

Молекулярная биология, 2022, T. 56, № 4, стр. 604-618
Эффективная вирусная доставка в нейрональную культуру генетических конструкций для моделирования и генной терапии GNAO1-энцефалопатии
Е. А. Лунев a, b, c, А. А. Шмидт a, С. Г. Васильева a, И. М. Савченко a, В. А. Логинов a, В. И. Марина b, Т. В. Егорова a, М. В. Бардина a, b, c, *
a Институт биологии гена Российской академии наук
119334 Москва, Россия
b ООО “Марлин Биотех”
354340 Сочи, Россия
c Центр высокоточного редактирования и генетических технологий для биомедицины, Институт биологии гена Российской академии наук
119334 Москва, Россия
* E-mail: maryana.bardina@gmail.com
Поступила в редакцию 31.01.2022
После доработки 25.02.2022
Принята к публикации 28.02.2022
- EDN: DEJCPY
- DOI: 10.31857/S0026898422040061
Аннотация
GNAO1-энцефалопатия – орфанное генетическое заболевание, которое характеризуется ранней младенческой эпилепсией, нарушением контроля над движениями и серьезной задержкой развития. Заболевание вызывается мутациями в гене GNAO1, приводящими к нарушению функций кодируемого этим геном белка Gαo1. Методы лечения GNAO1-энцефалопатии отсутствуют, а симптоматическая терапия неэффективна. Фенотипическая гетерогенность заболевания указывает на необходимость персонализированного подхода к разработке терапии пациентов с конкретным патогенным вариантом GNAO1, что требует изучения механизма заболевания на животных и клеточных моделях. В настоящей работе разработан подход к созданию клеточных моделей GNAO1-энцефалопатии на первичной культуре нейронов здоровой мыши, а также к тестированию препаратов генной терапии in vitro. С этой целью нами оптимизирована доставка трансгенов в Gαo1-экспрессирующие клетки с помощью аденоассоциированных вирусов (ААВ). Оценен тропизм пяти нейротропных серотипов ААВ (1, 2, 6, 9, DJ) к Gαo1-позитивным нейронам в культуре из цельного мозга мыши. Самый высокий потенциал в качестве средства доставки репортера показал серотип DJ, который заражал до 66% таргетных нейронов без выраженной цитотоксичности. AAВ-DJ также обеспечивал эффективную доставку и экспрессию генетических конструкций, кодирующих нормальную и мутантную форму Gαo1, а также коротких шпилечных РНК (shРНК) для супрессии эндогенного Gnao1 в нейронах мыши. Полученные нами результаты позволят продвинуться в изучении механизма патогенеза клинических вариантов GNAO1-энцефалопатии, а также тестировать конструкции для генной терапии данного заболевания в клеточных моделях.
ВВЕДЕНИЕ
Использование технологий высокопроизводительного секвенирования позволило обнаружить связь между мутациями в гене GNAO1 и орфанным неврологическим расстройством ‒ GNAO1-энцефалопатией [1, 2]. Это генетическое заболевание проявляется в младенчестве и характеризуется эпилептической энцефалопатией (OMIM 615473) и/или нарушением развития мозга с непроизвольными движениями (OMIM 617493). Ген GNAO1 экспрессируется преимущественно в головном мозге и кодирует белок Gαo1 [3, 4]. Функция белка Gαo1 слабо изучена, однако известно, что Gαo1 относится к семейству ингибиторных G-белков (Gi/o) и участвует в модуляции сигналинга в нейронах [5]. Описано 25 клинически значимых мутаций гена GNAO1 [5], все они проявляются в гетерозиготном состоянии и возникают de novo.
Способов лечения GNAO1-энцефалопатии не существует. Симптоматическая терапия включает антиконвульсанты и препараты для снятия гиперкинеза, однако они проявляют высокую эффективность не у всех пациентов [6, 7]. Ситуация осложняется фенотипической гетерогенностью заболевания, вызванной разным влиянием конкретных мутаций на функцию Gαo1 и опосредованную им нейрональную передачу. Так, описана группа мутаций GNAO1 (D174G, I279N, G40R), которые проявляются преимущественно младенческой эпилепсией; другая группа мутаций (E246K, R209H, R209C, R209G) приводит к нарушению двигательной активности [8–11]. Вероятно, эпилепсия развивается в результате мутаций, приводящих дефициту белка Gαo1 (так называемые LOF-мутации, oт англ. loss-of-function), а двигательные нарушения, напротив, ассоциированы с мутациями, усиливающими функцию Gαo1 (GOF-мутации, oт англ. gain-of-function) [12]. Примечательно, что наиболее часто встречающийся вариант GNAO1 c.607 G>A (p.G203R) приводит к развитию двойного фенотипа, т.е. у пациентов развиваются как эпилепсия, так и двигательные нарушения [13]. Фенотипическую гетерогенность необходимо принимать во внимание при подборе схемы терапии и применять персонализированный подход с учетом конкретного патогенного варианта GNAO1 и молекулярного механизма заболевания.
Патогенез GNAO1-энцефалопатии и ее отдельных вариантов необходимо детально изучать на животных и клеточных моделях. Однако создание персонализированных животных моделей в случае таких ультраредких заболеваний, как GNAO1-энцефалопатия (около 200 случаев в мире по данным https://gnao1.org/), является трудозатратным, а материал для получения пациент-специфических нейронов не всегда доступен. Альтернативой клеточной модели GNAO1-энцефалопатии могут служить нейроны здоровых мышей с доставкой в них трансгенов с помощью рекомбинантных аденоассоциированных вирусов (ААВ) (рис. 1). ААВ представляют собой эффективный инструмент для доставки генов в нейрональные клетки [14, 15]. Разнообразие доступных природных, рекомбинантных и синтетических серотипов позволяет трансдуцировать разные субпопуляции нейронов [15–17]. Для создания модели GNAO1-энцефалопатии с LOF-мутациями можно доставлять в нейроны мыши короткие шпилечные РНК (shРНК). Специфичные shРНК будут подавлять экспрессию эндогенного Gnao1 по механизму РНК-интерференции, воспроизводя дефицит нормального белка Gαo1. Патологию GNAO1-энцефалопатии с GOF- или доминантно-негативными мутациями (нарушающими функцию нормального белка) можно смоделировать, доставляя с помощью нейротропных ААВ белоккодирующую последовательность мутантного гена.
Рис. 1.
Общая схема применения ААВ-опосредованной доставки трансгенов в первичную культуру нейронов мыши для моделирования и тестирования препаратов генной терапии для GNAO1-энцефалопатии.
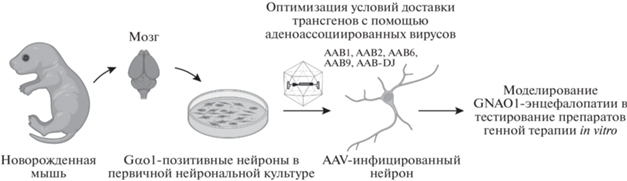
Рекомбинантные ААВ часто используются в качестве векторов при разработке генотерапевтических препаратов [18]. Высокая способность к трансдукции нейронов, низкая токсичность и иммуногенность позволяют использовать рекомбинантные ААВ в генной терапии заболеваний центральной нервной системы [19]. Генная терапия потенциально применима для коррекции GNAO1-энцефалопатии. Так, пациентам с LOF-мутациями подходит заместительная генная терапия с ААВ-опосредованной доставкой функциональной копии гена GNAO1 для восполнения дефицита белка Gαo1. В случае доминантно-негативных и GOF-мутаций можно применить аллель-селективное подавление экспрессии мутантного аллеля или подавление обоих аллелей в комбинации с доставкой нормального гена [20]. Кроме того, ААВ могут обеспечить доставку современных инструментов для редактирования нуклеиновых кислот независимо от типа мутации [21]. Первичное тестирование ААВ-векторов можно проводить на первичной культуре нейронов мыши (рис. 1).
В настоящей работе мы оптимизировали ААВ-опосредованную доставку трансгенов в нейроны мыши in vitro для моделирования GNAO1-энцефалопатии и скрининга препаратов для генной терапии. На первом этапе мы получили из мозга мышей первичную культуру Gαo1-экспрессирующих нейронов. Далее охарактеризовали ААВ пяти нейротропных серотипов (1, 2, 6, 9, DJ) с репортерным геном и показали, что ААВ-DJ обладает наибольшим тропизмом к Gαo1-положительным нейронам при отсутствии выраженной цитотоксичности. Кроме того, нами показано, что ААВ-DJ обеспечивает эффективную доставку трансгенного GNAO1 дикого типа и с мутацией G203R, а также shРНК для супрессии эндогенного Gnao1 в нейрональной культуре мыши.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Животные. Мыши линии CD-1 получены из разведения питомника “Столбовая”, Филиала Научного центра биомедицинских технологий ФМБА. Экспериментальные протоколы одобрены этической комиссией Института биологии гена РАН.
Пациенты. От родителей пациента с вариантом GNAO1 c.607 G> A получено письменное информированное согласие на проведение биопсии и участие в научных исследованиях.
Клонирование плазмидных конструкций. pAAV-GNAO1-wt-myc и pAAV-GNAO1-G203R-flag. Биоптат кожи пациента с GNAO1-энцефалопатией, вызванной гетерозиготным вариантом GNAO1 c.607 G>A, предоставлен НИКИ педиатрии им. Ю.Е. Вельтищева. Фибробласты кожи пациента использовали для амплификации белоккодирующей последовательности гена GNAO1 (CCDS10756.1) дикого типа и с мутацией c.607 G>A (G203R). Ампликоны (1065 п.н.) получали в ПЦР с парой праймеров (5'-ATGGGATGTACTCTGAGCGCA-3' и 5'-TCAGTACAAGCCGCAGCCC-3') и клонировали в коммерческий вектор pEGFP-C1 (“Clontech”, США) вместо EGFP безлигазным методом. Для этого вектор линеаризовали в ПЦР с праймерами 5'-GGCTGCGGCTTGTACTGATCCGGACTCAGATCTCGAGCTCAA-3' и 5'-GCGCTCAGAGTACATCCCATGGTGGCGACCGGTAGCG-3', сборку проводили на основе перекрывания концевых последовательностей ампликонов с использованием Т4 ДНК-полимеразы (“SibEnzyme”, Россия, E339). Полученные плазмиды обозначены pGNAO1 и pGNAO1-G203R.
На следующем этапе CDS GNAO1 и GNAO1-G203R встроили в вектор pAAV-eGFP вместо eGFP с добавлением на C-конец эпитопов myc и flag соответственно (pAAV-GNAO1-wt-myc и pAAV-GNAO1-G203R-flag). С этой целью GNAO1-wt-myc синтезировали на матрице pGNAO1 c использованием пары праймеров GNAO1_F (5'-GAAGTCACCATGGGATGTACTCTGAGCGCAGAGGAG-3') и Myc_R (5'-GATCCTCTTCTGAGATGAGTTTTTGTTCGTACAAGCCGCAGCCCCGGA-3'), вектор pAAV-eGFP готовили для вставки путем амплификации с праймерами GNAO1_R (5'-GTACATCCCATGGTGACTTCTTT-TTTGCTTTAGCAGGCTCTTTC-3') и Myc_F (5'-AAACTCATCTCAGAAGAGGATCTGTAATAATAACCGGGCAGGCCATGTCT-3'). Вставки GNAO1-G203R-flag синтезировали на матрице pGNAO1-G203R c праймерами GNAO1_F и Flag_R (5'-CTTGTCGTCATCGTCTTTGTAGTCGTACAAGCCGCAGCCCCGGA-3'), вектор готовили с праймерами GNAO1_R и Flag_F (5'-CAAAGACGATGACGACAAGTAATAATAACCGGGCAGGCCATGTCT-3'). Сборку проводили методом безлигазного клонирования, как описано выше.
Конструкции с shРНК. shРНК для подавления эндогенного транскрипта Gnao1 (NM_010308.3) подобраны с помощью интернет-портала Genetic Perturbation Platform (https://portals.broadinstitute.org/gpp/public/seq/search). Из 13 предсказанных shРНК выбрали три перспективные c высоким рейтингом (intrinsic score) для таргетных последовательностей: TRCN0000036487 (5'-CGCTCACCCAACAAAGAAATA-3'), TRCN0000097757 (5'-CGCTCACCCAACAAAGAAATT-3'); TRCN0000097756 (5'-CGAATAATATCCAGGTGGTAT-3'), далее shGnao1/1, shGnao1/2 и shGnao1/3 соответственно. Плазмида pLKO.1-shGnao1/1 приобретена в составе набора TRC Lentiviral Human Gnao1 shRNA (“Dharmacon”, США, RHS4533-EG2775). shGnao1/2, shGnao1/3, а также shEGFP (таргетная последовательность 5'-TACAACAGCCACAACGTCTAT-3') были клонированы в вектор pLKO.1 (Addgene plasmid #10878) [22], любезно предоставленный David Root, по протоколу сайта Addgene [23] c изменениями. Вставки клонировали с использованием пары одноцепочечных олигонуклеотидов (“Евроген”, Россия) c таргетными последовательностями. Пару олигонуклеотидов отжигали в буфере NEBuffer 2 (B7002S) для получения дуплекса с липкими концами для AgeI и EcoRI и фосфорилировали Т4-полинуклеотидкиназой (“Sibenzyme”). Вектор линеаризовали и одновременно дефосфорилировали, обрабатывая рестриктазами EcoRI и AgeI (“SibEnzyme”) и щелочной фосфатазой FastAP (“ThermoFisher,” CША) в буфере SE Orange (“SibEnzyme”) в течение 2 ч при 37°C, затем выделяли из 0.6%-ного агарозного геля. Лигирование проводили с помощью T4-ДНК-лигазы (“ThermoFisher Scientific”). Все полученные конструкции имели одинаковый дизайн shРНК (стебель 21 п.н. и шестинуклеотидная петля с сайтом рестрикции XhoI) и отличались только таргетными последовательностями. Контрольный вектор pLKO.1-shSCR c нетаргетирующей shРНК любезно предоставлен David Sabatini (Addgene plasmid, #1864).
После функциональной проверки экспрессионные кассеты из pLKO.1-shGnao1/1 и pLKO.1-shSCR, содержащие U6-промотор, shРНК и терминатор, клонировали в вектор pAAV-eGFP между 5'-ITR и CMV-промотором. Для этого экспрессионную кассету и вектор pAAV-eGFP амплифицировали с помощью пар праймеров (соответственно 5'-GGAGTTCCGGAACGTCTGCCCCCTTCACCGAGG-3', 5'-CGTTGGCTGCGAGTGATGACGAGCCATTTGTCTCGAGGTCGAGAATTCAAAA-3' и 5'-CACTCGCAGCCAACGACGCGGGCAGAGTTCCGCGTTACATAACTTACGGT-3', 5'-CAGACGTTCCGGAACTCCATA-TATGGGCTATGAACTAATG-3'). Клонирование проводили с использованием набора NEBuilder HiFi DNA Assembly (“NEB”, США, E5520S). Полученные векторы названы pAAV-shGnao1/1-eGFP и pAAV-shSCR-eGFP.
pMusGNAO1. Плазмиду с кодирующей областью Gnao1 мыши (CCDS22532.1) создавали по такой же технологии, как и pGNAO1. CDS Gnao1 амплифицировали из образцов мозга мыши с праймерами 5'-CGCTACCGGTCGCCACCATGGGATGTACGCTGAGCGC-3׳ и 5'-TCAGTTATCTAGATCCGGTGGATTCAGTACAAGCCGCAGCCC-3', вектор готовили с помощью праймеров 5'-ATCCACCGGATCTAGATAACTGATCATA-ATCA-3' и 5'-GGTGGCGACCGGTAGCG-3'.
Нуклеотидную последовательность векторов подтверждали секвенированием по Сэнгеру.
Проверка плазмидных конструкций в HEK293T. Сверхэкспрессию трансгенов и скрининг shРНК для подавления Gnao1 проводили в культуре клеток HEK293T (ATCC) путем трансфекции плазмид. Клетки культивировали в среде DMEM (“Gibco”, 11995) с добавлением 10% эмбриональной сыворотки крупного скота (“BioSera”, Франция, FB-1001/500) и пенициллина-стрептомицина (“ПанЭко”, Россия, А063). В день трансфекции клетки снимали с подложки 0.05%-ным раствором трипсина, осаждали, ресуспендировали в среде DMEM и рассевали в 24-луночный планшет в количестве 350 000 клеток на лунку. Плазмидную ДНК (250 нг), содержащую 2.5 нг pAAV-GNAO1 или смесь плазмид (2.5 нг pMusGnao1, 2.5 нг pEGFP-C1, 100 нг pLKO.1-shРНК), добавляли к раствору линейного полиэтиленимина (ЛПЭИ) (“Polysciences”, США, 23966-1) в среде DMEM в массовом соотношении 1 : 4 (ДНК : : ЛПЭИ). Трансфекционную смесь инкубировали в течение 20 мин и добавляли к клеткам. Через 72 ч после трансфекции клетки собирали и методом вестерн-блотинга определяли в них содержание белка Gαo1.
Наработка и очистка вирусов. Рекомбинантные ААВ получали с помощью системы AAV Helper-Free System (“Agilent”, США) в прикрепленной культуре HEK293T (ATCC) по ранее описанному протоколу [24]. Вирусы разных серотипов нарабатывали с использованием плазмидных конструкций, кодирующих белки Rep и Cap, – pAAV-RC1 (“Cell Biolabs”, США), pAAV-RC2 (“Agilent”), pAAV-RC6 (“Cell Biolabs”), pAAV-RC9 (“Penn Vector Core”) и pAAV-DJ (“Cell Biolabs”). В качестве векторной плазмиды использовали pAAV-eGFP, а также pAAV-GNAO1-wt-myc, pAAV-GNAO1-G203R-flag, pAAV-shSCR-eGFP или pAAV-shGnao1/1-eGFP. Вирусные частицы ААВ очищали с помощью универсального для всех серотипов метода, основанного на центрифугировании в градиенте плотности йодиксанола [24, 25]. Методом количественной ПЦР контролировали титр вирусных частиц на стадиях продукции, очистки и конечного концентрированного препарата [24]. Отсутствие белковых примесей подтверждали окрашиванием Кумасси в полиакриламидном геле согласно стандартному протоколу (Coomassie Brilliant blue, G250, “Helicon”, Россия).
Первичная нейрональная культура. Первичную нейрональную культуру получали из цельного головного мозга новорожденных мышей линии CD-1 согласно протоколу [26] с модификациями. Извлеченный головной мозг промывали в охлажденной среде DMEM (“Gibco”), содержащей 15 мM HEPES, и измельчали скальпелем до однородной массы. Измельченную ткань обрабатывали 0.25% раствором трипсин-EDTA (“ПанЭко”) с добавлением 10 нг/мкл ДНКазы I (“PanReac Applichem”, Испания) при 37oC в течение 20 мин. Затем добавляли ингибитор трипсина (“ПанЭко”, П071) и центрифугировали суспензию при 1000 g в течение 7 мин. Супернатант удаляли и ресуспендировали ткань в 10 мл HEPES-DMEM с последующим центрифугированием и удалением супернатанта. Промывку повторяли 2 раза, после чего клетки пропускали через клеточное сито с диаметром пор 70 мкм. Затем клетки центрифугировали при 1000 g в течение 5 мин и заменяли раствор на полную среду Neurobasal-A (“Gibco”), содержащую добавку B-27 (“Gibco”) и 2 мM аланил-глутамин (“ПанЭко”). Для проведения вестерн-блотинга клетки высевали в 12-луночные планшеты, для анализа цитотоксичности – в 96-луночные. Для анализа методом иммунофлуоресценции клетки высевали в 6-луночные культуральные планшеты на покровные стекла размером 24 × 24 мм (Menzel coverslips). Поверхность планшетов и стекол предварительно покрывали поли-D-лизином (“Sigma-Aldrich”, США). Клетки высевали при расчетной плотности ~100 000 клеток/см2 и инкубировали при 37oC в атмосфере 5% СО2. Изображения клеток получали на инвертированном микроскопе с фазовым контрастом Nikon Ti-E.
Вирусная трансдукция. Первичную нейрональную культуру заражали ААВ на 7-й день культивирования. К нейронам в полной нейробазальной среде добавляли очищенный препарат вируса из расчета 1 × 105 геномных копий (ГК) на клетку. После заражения клетки инкубировали в течение еще 7 дней до анализа образцов.
Цитотоксичность. Выживаемость клеток после трансдукции препаратами ААВ оценивали с использованием реагента PrestoBlue™ Cell Viability Reagent (“Invitrogen”, США, A13261). В качестве контроля использовали незараженные клетки. Анализ проводили в 96-луночном культуральном планшете согласно инструкции производителя. После добавления реагента клетки инкубировали при 37oC в течение 1.5 ч. Определяли поглощение при длине волны 570 нм на планшетном ридере Clariostar Plus (“BMG Labtech”, Германия). Полученные значения нормировали по поглощению при 600 нм. Данные обрабатывали с использованием программного обеспечении MARS (“BMG Labtech”).
Вестерн-блотинг. Белковый экстракт клеток HEK293T готовили в буфере RLB (“Promega”, США, E397A) согласно инструкциям производителя. Первичные нейроны мыши лизировали в охлажденном буфере (50 мМ Трис-HCl pH 7.4, 150 мМ NaCl, 5 мМ EDТА, 5 мМ EGТА, 1% Тритона Х-100, 0.1% натрия дезоксихолата и 0.1% додецилсульфата натрия) [27] и осветляли центрифугированием при 17000 g в течение 5 мин при 4°С. Концентрацию белка в образцах измеряли с помощью реагента Quick Start Bradford 1× Dye Reagent (“Bio-Rad”, США, 5000205) на планшетном ридере Clariostar Plus (“BMG Labtech”). Белковый экстракт (5–10 мкг) в буфере Лэммли подвергали электрофорезу в денатурирующих условиях в 12%-ном полиакриламидном геле. Белки переносили на нитроцеллюлозную мембрану, используя набор Trans-Blot Turbo RTA Transfer kit (“Bio-Rad”, 170-4270), на приборе Transfer-blot Turbo (“Bio-Rad”). Качество нанесения и эффективность переноса белков оценивали, окрашивая мембрану Ponceau S. Мембрану инкубировали с антителами кролика к Gαo1 мыши и человека (“Invitrogen”, PA5-30044; 1 : 5000), GFP (“Sigma”, США, G1544-100UG; 1 : 4500), βIII-тубулину (“Аbcam”, Великобритания, ab18207; 1 : 1000), антителами мыши к myc-эпитопу (“ThermoFisher Scientific”, MA1-980; 1 : 2000), GAPDH (G8795, “Sigma-Aldrich”; 1 : 20 000) или антителами козы к flag-эпитопу (“Abcam”, ab1257; 1 : 5000). Затем мембрану инкубировали с вторичными антителами, меченными пероксидазой хрена, против иммуноглобулинов кролика (“Bio-Rad”, Cat# 170-6515, 1 : : 3000), мыши (“Bio-Rad”, Cat# 170-6516; 1 : 3000) или козы (“Invitrogen”, #81-1620; 1 : 3000). Сигнал детектировали с помощью набора Clarity Western ECL substrate (“BioRad”, #170-5060) на приборе iBright 1500 (“ThermoFisher”). Анализ изображений проводили в программе Fiji [28].
Количественная ПЦР. Суммарную РНК выделяли из инфицированных нейронов с помощью набора ReliaPrep™ RNA Cell Miniprep System (“Promega”). Концентрацию РНК измеряли на спектрофотометре NanoDrop™ 8000 и обрабатывали ДНКазой I (“NEB”, M0303). Реакцию обратной транскрипции проводили с помощью набора MMLV RT kit (SK021, “Евроген”) c использованием смеси cлучайного (“Eвроген”, SB002) и олиго(dT)15 (“Eвроген”, SB001) праймеров в концентрации 1 мкM каждый. Полученную кДНК разводили в 5 раз в воде без нуклеаз (“Евроген”, PB007). Экспериментальный образец (5 мкл) добавляли к 20 мкл ПЦР мастер-микса (“Синтол”, Россия, M-428), содержащего специфические (400 нM) праймеры и зонды (200 нM), 2% DMSO и дополнительно 3 мM MgCl2. Gnao1 мыши детектировали с использованием праймеров 5'-TACTACCTGGACAGCCTGGA-3', 5'-GGATCCACTTCTTGCGTTCA-3' и зонда 5'-ROX-CGTCGGGGGCCAGC-BHQ2-3'. Gаpdh детектировали с использованием праймеров 5'-GGAGAAACCTGCCAAGTATGA-3', 5'-TCCTCAGTGTAGCCCAAGA-3' и зонда 5'-VIC-TCAAGAAGGTGGTGAAGCAGGCAT-BHQ1-3'. ПЦР в реальном времени проводили на приборе CFX96 Real-Time PCR Detection System (“Bio-Rad Laboratories”). Амплификацию проводили по следующей программе: денатурация при 94°С, 3 мин; далее 40 циклов – 94°С, 15 с и 60°С, 40 с; считывание флуоресцентного сигнала проводили в каналах VIC и ROX. Количество копий транскриптов Gnao1 определяли по стандартной кривой ДНК-стандарта (плазмида pMusGnao1 с известной концентрацией) и нормировали по уровню Gapdh.
Иммунофлуоресценция. Клетки фиксировали на покровных стеклах в течение 30 мин при комнатной температуре в 4%-ном параформальдегиде (“Panreac AppliChem”, 141451.1211) с 2% сахарозы D(+) (“Panreac AppliChem”, 57-50-1) в фосфатно-солевом буфере (“ПанЭко”, В-60201) с последующей пермеабилизацией в 0.2%-ном Тритон X-100 (“Amresco”, США, Am-O694-1.0) в течение 10 мин при комнатной температуре. Для блокирования неспецифического связывания антител клетки инкубировали в течение 1 ч при комнатной температуре в фосфатно-солевом буфере с 3% БСА (“ПанЭко”, РМ-Т1725.50) и 0.2% Тритон X-100.
Первичную культуру нейронов окрашивали поликлональными антителами кролика к βIII-тубулину (“Аbcam” ab18207 1 : 300), GFAP (“Abcam” ab7260; 1 : 300) или Gαo1 (“Invitrogen”, PA5-30044; 1 : : 300). В качестве вторичных антител использовали антитела козы против иммуноглобулинов кролика, меченные красителем Alexa Fluor 633 (“Invitrogen”, A 21072; 1 : 1000). Ядра клеток окрашивали красителем Hoechst 33342 (“ThermoFisher Scientific”, H3570, 1 : 1000). Стекла с окрашенными клетками покрывали средой для заключения (ProLong™ Glass Antifade Mountant, “ThermoFisher Scientific”, P3698). Изображения получали на инвертированном микроскопе Leica DMI6000 одновременно по двум каналам (Alexa 405, Alexa 633).
Тропизм ААВ анализировали с использованием поликлональных антител кролика к Gαo1 (“Invitrogen”, PA5-30044; 1 : 300) и антител козы против иммуноглобулинов кролика, меченных красителем Alexa Fluor 633 (“Invitrogen”, A 21072; 1 : 1000). Ядра клеток контрастировали красителем Hoechst 33342 (“Thermofisher Scientific”, H3570, 1 : 1000). Стекла помещали на предметные стекла в среду для заключения. Изображения получали на микроскопе THUNDER Imaging Systems (“Leica”) одновременно по трем каналам (Alexa 405, Alexa 488, Alexa 633). Для каждого образца отснято по 144 поля зрения.
Анализ колокализации. Для оценки тропизма ААВ разных серотипов определяли колокализацию трансгенного GFP с эндогенным Gαo1 мыши (mGαo1). Полученные изображения анализировали с помощью программного обеспечения CellProfiler 4.2.1 [29]. Для каждого изображения в трех каналах выполнена коррекция освещенности “CorrectIllumination” и удаление шума путем наложения фильтра Гаусса “GaussianFilter”. На первом этапе идентифицировали объекты в каждом из каналов. Синий канал – окраска ядер, зеленый канал – флуоресценция GFP, красный канал – окраска специфичными антителами к Gαo1. Путем сравнения слоев в синем и красном каналах определяли Gαo1-позитивные клетки. В дальнейшем определяли позитивные клетки по двум каналам, сравнивая слои с Gαo1- и GFP-позитивными клетками. Процент колокализации оценивали как отношение клеток, позитивных по двум каналам, к общему количеству Gαo1-позитивных клеток. Проанализировано не менее 1800 Gαo1-позитивных клеток каждого образца.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Gαo1-богатая нейрональная культура
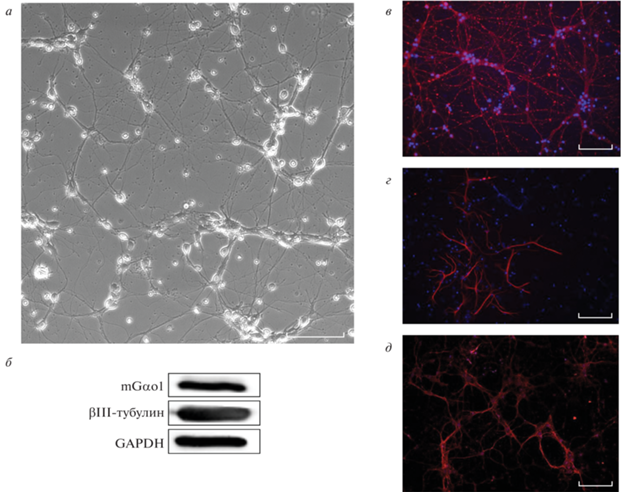
На первом этапе исследований перед нами стояла задача получения первичной нейрональной культуры мыши, богатой Gαo1-позитивными клетками. Согласно опубликованным данным, ген Gnao1 экспрессируется во всех отделах головного мозга грызунов, в том числе в коре больших полушарий, мозжечке, базальных ганглиях, обонятельной луковице [3, 30–32]. Для получения всего многообразия нейрональных клеток, в которых осуществляется Gαo1-опосредованная передача сигнала, мы выделяли нейроны из цельного мозга новорожденных мышей, адаптировав ранее описанный протокол [26]. Качество нейронов оценивали через 7 дней после выделения в культуру по морфологии клеток в фазовом контрасте (рис. 2а), а также методом вестерн-блотинга (рис. 2б) и иммунофлуоресценции (рис. 2в–д). Окрашивание антителами к маркеру βIII-тубулину показало, что культура состоит преимущественно из нейронов (рис. 2б, в), а антитела к маркеру GFAP (глиальный кислый фибриллярный белок) выявили лишь незначительное присутствие астроцитов (рис. 2г). В полученной первичной нейрональной культуре нами обнаружен существенный синтез белка Gαo1 мыши (mGαo1), который детектировали с помощью специфичных антител методами вестерн-блотинга и иммунофлуоресценции (рис. 2б, д). Таким образом, выделение нейронов из цельного мозга новорожденных мышей позволило получить культуру нейронов, богатую Gαo1-позитивными клетками. Эту культуру использовали в дальнейших экспериментах для вирусной доставки.
Рис. 2.
Оценка качества клеток и экспрессии Gαo1 в первичной нейрональной культуре, полученной из цельного мозга мышей. а – Фазовый контраст; б – анализ экспрессии mGαo1 и маркера нейронов βIII-тубулина методом вестерн-блотинга; контроль нагрузки – GAPDH. Иммунофлуоресцентное окрашивание культуры нейронов мыши антителами к нейрональному маркеру βIII-тубулину (в), глиальному маркеру GFAP (г) и mGαo1 (д). Окраска специфичными антителами показана красным, ядра контрастированы Hoechst 33342 (синий). На в и д показаны репрезентативные поля, на г показано поле с максимальным количеством GFAP-положительных клеток. Длина масштабного отрезка 100 мкм.
Тропизм серотипов ААВ к Gαo1-позитивным клеткам in vitro
На следующем этапе мы подобрали условия эффективной доставки трансгенов в Gαo1-позитивные нейроны мыши in vitro. С этой целью использовали контрольный вирусный вектор с репортерным геном eGFP под конститутивным CMV-промотором (рис. 3a). Для упаковки вектора pAAV-eGFP использовали капсиды серотипов 1, 2, 6, 9 и DJ, охарактеризованных ранее как нейротропные в исследованиях in vitro и in vivo [15, 16]. Эффективность продукции вирусных частиц в клетках, а также потери в процессе очистки, контролировали методом кПЦР (рис. 3б). Средняя продукция вирусных частиц разных серотипов в неочищенном клеточном лизате различалась до 8 раз и составила 3.8 × 1010 (ААВ1), 1.8 × 1010 (ААВ2), 3.7 × 1010 (ААВ6), 1.6 × 1011 (ААВ9) и 1.1 × 1011 (ААВ-DJ) геномных копий (ГК) на см2 площади культуральной поверхности. Наиболее высокой была эффективность продукции серотипов ААВ9 и DJ, а самой низкой – серотипа 2, что согласуется с опубликованными данными [33]. Средний выход вирусных частиц разных серотипов после всех стадий очистки варьировал в диапазоне 15–27%. Наибольшая эффективность очистки наблюдалась для серотипов 6 и 9 (27 и 22% соответственно, табл. 1). Наибольшими были потери вирусного материала (до 85%) при очистке ААВ1 и ААВ2. Все препараты ААВ концентрировали до титра ~2 × 1013 ГК/мл.
Рис. 3.
Получение и тестирование препаратов вируса различных серотипов, кодирующих белок-репортер. a ‒ Схема контрольного вирусного вектора, кодирующего репортерный белок eGFP. б ‒ Количество вирусных частиц разных серотипов в клеточном лизате и после стадий очистки методом кПЦР. Данные представлены в виде числа геномных копий (ГК)/см2 культуральной поверхности. n – Количество независимых экспериментов. в ‒ Вестерн-блотинг с лизатами первичной нейрональной культуры, трансдуцированной разными серотипами ААВ при множественности заражения 105 ГК/клетку. Показан уровень экспрессии eGFP через 7 дней после трансдукции. г ‒ Количественный обсчет результатов вестерн-блотинга. Показан относительный уровень экспрессии GFP в нейронах, трансдуцированных разными серотипами ААВ. Уровень экспрессии GFP нормирован по уровню экспрессии βIII-тубулина. д ‒ Анализ цитотоксичности разных серотипов в первичной нейрональной культуре мыши. Показан уровень жизнеспособности трансдуцированных клеток в процентах от нетрансдуцированной культуры.
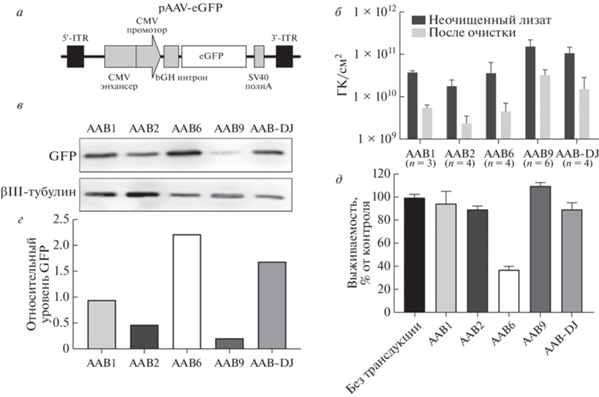
Таблица 1.
Эффективность очистки вирусных частиц разных серотипов, кодирующих репортерный белок eGFP
| Серотип ААВ | Число независимых экспериментов, n | Средний выход*, % | Стандартное отклонение, % |
|---|---|---|---|
| ААВ1 | 3 | 15 | 4 |
| ААВ2 | 4 | 15 | 8 |
| ААВ6 | 4 | 27 | 16 |
| ААВ9 | 6 | 22 | 2 |
| ААВ-DJ | 4 | 19 | 15 |
Первичную культуру нейронов мыши заражали вирусными частицами ААВ-eGFP серотипов 1, 2, 6, 9 и DJ при множественности инфекции 105 ГК на клетку. Общую эффективность заражения первичной нейрональной культуры разными серотипами ААВ оценивали по уровню экспрессии трансгена eGFP методом вестерн-блотинга (рис. 3в, г). Установлено, что относительный уровень экспрессии GFP через 7 дней после трансдукции был выше в культурах, трансдуцированных серотипами 6 и DJ. Наименьшим уровень экспрессии трансгена был в культуре, трансдуцированной ААВ9. Анализ выживаемости первичных нейронов не выявил значительных различий между культурами, трансдуцированными серотипами ААВ 1, 2, 9, DJ, и нетрансдуцированными клетками (рис. 3д). Однако обнаружено, что вирусные частицы с капсидом серотипа 6 проявляли существенную цитотоксичность в отношении первичной нейрональной культуры.
Чтобы выявить серотип ААВ с наибольшим тропизмом к субпопуляции Gαo1-позитивных нейронов, мы изучили колокализацию экспрессии GFP и mGαo1 (рис. 4). С этой целью определили процент Gαo1-позитивных клеток, зараженных ААВ-eGFP (рис. 4а). Все протестированные серотипы ААВ трансдуцировали Gαo1-позитивные клетки, однако с разной эффективностью. Наибольшим тропизмом к Gαo1-позитивным клеткам обладали серотипы ААВ6 и ААВ-DJ: 59 и 66% колокализации соответственно. Серотипы ААВ1, ААВ2 и ААВ9 имели более низкий тропизм ‒ колокализация с таргетными клетками составила только 19, 25 и 8% соответственно. Пример колокализации серотипа ААВ-DJ представлен на рис. 4б.
Рис. 4.
Анализ тропизма рекомбинантных аденоассоциированных вирусов разных серотипов на первичной нейрональной культуре. а ‒ Оценка количества трансдуцированных mGαo1-позитивных нейронов мыши. Процент колокализации (черный сегмент) оценивали как отношение mGαo1-позитивных клеток, экспрессирующих eGFP, к общему количеству mGαo1-позитивных клеток. Количество незараженных mGαo1-позитивных нейронов показано серым. В скобках указано количество клеток, участвующих в подсчете. б ‒ Пример колокализации сигнала от клеток, трансдуцированных ААВ-DJ, экспрессирующих GFP (зеленый), с сигналом от mGαo1-позитивных клеток, окрашенных специфичными антителами (красный). Ядра контрастированы Hoechst 33342 (cиний).
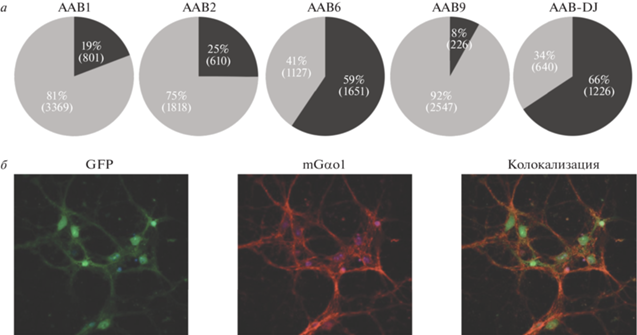
Таким образом, по результатам скрининга пяти нейротропных серотипов ААВ оптимальным для заражения mGαo1-позитивных нейронов мыши in vitro оказался ААВ-DJ. Серотип DJ при множественности 105 ГК на клетку обеспечивал эффективную доставку репортерного трансгена в целевые клетки и обладал низкой цитотоксичностью. Этот серотип использовали в дальнейшем для тестирования потенциальных генотерапевтических подходов и моделирования GNAO1-энцефалопатии.
Экспрессия Gαo1 человека и супрессия эндогенного Gαo1
На заключительном этапе мы проверили эффективность доставки с помощью ААВ-DJ генетических конструкций разных конфигураций, которые потенциально могут быть применены в генной терапии и моделировании GNAO1-энцефалопатии в нейронах мыши in vitro. Разработаны две группы экспрессионных ААВ-векторов (рис. 5а, б).
Рис. 5.
Скрининг плазмидных конструкций для сверхэкспрессии трансгенного hGαo1 или подавления mGαo1 мыши. а ‒ Схема вирусных векторов, кодирующих нормальную или мутантную форму белка hGαo1, меченного myc- или flag-эпитопом соответственно. б ‒ Схема вирусного вектора, кодирующего shРНК и GFP в качестве репортерного гена. в ‒ Результат вестерн-блотинга, показывающий экспрессию нормальной или мутантной формы hGαo1 в культуре HEK293T, трансфицированной разработанными ААВ-векторами. г ‒ Результат скрининга shРНК (shGnao1/1, shGnao1/2, shGnao1/3), таргетирующих транскрипт гена Gnao1. Конструкции pLKO.1-shРНК транзиентно экспрессировали в культуре клеток HEK293T совместно с плазмидой pGnao1, кодирующей mGαo1 мыши. Подавление анализировали методом вестерн-блотинга с антителами, специфичными к Gαo1, GFP. Контроль нанесения – GAPDH.
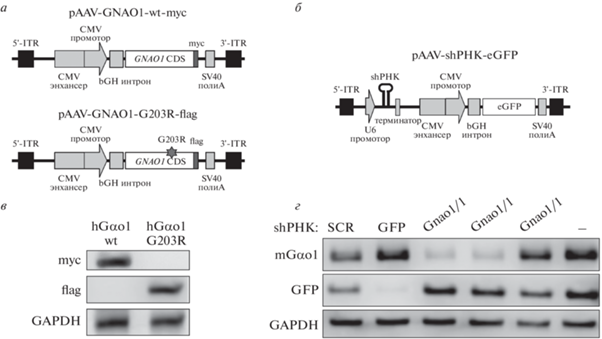
Первая группа ААВ-векторов несет в качестве трансгена кодирующие белок последовательности GNAO1. Вектор pAAV-GNAO1-wt-myc экспрессирует Gαo1 человека (hGαo1) дикого типа с myc-эпитопом (рис. 5а). Эту конструкцию можно использовать для тестирования заместительной генной терапии, применимой к пациентам с LOF-мутациями в гене GNAO1 [12]. С помощью вектора pAAV-GNAO1-G203R-flag можно доставлять мутантную форму трансгенного белка hGαo1-G203R для выяснения механизма патогенеза наиболее часто встречающегося варианта. Экспрессию обеих форм hGαo1 (нормальной и мутантной) ААВ-векторами подтвердили в контексте плазмидной трансфекции в HEK293T с помощью антител, специфичных к myc- и flag-эпитопам соответственно (рис. 5в).
Вторая группа ААВ-векторов кодирует shРНК (рис. 5б). Специфичные к Gnao1 shРНК подавляют экспрессию эндогенного mGαo1 на посттранскрипционном уровне посредством РНК-интерференции [20]. Эти векторы можно использовать для моделирования GNAO1-энцефалопатии, вызванной LOF-мутациями, которые приводят к дефициту белка. Кроме того, векторы с shРНК, специфичные к GNAO1 человека, можно использовать в генотерапевтических подходах, основанных на супрессии генов и эффективных при GOF-мутациях и мутациях с доминантно-негативным проявлением.
Чтобы подобрать shРНК для эффективного подавления mGαo1, мы протестировали три плазмидные конструкции с таргетирующими последовательностями к Gnao1 (shGnao1/1, shGnao1/2, shGnao1/3) в HEK293T cо сверхэкспрессией Gαo1 мыши (рис. 5г). В качестве негативных контролей использовали shРНК с нетаргетирующей последовательностью шпильки (shSCR) и последовательностью для подавления GFP (shGFP). Конструкции shGnao1/1 и shGnao1/2 эффективно подавляли продукцию mGαo1. Последовательность shGnao1/1 отобрали для создания ААВ-вектора (pAAV-shGnao1/1-eGFP), учитывая способность подавлять Gαo1 как мыши, так и человека.
C помощью описанных экспрессионных векторов, несущих трансгенный GNAO1 или shРНК (рис. 5а, б), мы получили очищенные препараты ААВ в капсиде DJ. Продукцию и эффективность очистки вирусных частиц оценивали, как в случае вирусов с GFP-репортером. Продукция всех вирусов серотипа DJ в лизате клеток HEK293T была сходной: 8.0 × 1010 (GNAO1-wt-myc), 9.6 × 1010 (GNAO1-G203R-Flag), 7.3 × 1010 (shSCR) и 1.0 × 1011 (shGnao1/1) ГК/см2 культуральной поверхности (рис. 6а). Выход указанных вирусных препаратов после очистки составил 17, 14, 20 и 12% соответственно. Вирусные препараты сконцентрировали до 2‒3 × 1013 ГК/мл, отсутствие белковых примесей проверяли, анализируя вирусные частицы в полиакриламидном геле с окрашиванием Кумасси (рис. 6б).
Рис. 6.
Получение и тестирование вирусных препаратов для сверхэкспрессии трансгенного hGαo1 или подавления эндогенного Gnao1 в первичной культуре нейронов из мозга мышей. а ‒ Оценка количества целевых ААВ в клеточном лизате и после стадий очистки методом кПЦР. б ‒ Анализ чистоты вирусных препаратов в полиакриламидном геле в денатурирующих условиях. Окрашивание Кумасси: отмечены три белка вирусного капсида (VP1, VP2, VP3). в ‒ Вестерн-блот-анализ экспрессии нормальной или мутантной формы hGαo1 в первичных нейронах, трансдуцированных вирусами ААВ-DJ-GNAO1-wt-myc и ААВ-DJ-GNAO1-G203R-flag. г ‒ Анализ подавления экспрессии эндогенного Gnao1 на уровне транскрипта методом кПЦР после заражения ААВ-DJ-shРНК первичной нейрональной культуры. Показан уровень экспрессии (%) от уровня в культуре, трансдуцированной нетаргетирующей контрольной шпилькой (shSCR). д ‒ Вестерн-блот-анализ подавления экспрессии mGαo1 в трансдуцированной первичной нейрональной культуре мыши.
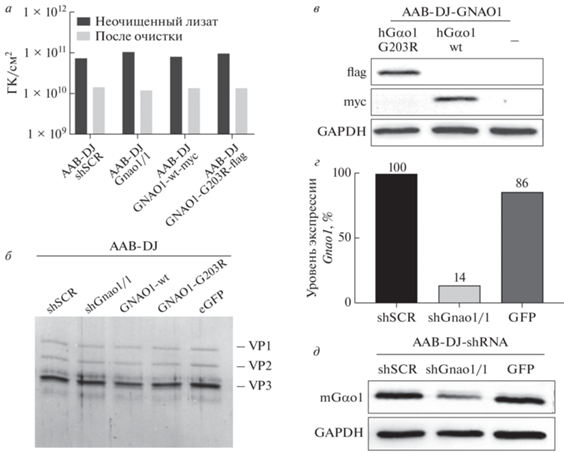
Вирусные препараты добавляли к mGαo1-богатой первичной культуре нейронов из цельного мозга мышей при множественности инфекции 105 ГК на клетку. Через 7 дней после инфекции анализировали уровень экспрессии трансгенного hGαo1 человека и эндогенного mGαo1 мыши. Как показано на рис. 6в, ААВ-DJ обеспечивает хорошо детектируемый уровень экспрессии трансгенов GNAO1-wt-myc и GNAO1-G203R-flag в нейронах на уровне белка. Используя ААВ-DJ для доставки shРНК, нам удалось добиться снижения экспрессии эндогенного Gnao1 в первичной нейрональной культуре на 85% на уровне РНК и вдвое на уровне белка mGαo1 (рис. 6г, д соответственно).
Таким образом, нами показано, что серотип ААВ-DJ подходит для эффективной доставки в первичные нейроны мыши трансгенов, обеспечивающих сверхэкспрессию Gαo1 человека, и shРНК для подавления эндогенного Gnao1. Подобные условия доставки могут быть использованы как для моделирования GNAO1-энцефалопатии, так и для тестирования подходов генной терапии в нейронах мыши in vitro.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Вирусная доставка генетических конструкций в клетки-мишени широко применяется в фундаментальных исследованиях [34], а также при разработке генотерапевтических препаратов [35]. Популярным вирусным вектором для доставки трансгенов в центральную нервную систему являются рекомбинантные ААВ [18].
В настоящей работе мы оптимизировали ААВ-опосредованную доставку трансгенов в нейрональной культуре, полученной из цельного мозга мышей, с целью проведения исследований по GNAO1-энцефалопатии (рис. 1). Нами проверены пять нейротропных серотипов (1, 2, 6, 9 и DJ) при множественности инфекции 105 вирусных геномов на клетку. ААВ6 и ААВ-DJ обеспечили самый высокий уровень экспрессии доставленного GFP-репортера (рис. 3в, г), а также самый высокий тропизм к Gαo1-позитивным нейронам (59 и 66% соответственно) (рис. 4). Установлено, что серотип ААВ6, в отличие от ААВ-DJ, проявляет существенную цитотоксичность в первичной нейрональной культуре мыши (рис. 3д).
Ранее были опубликованы результаты исследования, в котором сравнивали цитотоксичность и тропизм серотипов ААВ1, 2, 5, 6, 7, 8, 9 в первичной кортикальной культуре крысы [36]. С помощью иммунофлуоресцентного анализа показано, что серотипы ААВ2 и ААВ9 обеспечивают минимальную экспрессию GFP в трансдуцированной культуре, в то время как в клетках, трансдуцированных серотипами ААВ1 и ААВ6, наблюдается самая высокая интенсивность сигнала GFP. Эти данные согласуются с нашими результатами, полученными методом вестерн-блотинга (рис. 3г). Сообщается также о цитотоксичности ААВ6, хотя и менее выраженной, чем в наших экспериментах (рис. 3д). Мы предполагаем, что это связано с большей гетерогенностью нейрональной культуры, полученной из цельного мозга, чем кортикальной культуры. Royo и соавт. [37] также отметили повышенную токсичность ААВ6 в гиппокампальной культуре крысы в сравнении с серотипами 1, 2, 5, 7, 8 и 9. Изучив роль аминокислотной последовательности капсида ААВ6, установили, что внесение замены K531E в участок капсида, критичный для связывания гепарина, приводит к уменьшению цитотоксичности без снижения эффективности трансдукции нейронов [37]. Серотип ААВ-DJ обладает генно-инженерным капсидом, полученным сочетанием восьми природных серотипов [38]. В разных работах показана повышенная cпособность серотипа DJ трансдуцировать in vitro клеточные культуры, в том числе нейрональные [15]. Таким образом, исходя из опубликованных данных и результатов нашего исследования, мы делаем вывод о том, что ААВ-DJ является оптимальным серотипом для доставки трансгенов в Gαo1-богатую первичную нейрональную культуру, полученную из цельного мозга мышей.
С целью моделирования GNAO1-энцефалопатии, а также тестирования генотерапевтических конструкций, нами созданы ААВ-векторы для экспрессии hGαo человека и подавления эндогенного Gnao1 с помощью shРНК (рис. 5а, б). Показано, что серотип DJ обеспечивает эффективную доставку этих генетических конструкций в культуру mGαo1-экспрессирующих нейронов (рис. 6в‒д).
Нейроны мыши с ААВ-опосредованной экспрессией hGαo1-G203R могут служить клеточной моделью GNAO1-энцефалопатии с вариантом с.607 G>A (рис. 6в). Эта мутация наиболее часто встречается у пациентов с GNAO1-энцефалопатией [5]. По данным родительских организаций под наблюдением врачей в России находятся четыре таких пациента. При этом данные о влиянии мутации G203R на функциональную активность Gαo1 остаются противоречивыми [1, 12, 39]. Нейроны со сниженным с помощью shРНК уровнем эндогенного mGαo1 воспроизводят дефицит функционального белка у пациентов c LOF-мутациями (рис. 6г, д). На таких клеточных моделях можно изучать молекулярный механизм патологии GNAO1-энцефалопатии. В частности, можно исследовать общую функциональную активность ААВ-трансдуцированных нейронов (методом визуализации Са2+), возможные нарушения аденилатциклазного каскада, а также электрофизиологические характеристики, такие как сопряжение активности Gαo1-содержащих рецепторов с работой потенциалзависимых Са2+-каналов и активируемых G-белком GIRK-каналов. Подобные исследования помогут восполнить пробел в понимании молекулярных механизмов патологии [12], а также скорректировать схему терапии и разработать персонализированные подходы к терапии пациентов с GNAO1-энцефалопатией.
Полученные нами генетические конструкции могут стать основой для создания генотерапевтических препаратов. Так, конструкция pAAV-GNAO1-wt-myc потенциально может использоваться в заместительной терапии пациентов c LOF-мутациями и дефицитом нормального Gαo1 в тканях мозга (рис. 5а). ААВ-вектор с shGnao1/1 обладает таргетной последовательностью (TRCN0000036487), универсальной для супрессии Gαo1 как мыши, так и человека (рис. 5б). Следовательно, вектор pAAV-shGnao1/1 можно адаптировать для супрессии генного продукта с GOF-мутацией или доминантно-негативным проявлением. Аналогичная генотерапевтическая стратегия описана для других неврологических заболеваний [40–43]. В одном из вариантов этой стратегии (так называемая “Silence-and-replace” [20, 44]) shРНК подавляет экспрессию как мутантного, так и нормального аллеля, при этом параллельно доставляется функциональная копия гена в том же или в другом ААВ-векторе. Проверку экспрессии генотерапевтических конструкций для GNAO1-энцефалопатии можно проводить на первичных нейронах, используя ААВ-DJ в качестве инструмента для доставки.
Серотип ААВ-DJ может использоваться также в качестве средства доставки в новых подходах к генной терапии GNAO1-энцефалопатии, таких как CRISPR-Cas9, CRISPR-Cas13 и молекулярные редакторы оснований ДНК [21].
Таким образом, созданные в нашей работе генетические конструкции и оптимизированные протоколы с применением ААВ помогут продвинуться в понимании механизмов патогенеза GNAO1-энцефалопатии и могут лечь в основу генотерапевтических препаратов.
Мы благодарим за помощь в клонировании ряда плазмидных конструкций Усачёва Е.В. (Лаборатория трансляционной биомедицины НИЦЭМ им. Н.Ф. Гамалеи), Лосеву Е.М. и Лучкину Е.А. (OOO “Марлин Биотех”) за помощь в клонировании и первичную проверку плазмидных векторов, Веляева О.А. (OOO “Марлин Биотех”) за продукцию и очистку плазмидных векторов, Хаматову А.Ю. (OOO “Марлин Биотех”) за определение титра вирусных препаратов методом кПЦР.
Работа проведена с использованием оборудования ИБР им. Н.К. Кольцова РАН. Мы благодарим за предоставленное оборудование Центр высокоточного редактирования генома и генетических технологий для биомедицины (Институт биологии гена), развиваемый при поддержке Министерства науки и высшего образования Российской Федерации.
Все процедуры, выполненные в данной работе, соответствуют этическим стандартам институционального комитета по исследовательской этике и Хельсинкской декларации 1964 года и ее последующим изменениям или сопоставимым нормам этики. Экспериментальные протоколы одобрены этической комиссией Института биологии гена РАН (Москва, Россия). От родителей пациента с вариантом GNAO1 c.607 G>A получено письменное информированное согласие на проведение биопсии и участие в научных исследованиях.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Nakamura K., Kodera H., Akita T., Shiina M., Kato M., Hoshino H., Terashima H., Osaka H., Nakamura S., Tohyama J., Kumada T., Furukawa T., Iwata S., Shiihara T., Kubota M., Miyatake S., Koshimizu E., Nishiyama K., Nakashima M., Tsurusaki Y., Miyake N., Hayasaka K., Ogata K., Fukuda A., Matsumoto N., Saitsu H. (2013) De novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am. J. Hum. Genet. 93, 496–505.
EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, and Epi4K Consortium (2014) De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 95, 360–370.
Worley P.F., Baraban J.M., Van Dop C. (1986) Go, a guanine nucleotide-binding protein: immunohistochemical localization in rat brain resembles distribution of second messenger systems. Proc. Natl. Acad. Sci. USA. 83, 4561–4565.
Jiang M., Bajpayee N.S. (2009) Molecular mechanisms of go signaling. Neurosignals. 17, 23–41.
Feng H., Khalil S., Neubig R.R., Sidiropoulos C. (2018) A mechanistic review on GNAO1-associated movement disorder. Neurobiol. Dis. 116, 131–141.
Ananth A.L., Robichaux-Viehoever A., Kim Y.M., Hanson-Kahn A., Cox R., Enns G.M., Strober J., Willing M., Schlaggar B.L, Wu Y.W., Bernstein J.A. (2016) Clinical course of six children with GNAO1 mutations causing a severe and distinctive movement disorder. Pediatr. Neurol. 59, 81–84.
Danti F.R, Serena Galosi S., Romani M., Montomoli M., Carss K.J., Raymond F.L., Parrini E., Bianchini C., McShane T., Dale R.C., Mohammad S.S., Shah U., Mahant N., Ng J., McTague A., Samanta R., Vadlamani G., Valente E.M., Leuzzi V., Kurian M.A., Guerrini R. (2017) GNAO1 encephalopathy: broadening the phenotype and evaluating treatment and outcome. Neurol. Genet. 3, 143.
Menke L.A., Engelen M., Alders M., Odekerken V.J.J., Baas F., Cobben J.M. (2016) Recurrent GNAO1 mutations associated with developmental delay and a movement disorder. J. Child. Neurol. 31, 1598–1601.
Saitsu H., Fukai R., Ben-Zeev B., Sakai Y., Mimaki M., Okamoto N., Suzuki Y., Monden Y., Saito H., Tziperman B., Torio M., Akamine S., Takahashi N., Osaka H., Yamagata T., Nakamura K., Tsurusaki Y., Nakashima M., Miyake N., Shiina M., Ogata K., Matsumoto N. (2015) Phenotypic spectrum of GNAO1 variants: epileptic encephalopathy to involuntary movements with severe developmental delay. Eur. J. Hum. Genet. 24, 129–134.
Roddy D.W., Farrell C., Doolin K., Roman E., Tozzi L., Frodl T., O’Keane V., O’Hanlon E. (2019) The hippocampus in depression: more than the sum of its parts? Advanced hippocampal substructure segmentation in depression. Biol. Psychiatry. 85, 487–497.
Kim S.Y., Shim Y., Ko Y.J., Park S., Jang S.S., Lim B.C., Ki Joong Kim K.J., Chae J. (2020) Spectrum of movement disorders in GNAO1 encephalopathy: in-depth phenotyping and case-by-case analysis. Orphanet. J. Rare. Dis. 15, 1–6.
Feng H., Sjögren B., Karaj B., Shaw V., Gezer A., Neubig R.R. (2017) Movement disorder in GNAO1 encephalopathy associated with gain-of-function mutations. Neurology. 89, 762–770.
Arya R., Spaeth C., Gilbert D.L., Leach J.L., Holland K.D. (2017) GNAO1-associated epileptic encephalopathy and movement disorders: c.607G>A variant represents a probable mutation hotspot with a distinct phenotype. Epileptic. Disord. 19, 67–75.
Bedbrook C.N., Deverman B.E., Gradinaru V. (2018) Viral strategies for targeting the central and peripheral nervous systems. Annu. Rev. Neurosci. 41, 323–348.
Haery L., Deverman B.E., Matho K.S., Cetin A., Woodard K., Cepko C., Guerin K.I., Rego M.A., Ersing I., Bachle S.M., Joanne Kamens J., Fan M. (2019) Adeno-associated virus technologies and methods for targeted neuronal manipulation. Front. Neuroanat. 13. https //doi.org /10.3389 /fnana.2019.0093
Hammond S.L., Leek A.N., Richman E.H., Tjalkens R.B. (2017) Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection. PLoS One. 12, e0188830.
Kondratov O., Kondratova L., Mandel R.J., Coleman K., Savage M.A., Gray-Edwards H.L., Ness T.J., Rodriguez-Lebron E., Bell R.D., Rabinowitz J., Gamlin P.D., Zolotukhin S. (2021) A comprehensive study of a 29-capsid AAV library in a non-human primate central nervous system. Mol. Ther. 29, 2806–2820.
Wang D., Tai P.W.L., Gao G. (2019) Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug. Discov. 18, 358–378.
Hocquemiller M., Giersch L., Audrain M., Parker S., Cartier N. (2016) Adeno-associated virus-based gene therapy for CNS diseases. Hum. Gene. Ther. 27, 478–496.
Borel F., Kay M.A., Mueller C. (2014) Recombinant AAV as a platform for translating the therapeutic potential of RNA interference. Mol. Ther. 22, 692–701.
Wang D., Zhang F., Gao G. (2020) CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell. 181, 136–150.
Moffat J., Grueneberg D.A., Yang X., Kim S.Y., Kloepfer A.M., Hinkle G., Piqani B., Eisenhaure T.M., Luo B., Grenier J.K., Carpenter A.E., Foo S.Y., Stewart S.A., Stockwell B.R., Hacohen N., Hahn W.C., Lander E.S., Sabatini D.M., Root D.E. (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 124, 1283–1298.
Addgene: Protocol pLKO.1–TRC Cloning Vector. https://www.addgene.org/protocols/plko/ (accessed January 31, 2022)
Starikova A.V., Skopenkova V.V., Polikarpova A.V., Reshetov D.A, Vassilieva S.G, Velyaev O.A., Shmidt A.A., Savchenko I.M., Soldatov V.O., Egorova T.V., Bardina M.V. (2022) Therapeutic potential of highly functional codon-optimized microutrophin for muscle-specific expression. Sci. Rep. 12, 1–13.
Buclez P.O., Dias Florencio G., Relizani K., Beley C., Garcia L., Benchaouir R. (2016) Rapid, scalable, and low-cost purification of recombinant adeno-associated virus produced by baculovirus expression vector system. Mol. Ther. Methods. Clin. Dev. 3, 16035.
Nikitin E.S., Bal N.V., Malyshev A., Ierusalimsky V.N., Spivak Y., Balaban P.M., Volgushev M. (2017) Encoding of high frequencies improves with maturation of action potential generation in cultured neocortical neurons. Front. Cell. Neurosci. 11, 28.
Geissler M., Gottschling C., Aguado A., Rauch U., Wetzel C.H., Hatt H., Faissner A. (2013) Primary hippocampal neurons, which lack four crucial extracellular matrix molecules, display abnormalities of synaptic structure and function and severe deficits in perineuronal net formation. J. Neurosci. 33, 7742.
Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J., White D.J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9, 676–682.
Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J., Golland P., Sabatini D.M. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, 1–11.
Masuho I., Mototani Y., Sahara Y., Asami J., Nakamura S., Kozasa T., Inoue T. (2008) Dynamic expression patterns of G protein-regulated inducer of neurite outgrowth 1 (GRIN1) and its colocalization with Gαo implicate significant roles of Gαo-GRIN1 signaling in nervous system. Dev. Dyn. 237, 2415.
Choi J.M., Kim S.S., Choi C.Il., Cha H.L., Oh H.H., Ghil S., Lee Y.D., Birnbaumer L. (2016) Development of the main olfactory system and main olfactory epithelium-dependent male mating behavior are altered in Go-deficient mice. Proc. Natl. Acad. Sci. USA. 113, 10974–10979.
GNAO1 protein expression summary – The Human Protein Atlas. https://www.proteinatlas.org/ENSG00000087258-GNAO1 (accessed January 25, 2022)
Vandenberghe L.H., Xiao R., Lock M., Lin J., Korn M., Wilson J.M. (2010) Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum. Gene Ther. 21, 1251–1257.
Nectow A.R., Nestler E.J. (2020) Viral tools for neuroscience. Nat. Rev. Neurosci. 21, 669–681.
Bulcha J.T., Wang Y., Ma H., Tai P.W.L., Gao G. (2021) Viral vector platforms within the gene therapy landscape. Signal Transduct. Target Ther. 6, 1–24.
Howard D.B., Powers K., Wang Y., Harvey B.K. (2008) Tropism and toxicity of adeno-associated viral vector serotypes 1, 2, 5, 6, 7, 8, and 9 in rat neurons and glia in vitro. Virology. 372, 24–34.
Royo N.C., Vandenberghe L.H., Ma J.Y., Hauspurg A., Yu L.Y., Maronski M., Johnston J., Dichter M.A., Wilson J.M., Watson D.J. (2008) Specific AAV serotypes stably transduce primary hippocampal and cortical cultures with high efficiency and low toxicity. Brain Res. 1190, 15.
Grimm D., Lee J.S., Wang L., Desai T., Akache B., Storm T.A., Kay M.A. (2008) In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 82, 5887.
Muntean B.S., Masuho I., Dao M., Sutton L.P., Zucca S., Iwamoto H., Patil D.N., Wang D., Birnbaumer L., Blakely R.D., Grill B., Martemyanov K.A. (2021) Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders. Cell Rep. 34, 108718.
Aimiuwu O.V., Fowler A.M., Sah M., Teoh J.J., Kanber A., Pyne N.K., Petri S., Chana Rosenthal-Weiss C., Mu Yang M., Scott Q., Harper S.Q., Wayne N., Frankel W.N. (2020) RNAi-based gene therapy rescues developmental and epileptic encephalopathy in a genetic mouse model. Mol. Ther. 28, 1706–1716.
Choudhury S.R., Harris A.F., Cabral D.J., Keeler A.M., Sapp E., Ferreira J.S., Gray-Edwards H.L., Johnson J.A., Johnson A.K., Su Q., Stoica L., DiFiglia M., Aronin N., Martin D.R., Gao G., Sena-Esteves M. (2016) Widespread central nervous system gene transfer and silencing after systemic delivery of novel AAV-AS vector. Mol. Ther. 24, 726–735.
Martier R., Liefhebber J.M., García-Osta A., Miniarikova J., Cuadrado-Tejedor M., Espelosin M., Ursua S., Petry H., van Deventer S., Evers M.M., Konstantinova P. (2019) Targeting RNA-mediated toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol. Ther. Nucl. Acid. 16, 26–37.
Nobre R.J., Lobo D.D., Henriques C., Duarte S.P., Lopes S.M., Silva A.C., Lopes M.M., Mariet F., Schwarz L.K., Baatje M.S., Ferreira V., Vallès A., Almeida L., Evers M.M., Toonen L.J.A. (2021) MiRNA-mediated knockdown of ATXN3 alleviates molecular disease hallmarks in a mouse model for spinocerebellar ataxia type 3. Nucl. Acid Ther. https://doi.org/10.1089/nat.2021.0020
Strings-Ufombah V., Malerba A., Kao S.C., Harbaran S., Roth F., Cappellari O., Lu-Nguyen N., Takahashi K., Mukadam S., Kilfoil G., Claudia Kloth C., Roelvink P., Dickson G., Trollet C., Suhy D. (2021) BB-301: a silence and replace AAV-based vector for the treatment of oculopharyngeal muscular dystrophy. Mol. Ther. Nucl. Acids. 24, 67–78.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология