shore), светло-серым – в отдаленных областях (shelf), белым – в прочих областях (sea).
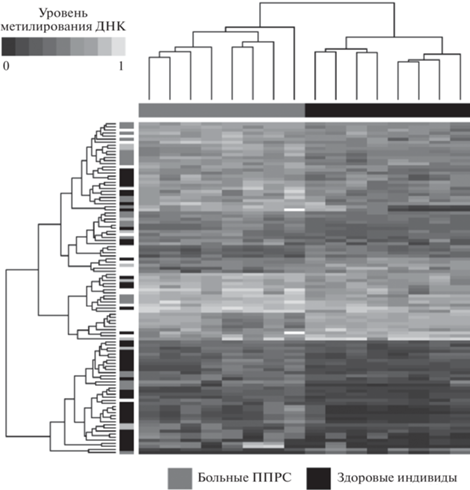
Характеристика выявленных ДМС представлена в табл. 1. Большая их часть (81%) оказалась гиперметилированной у больных при сравнении с контрольной группой. Более половины всех ДМС (66%) располагаются в области генов (всего выявлено 53 таких гена). Из них ген OR2L13 содержал восемь ДМС, ген IGF2BP1 – три ДМС, а гены ERICH1, HKR1, IGSF9B, LCLAT1, NPAS3, RHOJ, SP140, HCG4B и HLA-H – по два ДМС. Уровни метилирования всех ДМС в области генов C11orf58, CCDC172, CLRN3, RASA3, PTCD3, SP140, TONSL, FAM157B были снижены у больных ППРС по сравнению с контрольной группой. У больных ген HLA-H содержал один гипометилированный и один гиперметилированный ДМС, а остальные 44 гена содержали гиперметилированые ДМС.
Таблица 1.
ДМС, выявленные при анализе метилирования ДНК CD4+ Т-лимфоцитов, у больных ППРС и здоровых индивидов
| Общее количество ДМС | 108 (100%) |
| ДМС, гиперметилированные у больных ППРС | 88 (81%) |
| ДМС в области генов | 71 (66%) |
| Гены, содержащие ДМС | 53 |
| Гены, содержащие ДМС, гиперметилированные у больных ППРС | 44 (83%) |
| Белоккодирующие гены, содержащие ДМС | AGA; ASRGL1; C11orf58; C1orf35; CBFA2T3; CCDC172; CCDC85C; CDK11B; CLRN3; CORO1C; CRTAC1; DCLRE1C; ENOSF1; ERICH1(2); FLI1; FYTTD1; HKR1(2); HLA-DRB5; IGDCC4; IGF2BP1(3); IGSF9B(2); IMMT; KIAA0513; LCLAT1(2); LIME1; MACROH2A1; NKAIN1; NPAS3(2); OR2L13(8); PLEKHA2; PRTFDC1; PTH1R; RASA3; RHOJ(2); SERPINA11; SIM2; SLC22A16; SLC35E4; SLC46A2; PEAK3; PTCD3; SP140(2); SPAG1; TGFBR3; TMEM44; TONSL; TULP2; UBXN7; XKR5 |
| Белокнекодирующие гены, содержащие ДМС | FAM157B; GUSBP4; HCG4B(2); HLA-H(2)* |
Примечание. Критерии отбора ДМС: p < 0.01, |Δβ| > 0.1. Жирным отмечены гены, ДМС в которых имеют пониженный уровень метилирования у больных ППРС. В скобках около названия гена указано число ДМС в гене, если их более 1. * В гене выявлен один ДМС с повышенным и один с пониженным уровнем метилирования.
Выявлены различия в уровне метилирования 16 ДМС в сравниваемых группах более чем на 20% (13 из них расположены в восьми генах). Гиперметилированными на 20–37% были ДМС cg08260406, cg20507276 и cg08944170 (ген OR2L13), cg07157030 (RHOJ), cg19980771 (SLC22A16), cg17749961 и cg12454169 (LCLAT1), cg15070894 и cg18786623 (HCG4B), cg01053087 и cg05875700 (ERICH1). Сg04824555 (FAM157B) и cg02938066 (TONSL) были гипометилированными у больных на 21 и 22% соответственно.
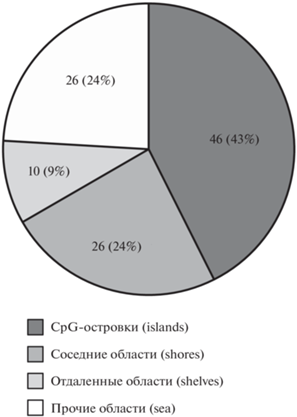
Для оценки возможного влияния выявленных ДМС на экспрессию генов, в области которых они расположены, проведен анализ их локализации по отношению к функционально значимым скоплениям CpG-сайтов. Как видно из рис. 2, более половины ДМС находятся в функционально наиболее важных областях: в CpG-островках (43%) и в соседних с ними областях (24%).
Рис. 2.
Локализация ДМС, выявленных при анализе метилирования ДНК CD4+ Т-лимфоцитов больных ППРС и здоровых индивидов, по отношению к функционально значимым скоплениям CpG-сайтов. В секторах диаграммы указано абсолютное число ДМС (% от общего числа ДМС).
Проведен статистический анализ перепредставленности известных биологических процессов (GO Biological Process) генами, содержащими ДМС. Как можно видеть из табл. 2, наиболее значимое обогащение наблюдается для процессов “Процессирование и презентация антигена” (GO:0019882) и “Развитие иммунной системы” (GO:0002520). Остальные идентифицированные процессы относятся к глобальным механизмам поддержания жизнедеятельности клетки: “Внутриклеточный метаболизм аминокислот” (GO:0006520), “Регуляция удержания в клетке ионов Ca2+” (GO:0051282), “Регуляция адгезии клеток к внеклеточному матриксу” (GO:0001952), “Транспорт мРНК” (GO:0051028), “Регуляция морфогенеза клеток” (GO:0022604).
Таблица 2.
Перепредставленность генов, содержащих ДМС, в известных биологических процессах (по данным базы Gene Onthology)
| Процесс (ID в базе Gene Onthology) | Гены, входящие в набор, общее коли-чество | ДМС-содержащие гены, вовлеченные в процесс | Обога-щение, % | Значение р |
||
|---|---|---|---|---|---|---|
| фактическое количество | ген | ожидаемое количество | ||||
| Процессинг и презентация антигена (GO:0019882) | 94 | 2 | HLA-DRB5, HLA-H | 0.23 | 8.76 | 0.023 |
| Развитие иммунной системы (GO:0002520) | 665 | 5 | CBFA2T3, DCLRE1C, FLI1, SLC46A2, TGFBR3 | 1.61 | 3.1 | 0.023 |
| Внутриклеточный метаболизм аминокислот (GO:0006520) | 276 | 3 | AGA, ASRGL1, ENOSF1 | 0.67 | 4.48 | 0.030 |
| Регуляция удержания в клетке ионов Ca2+ (GO:0051282) | 119 | 2 | LIME1, RASA3 | 0.29 | 6.92 | 0.035 |
| Регуляция адгезии клеток к внеклеточному матриксу (GO:0001952) | 125 | 2 | CORO1C, PLEKHA2 | 0.3 | 6.59 | 0.038 |
| Транспорт мРНК (GO:0051028) | 137 | 2 | FYTTD1, IGF2BP1 | 0.33 | 6.01 | 0.045 |
| Регуляция морфогенеза клеток (GO:0022604) | 310 | 3 | CORO1C, PEAK3, RHOJ | 0.75 | 3.99 | 0.040 |
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В настоящей работе мы впервые идентифицировали различия профилей метилирования ДНК в популяции CD4+ Т-лимфоцитов, ключевой для патогенеза РС, у больных ППРС и здоровых индивидов контрольной группы. Более половины выявленных ДМС находится в области известных генов и входят в состав CpG-островков и соседних с ними областей, что говорит о высокой функциональной значимости этих ДМС в патологическом процессе.
При ППРС наблюдается повышение уровня метилирования ДНК CD4+ Т-лимфоцитов (81% ДМС гиперметилированы), что полностью согласуется с данными, полученными нами ранее при анализе ДНК из МНК (86% ДМС гиперметилированы) [8]. Подобное повышение уровней метилирования ДНК иммунных клеток, по-видимому, отличает ППРС от более распространенного РРС, при котором гиперметилированы 43% CpG-сайтов в ДНК МНК [8] и 41–50% – в CD4+ T-лимфоцитах [9, 19, 20].
Основной областью генома, определяющей наследственную предрасположенность к РС независимо от его клинической формы, признан локус HLA хромосомы 6 [21]. В настоящей работе мы выявили гиперметилирование ДНК трех генов, расположенных в этом локусе: HLA-DRB5, кодирующего β-цепь молекул HLA II, псевдогена HLA-H и белокнекодирующего гена HCG4B с неизвестной функцией. Дифференциальное метилирование генов HLA-DRB5 и HCG4B ранее уже обнаруживали при сравнении уровней метилирования ДНК CD4+ Т-лимфоцитов больных РРС и здоровых индивидов [9, 20]. Дифференциальное метилирование различных генов локуса HLA показано ранее и в других популяциях лимфоцитов больных РРС [10, 22] и ППРС [8] при сравнении с контрольной группой. В то же время, прямое сравнение не выявило различий в уровнях метилирования генов локуса HLA при этих двух формах РС [23]. Это еще раз подтверждает ключевую роль локуса HLA в патогенезе РС независимо от его формы и указывает на вовлечение эпигенетических механизмов в регуляцию экспрессии генов локуса HLA при развитии заболевания.
Мы сравнили спектры генов, содержащих ППРС-специфические ДМС, впервые полученные методом полногеномного профилирования ДНК из CD4+ Т-лимфоцитов (настоящее исследование), и из МНК [8]. Найдены четыре гена, повышение уровня метилирования которых наблюдали в обоих исследованиях: OR2L13, IGSF9B, HKR1 и TMEM44.
В промоторной области первого из этих генов, OR2LI3, обнаружено сразу восемь ДМС (см. табл. 1); несколько из них гиперметилированы более чем на 20%. Этот ген кодирует один из белков обширного семейства обонятельных рецепторов, сопряженных с G-белками. Рецепторы этого семейства широко экспрессируются в самых разных тканях и органах, включая ЦНС, и могут участвовать в выполнении множества различных функций [24]. Хотя ген OR2L13 к настоящему моменту практически не изучен, обнаружена его связь с несколькими заболеваниями ЦНС. Показано, что при развитии болезни Паркинсона в ЦНС снижается экспрессия OR2L13 [25], а у больных аутизмом наблюдается гиперметилирование гена в цельной крови [26].
Продукт второго гена, IGSF9B, представляет собой трансмембранный белок с высоким уровнем экспрессии в интернейронах и связанный с регуляцией развития тормозных синапсов [27]. Интересно, что SNP rs10894768, расположенный во втором экзоне, ассоциирован с тяжестью РС, а сам ген IGSF9B экспрессируется в астроцитах и макрофагах из хронически активных очагов демиелинизации [28]. Данные о функциях генов HKR1 и TMEM44 человека и их связи с аутоиммунными или нейродегенеративными заболеваниями в настоящий момент отсутствуют.
Отдельного упоминания заслуживают гены ERICH1 и LCLAT1. В нашем исследовании уровни метилирования ДМС в областях обоих этих генов у больных ППРС оказались более чем на 20% выше, чем у здоровых индивидов контрольной группы. Интересно, что, напротив, наблюдали снижение уровня метилирования и ERICH1, и LCLAT1 в CD4+ клетках больных РРС [9, 20], что может указывать на вовлечение эпигенетической регуляции экспрессии этих генов в формирование клинической формы РС. Функция ERICH1 остается пока неизученной. Показано участие продукта гена LCLAT1 в модификации фосфолипидов, в том числе митохондриального кардиолипина. Снижение экспрессии LCLAT1 коррелирует с повышением продукции активных форм кислорода и индукцией апоптоза [29].
Cтатистический анализ перепредставленности генов, содержащих ДМС, в основных биологических процессах показал, что часть из них вовлечена в процессинг и презентацию антигена (HLA-DRB5, HLA-H), развитие иммунной системы (CBFA2T3, DCLRE1C, FLI1, SLC46A2, TGFBR3), а также в регуляцию адгезии клеток к внеклеточному матриксу (CORO1C, PLEKHA2), во многом определяющую миграцию лейкоцитов. Остальные гены, содержащие ДМС, по-видимому, участвуют в патогенезе ППРС на уровне общих интегральных систем регуляции жизнедеятельности клетки.
В целом, проведенный нами анализ позволил идентифицировать паттерн метилирования ДНК, характерный для ППРС. Полученные нами данные, после соответствующего подтверждения на независимых выборках, могут помочь в углублении знаний об особенностях патогенеза ППРС, что, в свою очередь, может быть использовано при разработке новых эффективных препаратов для терапии этого заболевания.
Исследование выполнено в рамках Государственного задания ФГАОУ ВО РНИМУ им. Н.И. Пирогова МЗ РФ №121040600400-8.
От всех включенных в исследование индивидов получено информированное согласие на участие в эксперименте. Проведение исследования одобрено этическим комитетом ФГБОУ ВО РНИМУ им. Н.И. Пирогова МЗ РФ.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
International Multiple Sclerosis Genetics Consortium. (2019) Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science. 365(6460), eaav7188.
Oksenberg J.R. (2013) Decoding multiple sclerosis: an update on genomics and future directions. Expert. Rev. Neurother. 13(12 Suppl), 11–19.
Lublin F.D., Reingold S.C., Cohen J.A., Cutter G.R., Sørensen P.S., Thompson A.J., Wolinsky J.S., Balcer L.J., Banwell B., Barkhof F., Bebo B. Jr., Calabresi P.A., Clanet M., Comi G., Fox R.J., Freedman M.S., Goodman A.D., Inglese M., Kappos L., Kieseier B.C., Lincoln J.A., Lubetzki C., Miller A.E., Montalban X., O’Connor P.W., Petkau J., Pozzilli C., Rudick R.A., Sormani M.P., Stüve O., Waubant E., Polman C.H. (2014) Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 83(3), 278–286.
Koch M., Kingwell E., Rieckmann P., Tremlett H. (2009) The natural history of primary progressive multiple sclerosis. Neurology. 73(23), 1996–2002.
Correale J., Gaitán M.I., Ysrraelit M.C., Fiol M.P. (2017) Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain. 140(3), 527–546.
Debouverie M., Pittion-Vouyovitch S., Louis S., Guillemin F., LORSEP Group. (2008) Natural history of multiple sclerosis in a population-based cohort. Eur. J. Neurol. 15(9), 916–921.
Choy M.K., Movassagh M., Goh H.G., Bennett M.R., Down T.A., Foo R.S. (2010) Genome-wide conserved consensus transcription factor binding motifs are hyper-methylated. BMC Genomics. 11, 519.
Кулакова О.Г., Кабилов М.Р., Данилова Л.В., Попова Е.В., Батурина О.А., Царева Е.Ю., Баулина Н.М., Киселев И.С., Бойко А.Н., Фаворов А.В., Фаворова О.О., Власов В.В. (2016) Полногеномный анализ метилирования ДНК мононуклеарных клеток крови больных различными формами рассеянного склероза. Acta Naturae. 8(2), 39–47.
Maltby V.E., Lea R.A., Sanders K.A., White N., Benton M.C., Scott R.J., Lechner-Scott J. (2017) Differential methylation at MHC in CD4+ T cells is associated with multiple sclerosis independently of HLA-DRB1. Clin. Epigenetics. 9, 71.
Maltby V.E., Graves M.C., Lea R.A., Benton M.C., Sanders K.A., Tajouri L., Scott R.J., Lechner-Scott J. (2015) Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin. Epigenetics. 7, 118.
Huynh J.L., Garg P., Thin T.H., Yoo S., Dutta R., Trapp B.D., Haroutunian V., Zhu J., Donovan M.J., Sharp A.J., Casaccia P. (2014) Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat. Neurosci. 17(1), 121–130.
Marabita F., Almgren M., Sjöholm L.K., Kular L., Liu Y., James T., Kiss N.B., Feinberg A.P., Olsson T., Kockum I., Alfredsson L., Ekström T.J., Jagodic M. (2017) Smoking induces DNA methylation changes in multiple sclerosis patients with exposure-response relationship. Sci. Rep. 7(1), 14589.
Macartney-Coxson D., Cameron A.M., Clapham J., Benton M.C. (2020) DNA methylation in blood-potential to provide new insights into cell biology. PLoS One. 15(11), e0241367.
Legroux L., Arbour N. (2015) Multiple sclerosis and T lymphocytes: an entangled story. J. Neuroimmune Pharmacol. 10(4), 528–546.
Milo R., Miller A. (2014) Revised diagnostic criteria of multiple sclerosis. Autoimmun. Rev. 13(4–5), 518–524.
Davis S., Du P., Bilke S., Triche, Jr. T., Bootwalla M. Methylumi: Handle Illumina methylation data. R package version 2.38.0. https://bioconductor.org/packages/ release/bioc/html/methylumi.html (accessed Sep. 20, 2021).
Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl. Acids Res. 43(7), e47.
Thomas P.D., Campbell M.J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., Narechania A. (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13(9), 2129–2141.
Bos S.D., Page C.M., Andreassen B.K., Elboudwarej E., Gustavsen M.W., Briggs F., Quach H., Leikfoss I.S., Bjølgerud A., Berge T., Harbo H.F., Barcellos L.F. (2015) Genome-wide DNA methylation profiles indicate CD8+ T cell hypermethylation in multiple sclerosis. PLoS One. 10(3), e0117403.
Graves M.C., Benton M., Lea R.A., Boyle M., Tajouri L., Macartney-Coxson D., Scott R.J., Lechner-Scott J. (2014) Methylation differences at the HLA-DRB1 locus in CD4+ T-cells are associated with multiple sclerosis. Mult. Scler. 20(8), 1033–1041.
Hollenbach J.A., Oksenberg J.R. (2015) The immunogenetics of multiple sclerosis: a comprehensive review. J. Autoimmun. 64, 13–25.
Kular L., Liu Y., Ruhrmann S., Zheleznyakova G., Marabita F., Gomez-Cabrero D., James T., Ewing E., Lindén M., Górnikiewicz B., Aeinehband S., Stridh P., Link J., Andlauer T.F.M., Gasperi C., Wiendl H., Zipp F., Gold R., Tackenberg B., Weber F., Hemmer B., Strauch K., Heilmann-Heimbach S., Rawal R., Schminke U., Schmidt C.O., Kacprowski T., Franke A., Laudes M., Dilthey A.T., Celius E.G., Søndergaard H.B., Tegnér J., Harbo H.F., Oturai A.B., Olafsson S., Eggertsson H.P., Halldorsson B.V., Hjaltason H., Olafsson E., Jonsdottir I., Stefansson K., Olsson T., Piehl F., Ekström T.J., Kockum I., Feinberg A.P., Jagodic M. (2018) DNA methylation as a mediator of HLA-DRB1*15:01 and a protective variant in multiple sclerosis. Nat. Commun. 9(1), 2397.
Handel A.E., De Luca G.C., Morahan J., Handunnetthi L., Sadovnick A.D., Ebers G.C., Ramagopalan S.V. (2010) No evidence for an effect of DNA methylation on multiple sclerosis severity at HLA-DRB1*15 or HLA-DRB5. J. Neuroimmunol. 223(1–2), 120–123.
Feldmesser E., Olender T., Khen M., Yanai I., Ophir R., Lancet D. (2006) Widespread ectopic expression of olfactory receptor genes. BMC Genomics. 7, 121.
Garcia-Esparcia P., Schlüter A., Carmona M., Mo-reno J., Ansoleaga B., Torrejón-Escribano B., Gustincich S., Pujol A., Ferrer I. (2013) Functional genomics reveals dysregulation of cortical olfactory receptors in Parkinson disease: novel putative chemoreceptors in the human brain. J. Neuropathol. Exp. Neurol. 72(6), 524–539.
Wong C.C., Meaburn E.L., Ronald A., Price T.S., Jeffries A.R., Schalkwyk L.C., Plomin R., Mill J. (2014) Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry. 19(4), 495–503.
Woo J., Kwon S.K., Nam J., Choi S., Takahashi H., Krueger D., Park J., Lee Y., Bae J.Y., Lee D., Ko J., Kim H., Kim M.H., Bae Y.C., Chang S., Craig A.M., Kim E. (2013) The adhesion protein IgSF9b is coupled to neuroligin 2 via S-SCAM to promote inhibitory synapse development. J. Cell. Biol. 201(6), 929–944.
Gil-Varea E., Urcelay E., Vilariño-Güell C., Costa C., Midaglia L., Matesanz F., Rodríguez-Antigüedad A., Oksenberg J., Espino-Paisan L., Dessa Sadovnick A., Saiz A., Villar L.M., García-Merino J.A., Ramió-Torrentà L., Triviño J.C., Quintana E., Robles R., Sánchez-López A., Arroyo R., Alvarez-Cermeño J.C., Vidal-Jordana A., Malhotra S., Fissolo N., Montalban X., Comabella M. (2018) Exome sequencing study in patients with multiple sclerosis reveals variants associated with disease course. J. Neuroinflammation. 15(1), 265.
Huang L.S., Mathew B., Li H., Zhao Y., Ma S.F., Noth I., Reddy S.P., Harijith A., Usatyuk P.V., Berdyshev E.V., Kaminski N., Zhou T., Zhang W., Zhang Y., Rehman J., Kotha S.R., Gurney T.O., Parinandi N.L., Lussier Y.A., Garcia J.G., Natarajan V. (2014) The mitochondrial cardiolipin remodeling enzyme lysocardiolipin acyltransferase is a novel target in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 189(11), 1402–1415.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология