

Молекулярная биология, 2023, T. 57, № 1, стр. 127-138
Метод комплексного формирования предикторов для применения алгоритмов машинного обучения в задаче предсказания структуры и функций белка
Ю. В. Мильчевский a, *, В. Ю. Мильчевская a, b, Ю. В. Кравацкий a, c
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Institute of Medical Statistics and Bioinformatics, Faculty of Medicine, University of Cologne
50931 Cologne, Germany
c Центр высокоточного редактирования и генетических технологий для биомедицины,
Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: milch@eimb.ru
Поступила в редакцию 10.06.2022
После доработки 31.07.2022
Принята к публикации 01.09.2022
- EDN: AWNZLZ
- DOI: 10.31857/S0026898423010093
Аннотация
Повышение точности предсказания структуры и функций белков в последнее время связано в основном с применением и совершенствованием методов машинного обучения. Кодирование информации, содержащейся в последовательности аминокислот, ‒ первый этап предсказания структуры, и поэтому оно играет фундаментальную роль в конечном успехе этих методов. Мы предлагаем единую методику генерации предикторов сложного вида, позволяющую формализовать предположения о факторах, которые влияют на структуру и функцию белка. Кроме того, в рамках этой задачи предложен подход к созданию и использованию баз данных структурных свойств, предоставляющих новые возможности для статистического описания и анализа структурных свойств. Предложенные методы позволяют создавать и тестировать наборы предикторов (описывающих факторы, которые влияют на структуру и функцию белка) как для конкретных задач, так и универсальных. Статистические методы построения моделей, которые мы используем, позволяют отбирать статистически значимые предикторы и улучшать таким образом предсказательные модели. На классическом примере предсказания вторичной структуры белка мы показали эффективность данного подхода, получив точность предсказания для трех классов DSSP: Q3 = 81.3%. Предложенный метод реализован в виде мультиплатформенной программы на языке С++ для командной строки. Исходный код и использованные в этой работе данные расположены по ссылке https://github.com/Milchevskiy/protein-encoding-projects
ВВЕДЕНИЕ
Согласно термодинамической гипотезе Anfinsen [1], информация о структуре и функциях белка содержится в его аминокислотной последовательности. Так, из этой последовательности может быть извлечена информация о физико-химических свойствах белка, контактные потенциалы аминокислот в цепи, эволюционные данные и другая информация. На основе этих данных строят предсказания структуры и свойств белков.
В последние годы наблюдали значительный прогресс в методах предсказании структуры и функций белка на основе первичной структуры. Существенные продвижения произошли в таких задачах, как предсказание вторичной [2] и локальной [3] структуры белка, белковых контактов [4], белоксвязывающих участков [5] и др. Прогресс в этих задачах достигнут в основном за счет использования методов машинного обучения, особенно глубокого обучения [6]. Фактически, в текущий момент высокая точность предсказания структуры и функций белка достижима только методами машинного обучения. Подготовка данных для обучения предсказательных моделей ‒ наиболее трудоемкая задача, по-видимому, как в методическом, так и в алгоритмическом смысле. Суть проблемы в том, что генерация входных данных для машинного обучения в задачах предсказания структур и функций белков ‒ нестандартная процедура, которая обычно реализуется в контексте конкретной задачи. Кроме того, огромный массив данных о свойствах аминокислот содержится в разрозненных источниках.
Чтобы получить адекватные результаты, основанные на машинном обучении, обычно необходимы три составляющих: репрезентативные обучающие выборки, мощные алгоритмы машинного обучения и эффективное представление объектов через набор предикторов. Последние достижения в предсказании структуры белков связаны с увеличением объема доступных данных о структуре и функции белков, а также с революционным прогрессом в развитии алгоритмов глубокого машинного обучения [2]. В то же время методы представления белковых данных (amino acid encoding method, кодирование последовательности аминокислот) не привлекали пристального внимания исследователей.
Экстенсивные методы совершенствования предсказаний, связанные с ростом объемов доступных баз данных, а также с улучшением методов собственно машинного обучения, практически исчерпали себя. Мы полагаем, что повышение точности предсказаний лежит в области развития подходов к кодированию биологических данных в предикторы для последующего использования в предсказательных моделях. Создаваемый набор предикторов должен давать возможность формулировать и тестировать наши предположения о природе связи между последовательностью и структурой через физико-химические свойства, определяемые этой последовательностью.
МАТЕРИАЛЫ И МЕТОДЫ
В последние десятилетия предложены различные алгоритмы кодирования аминокислотных последовательностей в предикторы [7, 8]. Наиболее широко для формирования предикторов используют следующие кодировки: one-hot encoding, position specific scoring matrix (PSSM), а также физико-химические кодировки свойств аминокислот в белковой цепи. В дополнение к этим предложены такие варианты, как кодирование, оцененное по энергиям контакта между остатками [9] и полученное в результате выравнивания структуры белка [10] и из контекста последовательности [11]. Подробный обзор и классификация современных методов формирования предикторов для белковых последовательностей представлены в обзоре X. Jing и др. [12].
Cформулированные ранее подходы разнородны. Единый подход, включающий в себя формирование предикторов на основе всей доступной информации по аминокислотной последовательности (физической, химической, биологической), отсутствует. Например, в базе AAindex [13] собрана информация о более чем 550 физико-химических свойствах аминокислот. Подавляющее большинство этих свойств не использовали при предсказаниях методами машинного обучения.
Мы предлагаем единый алгоритм генерации предикторов сложного вида, позволяющий формализовать предположения о факторах, влияющих на структуру и функцию белков. Этот алгоритм включает, в том числе, генерацию регулярных выражений для создания предикторов, создание и использование баз данных структурных свойств и формирование предикторов непосредственно из физико-химических свойств.
Кроме того, метод позволяет создавать комбинированные предикторы, составленные из всех выше перечисленных типов. Ниже мы подробно описываем типы создаваемых предикторов, методы их создания и анализа в процессе построения предварительных моделей с помощью пошагового регрессионного анализа и пошагового дискриминантного анализа.
Предикторы, основанные на базе свойств аминокислот AAindex. Как уже упоминали, в базе данных AAindex [13] систематизировано более 550 физико-химических свойств аминокислот. Кроме них эта база данных также содержит различные матрицы аминокислотных мутаций (amino acid mutation matrices) и потенциалы попарных контактов (pair-wise contact potentials). По предлагаемой нами методике принцип формирования предикторов для всех этих данных одинаков.
Простейшим способом генерации предикторов для характеристики некоторого свойства белковой цепи (например, гидрофобности) является сопоставление последовательности набору соответствующих каждой аминокислоте значений гидрофобности. Однако наивно предполагать, что свойства аминокислотной последовательности описываются линейно гидрофобностью отдельных аминокислот.
Проиллюстрируем генерацию набора предикторов по предсказанию локальной структуры белка [14]. Рассмотрим фрагмент последовательности из n остатков. Каждая аминокислота $\{ {{a}_{i}},\,\,i = 1, \ldots n\} $ имеет определенное значение гидрофобности $\{ {{H}_{i}},\,\,i = 1, \ldots n\} $. Далее к этому массиву значений свойств применимо функциональное преобразование:
(1)
$\begin{gathered} F({{H}_{1}},{{H}_{2}},...{{H}_{n}}) = \\ = \sqrt {{{{\left( {\sum\limits_{k\,\, = \,\,1}^n {{{H}_{k}}\cos \left( {k\frac{{2\pi }}{T}} \right)} } \right)}}^{2}} + {{{\left( {\sum\limits_{k\,\, = \,\,1}^n {{{H}_{k}}\sin \left( {k\frac{{2\pi }}{T}} \right)} } \right)}}^{2}}} , \\ \end{gathered} $Если рассматриваемый участок находится в периодической (например, спиральной) конформации, то величина, рассчитанная по формуле (1), будет максимальна для периода T этой спирали. Остается “угадать” этот период, а также минимальное число остатков n, на котором сформированный предиктор будет значимым, т.е. отличаться от случайного шума по заданным статистическим критериям. Способы задания статистических критериев подробно описаны ниже в подразделе “пошаговый регрессионный анализ”. Далее, в базе AAindex представлено более 30 свойств, называемых шкалами гидрофобности. Какую из них выбрать? Расчетные методы, которые мы использовали для отладки модели, позволяют количественно сравнивать значимость множества предикторов. В рассматриваемом случае мы собрали в набор предикторы, построенные по формуле (1), для всех шкал гидрофобности и при различных диапазонах параметров T и n. Применение пошагового регрессионного анализа позволило выявить наиболее значимые предикторы и отбросить несущественные, что представляет основной этап усовершенствования модели.
В общем виде схема генерации предикторов, основанных на физико-химических свойствах, предполагает этап функциональной трансформации:

Алгоритм предполагает также возможность создания исследователями собственных функций функциональной трансформации, отражающих их гипотезы о возможных факторах, влияющих на структуру и функции белков.
Регулярные выражения как предикторы. Одна из широко используемых кодировок ‒ так называемая “binary encoding” [15]. В ней последовательность кодируется числами 0 и 1. В простейшем случае так называемого “one-hot encoding” каждую позицию в аминокислотной последовательности описывает бинарный вектор (0 и 1) размерности 20. Такой вектор содержит только одну единицу в позиции (поэтому он назван one-hot), соответствующей рассматриваемой аминокислоте, тогда как все остальные заполнены нулями. Из-за большой размерности и разреженности представления в “one-hot encoding” используют также вырожденные представления. Вырождение состоит в том, что 20 стандартных аминокислот распределяются по группам. Например, М. Dayhoff [16] опираясь на статистику точечных мутаций, распредел стандартные аминокислоты по шести группам: [H, R, K], [D, E, N, Q], [C], [S, T, P, A, G], [M, I, L, V], [F, Y, W].
Предлагаемый нами метод обладает возможностью генерировать предикторы для заранее выбранных распределений аминокислот по группам для более общей ситуации: когда рассматривают не отдельные аминокислоты, а некоторый участок последовательности (так называемое word, или “cлово”) заданной длины, при этом для каждого положения этого участка определяют свои правила разделения на группы. Эта задача представляет задание так называемых регулярных выражений. В качестве примера в табл. 1 приведен вариант распределения по группам для тетрапептидов. Такой способ уменьшения размерности задачи называют “заданием редуцированных алфавитов” [14].
Таблица 1.
Пример распределения аминокислот по группам внутри тетрапептида
| Сдвиг | Распределение аминокислот по группам эквивалентности |
|---|---|
| –2 | [ARNDCQEHILKMFPSTWYV] [G] [P] [O] |
| –1 | [HRK] [DENQ] [C] [STPAG] [MILV] [FYW] [O] |
| 0 | [A] [R] [N] [D] [C] [Q] [E] [H] [I] [L] [K] [M] [F] [P] [S] [T] [W] [Y] [V] [G] [P] |
| 1 | [ARNDCQEHILKMFPSTWYV] [G] [P] [O] |
В положении левее на 2 остатка от текущего (“–2” – сдвиг) положения все аминокислоты, кроме глицина и пролина, считают неразличимыми. В положении “-1” предполагают эквивалентность Н, К, R, затем D, E, N, Q и т.д. В текущем положении “0” все аминокислоты различаются. В табл. 1 приведена также виртуальная аминокислота “О”, которая нужна для представления фрагментов, находящихся на концах последовательности. Например, слово “OOMA” означает виртуальный тетрапептид, содержащий две аминокислоты, M и A, на N-конце белка. Чтобы описать положение последовательности по правилам, приведенным в табл. 1, необходимо 4 × 6 × × 20 × 4 = 1920 предикторов. Это все еще разреженный массив, однако он существенно отличается по размеру от one-hot массива, который состоял бы из всех возможных тетрапептидов и имел размер 204 = 160 000. Более сжатый характер массива и его существенно меньший размер позволяют использовать статистические методы для связи последовательности со структурой и функциями белка. В частности, в следующем разделе описано использование предикторов, построенных на статистических характеристиках встречаемости структурных элементов.
Предикторы, построенные на статистических характеристиках встречаемости структурных элементов. Создание базы данных структурных элементов. В качестве примера рассмотрим применение статистики встречаемости структурных элементов – 16 пентапептидов из работы А. de Brevern и др. [17]. Этот набор структурных фрагментов (Protein Blocks ‒ PB) широко используют для более детального описания конформации белковой цепи (по сравнению с общепринятой разметкой вторичной структуры DSSP [18]). Расстояние между фрагментами структуры главной цепи в терминах RMSD описывает формула:
(2)
$\begin{gathered} \operatorname{RMSD} ({{V}_{1}},{{V}_{2}}) = \\ = \min \left( {\sqrt {\frac{{\sum\limits_{i\,\, = \,\,1}^{3M} {{{{({{x}_{{i,1}}}\, - \,{{x}_{{i,2}}})}}^{2}}\, + \,{{{({{y}_{{i,1}}}\, - \,{{y}_{{i,2}}})}}^{2}}\, + \,{{{({{z}_{{i,1}}}\, - \,{{x}_{{i,2}}})}}^{2}}} }}{{3M}}} } \right), \\ \end{gathered} $Рассмотрим один из этих структурных элементов: $P{{B}_{j}},j \in \{ 1,...,16\} $ – и произвольный фрагмент последовательности длиной 5 остатков. ${{N}_{{{\text{occ}}}}}(seq)$ – частота встречаемости последовательности seq среди последовательностей с известной структурой (например, обучающая выборка). Пусть ${{\bar {\mu }}_{j}}(seq)$ – среднее расстояние (в терминах RMSD) между атомами главной цепи пентапептидов, образующими последовательность seq, и атомами главной цепи j-го блока $P{{B}_{j}}$. Далее, ${{\bar {\mu }}_{j}}$ – среднее расстояние между $P{{B}_{j}}$ и всеми пентапептидами обучающей выборки. Соответственно $s_{j}^{2}(seq)$ и $s_{j}^{2}$ – дисперсии расстояния для пентапептидов, имеющих последовательность seq и для всех пентапептидов выборки ${{t}_{j}} = \frac{{{{{\bar {\mu }}}_{j}} - {{{\bar {\mu }}}_{j}}(seq)}}{{{{s}_{j}}(seq)}}$, где N – размер обучающей выборки, а ${{s}_{j}}(seq) = \frac{{{{\sigma }^{2}}(seq)}}{{{{N}_{{{\text{occ}}}}}(seq)}} + \frac{{s_{j}^{2}}}{N}$.
Поскольку $N \gg {{N}_{{{\text{occ}}}}} > 1$, $s_{j}^{2}(seq) = \frac{{{{\sigma }^{2}}(seq)}}{{{{N}_{{{\text{occ}}}}}(seq)}}$. Следовательно,
(3)
${{t}_{j}} \approx \frac{{{{{\bar {\mu }}}_{j}} - {{{\bar {\mu }}}_{j}}(seq)}}{{{{\sigma }_{j}}(seq)}}\sqrt {{{N}_{{{\text{occ}}}}}} ,$В общем виде схему, описывающую формирование предикторов, построенных на статистических характеристиках встречаемости структурных элементов, можно представить следующим образом:
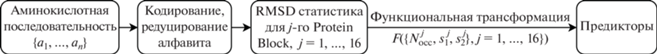
Чтобы применить формулу (3), необходимо составить базу данных встречаемости белковых структур для получения величин ${{\bar {\mu }}_{j}},{{\bar {\mu }}_{j}}(seq),$ ${{N}_{{{\text{occ}}}}}(seq),\sigma (seq)$.
Функциональная трансформация, задаваемая формулой (3), не единственное преобразование, которое можно использовать для генерации предикторов, связанных со встречаемостью структурных элементов, но для всех таких предикторов нужно хранить именно указанный выше набор величин. Приведенные выше параметры сохраняют в базах данных свойств структурных элементов. При построении предсказательной модели можно использовать множество таких баз данных, различающихся редуцированными алфавитами, длиной фрагмента последовательности и метрикой, задающей расстояние между фрагментами. Например, вместо метрики RMSD (root mean square deviation) можно использовать RMSDA (root mean square deviation of angular values) [17], либо другую метрику.
Комбинированные предикторы. В некоторых случаях может возникнуть необходимость использования предикторов, учитывающих одновременно несколько физико-химических или функциональных свойств белковой последовательности.
Комбинированные предикторы представляют дополнительные возможности для создания предикторов сложного вида. Они могут быть составлены по заранее заданному сценарию из нескольких описанных выше предикторов и условий. Чтобы реализовать эту возможность, каждому предиктору присваивается свое имя, по которому к нему обращаются для задания комбинированного предиктора. Простейший случай ‒ перемножение двух или нескольких предикторов. Для логических переменных это означает одновременное выполнение условий, определяемых для каждого из предикторов. В качестве примера рассмотрим три булевых предиктора:
1. Присутствие двух алифатических аминокислот на расстоянии от –6 до –2 от текущего положения.
2. Присутствие двух алифатических аминокислот на расстоянии от +3 до +6 от текущего положения.
3. Присутствие как пролина, так и глицина на расстоянии от –2 до +3.
Логическим произведением этих трех предикторов будет комбинированный предиктор, который устойчив к единичным вставкам и делециям. Комбинированные предикторы, составленные подобным образом, мы использовали для описания и последующего поиска β-шпилек.
В других случаях можно задавать различные логические условия, например результирующий предиктор, составленный из двух предикторов, который равен первому предиктору, если второй лежит в заданных пределах, в противном случае равен нулю.
Также возможно задание вложенных процедур генерации предикторов, в которых используют комбинированные предикторы, составленные на предыдущих шагах.
Расчетные методы для построения предсказательной модели. Чтобы оценить качество сформированной системы предикторов и для ее усовершенствования, предложено использование оригинальных модификаций регрессионного и дискриминантного анализов.
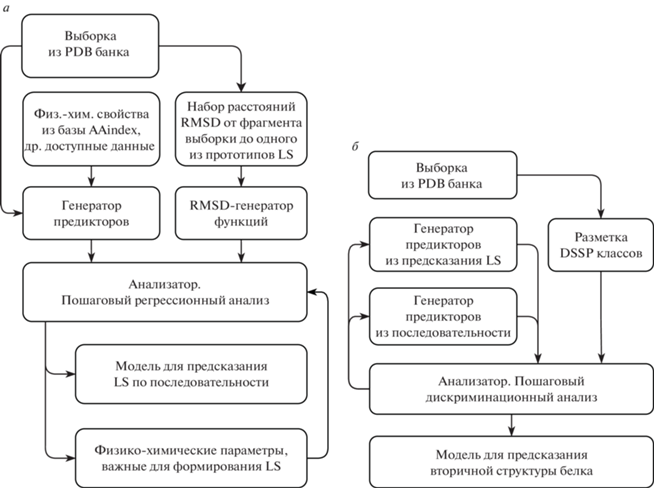
Пошаговый регрессионный анализ. Линейный регрессионный анализ [19] для задач предсказания структуры и функций белка нуждается в модификации, которая позволяет добавлять (для улучшения качества предсказания) или удалять (для упрощения регрессионной функции) предикторы. Пошаговая регрессия позволяет частично автоматизировать процедуру получения набора регрессионных функций без существенных дополнительных вычислительных затрат. Предикторы, включенные в итоговую регрессионную функцию, должны удовлетворять определенным критериям. Мы использовали подход, основанный на статистике Фишера (F-статистики) [20], для проверки значимости регрессионного коэффициента предиктора при принятии решения о включении или исключении его из регрессионной модели. Состав предикторов, включенных в итоговую регрессионную модель, зависит от пороговых значений F-статистики исключения и F-статистики включения. Следовательно, пороговые значения F-статистики влияют на состав и, таким образом, на качество предсказательной модели. Схема используемого в нашей методике пошагового регрессионного анализа изображена на рис. 1a.
Рис. 1.
Схемы формирования статистической модели для предсказания: a ‒ локальной структуры (LS) по аминокислотной последовательности белка; б ‒ вторичной структуры белка с использованием пошагового дискриминационного анализа.
Реализованный в алгоритме (и программном пакете) пошаговый регрессионный анализ обладает следующими возможностями:
1. Определение оптимального порогового значения F-статистики. Пороговые значения F-статистики и оптимальный набор предикторов выбирают с помощью метода перекрестной проверки (k-fold cross validation). Иначе говоря, для задачи, содержащей N объектов, N раз создают регрессионную модель, на каждом шаге из нее исключают один из объектов и предсказывают значение зависимой переменной для этого объекта. Итог этой процедуры – массивы предсказанных и экспериментальных значений зависимых переменных. Коэффициент корреляции Пирсона между этими массивами выбран мерой качества модели. Повторяя эту процедуру с различными пороговыми значениями F-статистики, мы выбирали ту модель, для которой коэффициент корреляции максимален. Анализ состава предикторов в окончательной модели и сравнения их статистической значимости дает ценную информацию о факторах, определяющих локальную структуру.
2. Возможность одновременно рассчитывать не одну зависимую переменную. В задаче предсказания локальной структуры начальный набор предикторов (до отбора удовлетворяющих пороговым значениям F-статистики) одинаков для каждой из зависимых переменных (например, для шаблонов локальной структуры, таких как α‑спираль, β-слой и т.д.). Подобная ситуация может встречаться во многих задачах. Самым затратным в смысле числа вычислительных операций (требующих на 2 порядка больше операций в случае реальных задач, чем все остальные) является этап вычисления матрицы перекрестных произведений, пропорциональный M × (M + 6)N [20], где M – число предикторов в начальном наборе, N – число объектов. Мы модифицировали стандартный метод, вычисляя расширенную матрицу перекрестных произведений размером (M + P)(M + P) один раз. Впоследствии эту матрицу используют для получения регрессионных функций для всех P зависимых переменных. Эта модификация обеспечивает радикальное повышение производительности.
Пошаговый дискриминантный анализ. Применение пошагового дискриминантного анализа [20] имеет много общего с применением пошагового регрессионного. В этом случае с помощью F-статистики также определяют набор статистически значимых предикторов, но не для предсказания зависимой переменной, а для классификации наблюдаемых объектов по группам. Алгоритмы формирования значимых предикторов в стандартном пошаговом методе дискриминантного анализа и пошаговом регрессионном анализе практически идентичны. Так же, как и в регрессионном анализе, происходит определение оптимального набора предикторов с помощью метода перекрестной проверки (k-fold cross validation). В качестве меры качества предсказательной модели выбрана доля правильно классифицированных (предсказанных) объектов. Схема используемого в нашей методике пошагового дискриминационного анализа изображена на рис. 1б.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Описанные в предыдущих разделах подходы реализованы в виде пакета программ для генерации и тестирования наборов предикторов описанными выше статистическими методами.
Иллюстрация применения нашего пакета программ к реальным задачам представляет обращение к классической задаче предсказания вторичной структуры белка. При этом использовали все указанные выше типы предикторов. В частности, мы выполняли создание и отладку предсказательной модели c применением вычислительных процедур, которые обеспечивают оценку вклада отдельных предикторов. Отладка модели ‒ итеративная процедура: на каждой итерации строят предсказательную модель, затем редактируют набор предикторов, исходя из статистической значимости каждого предиктора и качества полученной модели. При создании модели использовали последовательно пошаговые регрессионный и дискриминантный анализы. Технически современные компьютеры позволяют задавать большое число предикторов для обучения модели (до 10 000 и более). Процедура выявления значимых предикторов позволяет шаг за шагом выявлять имеющиеся закономерности, модифицировать и комбинировать предикторы, улучшая при этом модель.
Создание модели предсказания состоит из двух этапов. На первом этапе использована регрессионная модель для предсказания классификации по обобщенным координатам для 16 базовых структур (protein blocks) [14] (рис. 1a), затем эти обобщенные координаты использовали как предикторы (вместе с дополнительными предикторами) для построения модели с помощью пошагового дискриминантного анализа (рис. 1б).
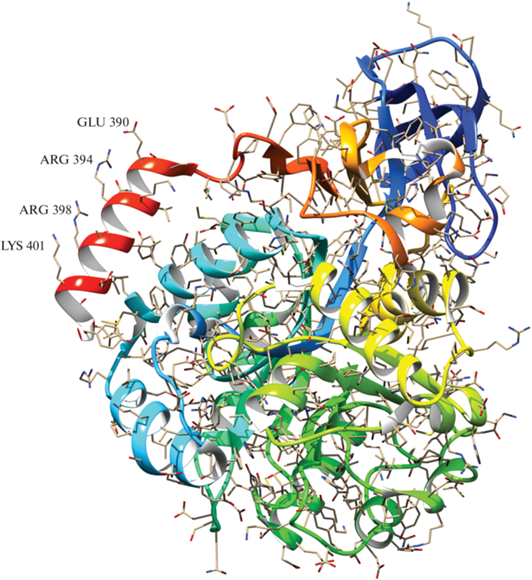
Проиллюстрируем первый этап создания предсказательной модели на примере двух наиболее важных наборов предикторов. Первый из них основан исключительно на физико-химических свойствах аминокислот из базы данных AAindex, использующихся для функционального преобразования по формуле (1). В процессе отладки таких предикторов опробованы не только все 30 шкал гидрофобности из этой базы, но и все остальные свойства из этой базы. Оказалось, что наиболее значимы предикторы, составленные по шкале WERD780101, которая формально не относится к гидрофобности, а характеризует склонность аминокислот быть внутри глобулы белка [21]. При этом среди всех опробованных значений периода выявлен период, равный 3.6 остаткам, что соответствует α-спиральной конформации. Оптимальная длина фрагмента, на которой этот предиктор наиболее статистически весóм, составляет 8 остатков. На рис. 2 видно, что гидрофильные аминокислоты ориентированы к водной поверхности белка через 3–4 остатка.
Рис. 2.
Формирование предиктора, связанного с периодическими вариациями гидрофобности, вдоль цепи в белке 1P1M.
Второй тип предикторов менее очевиден, однако для предсказания многих из 16 структурных классов де Бреверна (de Brevern) [17] предикторы этого типа обладают наибольшей статистической значимостью. На примере этого типа предикторов мы иллюстрируем поиск адекватного функционального преобразования, отражающего скрытые закономерности. При этом мы исходили из предположения, что если для всех фрагментов (в данном случае пентапептидов), имеющих последовательность seq, характерна конформация, близкая к одной из 16 шаблонных структур де Бреверна, то среднее расстояние (RMSD) между этими фрагментами и этой шаблонной структурой будет меньше, чем среднее расстояние между этой же шаблонной структурой и фрагментами всей выборки. Это расстояние может быть записано так:
(4)
$F(k,seq) = \frac{{\sum\limits_i^N {{{R}_{k}}} }}{N} - \frac{{\sum\limits_i^{{{N}_{{{\text{occ}}}}}(seq)} {{{R}_{k}}(seq)} }}{{{{N}_{{{\text{occ}}}}}(seq)}},$Но как можно сравнивать случаи, когда ${{N}_{{{\text{occ}}}}}(seq) = 1$ и когда ${{N}_{{{\text{occ}}}}}(seq) = 100$? Как добиться того, чтобы вклад этого предиктора в регрессионную функцию был линейным? Выполнили тестирование различных функциональных преобразований и в итоге предикторы, задаваемые формулой (5), стали наиболее статистически значимыми:
(5)
$\begin{gathered} F(k,seq) = \ln [1 + \ln (1 + {{N}_{{{\text{occ}}}}}(seq))] \times \\ \times \,\,\left( {\frac{{\sum\limits_i^N {{{R}_{i}}(k)} }}{N} - \frac{{\sum\limits_i^{{{N}_{{{\text{occ}}}}}(seq)} {{{R}_{i}}(k,seq)} }}{{{{N}_{{{\text{occ}}}}}(seq)}}} \right). \\ \end{gathered} $Алфавит не обязательно имеет длину 5 остатков, совпадающую с длиной шаблонных структур де Бреверна. В данной задаче в процессе отладки модели мы подбирали редуцированные алфавиты длиной до 15 остатков, используя различные способы распределения аминокислот по эквивалентным группам. В частности, самый значимый предиктор для предсказания конформации β-слоя (protein block “D” де Бреверна) получен по формуле (5) для случая редуцированного алфавита для фрагмента длиной 7 остатков (табл. 2).
Таблица 2.
Распределения аминокислот по группам для фрагмента длиной 7 остатков
| Сдвиг | Распределение аминокислот по группам эквивалентности |
|---|---|
| –3 | [A] [R] [N] [D] [C] [Q] [E] [H] [I] [L] [K] [M] [F] [P] [S] [T] [W] [Y] [V] [G] [P] |
| –2 | [A] [R] [N] [D] [C] [Q] [E] [H] [I] [L] [K] [M] [F] [P] [S] [T] [W] [Y] [V] [G] [P] |
| –1 | [ALMC] [VIFYWKRHDENQST] [GP] [O] [X] |
| 0 | [ALMC] [VIFYWKRHDENQST] [GP] [X] |
| 1 | [ALMC] [VIFYWKRHDENQST] [GP] [O] [X] |
| 2 | [A] [R] [N] [D] [C] [Q] [E] [H] [I] [L] [K] [M] [F] [P] [S] [T] [W] [Y] [V] [G] [P] |
| 3 | [A] [R] [N] [D] [C] [Q] [E] [H] [I] [L] [K] [M] [F] [P] [S] [T] [W] [Y] [V] [G] [P] |
Обучающая выборка
Мы использовали стандартный метод создания обучающих выборок PISCES30 [22]. Отбор белковых цепей из PDB выполнен по следующим параметрам: разрешение не хуже 2.5 Å, R-фактор не менее 1.0 и гомология последовательностей не более 30%. В итоге получена обучающая выборка, состоящая из 17148 негомологичных белковых цепей, структуры которых определены с помощью рентгеноструктурного анализа.
Предсказание вторичной структуры
Обучающая выборка для оценки эффективности модели предсказания разделена на две части в соотношении 4 : 1. Бóльшую часть использовали для обучения, меньшую – для оценки качества предсказания.
Предсказание выполняли для трех DSSP классов (α-спираль “H”, β-слой “E”, неупорядоченная структура “–”). Качество предсказательной модели можно оценить при помощи матрицы ошибок (confusion matrix), диагональные элементы которой содержат число правильно предсказанных конформаций соответствующего класса, а элементы каждой строки характеризуют распределение предсказаний по классам. В табл. 3 приведены результаты, полученные для контрольной выборки. В частности, класс H (α-спираль) встретился 307 026 раз (281 863 + 5 + 25 158). Из них этот класс верно предсказали 281 863 раз, 5 раз ошибочно как β-слой и 25 158 раз неверно как неупорядоченную структуру.
Таблица 3.
Матрица ошибок для трех классов DSSP
| DSSP-класс | Содержание ошибок | |||||
|---|---|---|---|---|---|---|
| число | % | |||||
| Н | E | – | Н | E | – | |
| Н | 281 863 | 5 | 25 158 | 91.80 | 0.001 | 8.19 |
| E | 212 | 139 392 | 42 698 | 0.12 | 76.46 | 23.42 |
| – | 26 264 | 44 671 | 239 587 | 8.46 | 14.39 | 77.16 |
Чтобы дополнительно проконтролировать качество предсказательной модели, использовали классическую выборку пептидов СВ513 [23], которую широко применяли другие авторы для анализа эффективности методов предсказания и сравнения этих методов между собой. Выборка представляет собой набор 513 белковых цепей (и частей белковый цепей), подобранных специально для перекрестной проверки методов предсказания вторичной структуры так, чтобы минимизировать влияние внутренней гомологии. Также мы выполнили сравнение точности предсказаний нашего метода с другими современными методами предсказания структур, такими как GApred [24], SPIDER2 [25], Jpred4 [26], FSVM [27], SSpro5 [28]. Результаты предсказания, выполненные предлагаемым нами подходом, и их сравнение с другими методами (данные других программ получены из работы [24]) приведены в табл. 4.
Таблица 4.
Сравнение точности предсказаний трех DSSP-классов, выполненное для выборки CB513 различными методами предсказания
| Метод | DSSP-класс | |||
|---|---|---|---|---|
| Q3 | Н | E | – | |
| GApred | 85.3 | 83.3 | 85.5 | 84.8 |
| SPIDER2 | 73.5 | 72.4 | 79.3 | 78.7 |
| Jpred4 | 65.6 | 60.2 | 63.4 | 67.1 |
| FSVM | 83.0 | 82.0 | 80.0 | 79.0 |
| SSpro5 | 82.0 | 78.3 | 83.0 | 80.5 |
| Наш метод | 81.4 | 91.2 | 76.3 | 76.2 |
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Полученное качество предсказания для трех DSSP-классов составляет Q3 = 82.62% для контрольной подвыборки из нашей обучающей выборки и 81.44% для выборки СВ513 [23]. Этот результат говорит о высоком качестве предсказания. Лучшие, так называемые state-of-the-art, методы предсказания обеспечивают качество Q3 в диапазоне 80‒85% [29‒32], хотя в этой работе предсказание выполняли без использования методов машинного обучения и данных о гомологичных белках (фактически, результат можно назвать предварительным для последующего применения методов машинного обучения).
Очевидно (табл. 3 и 4), что наилучшая точность предсказаний (91%) получена для DSSP-класса H (α-спираль). Это связано с тем, что класс Н наиболее структурно однороден и почти полностью совпадает с предсказанием класса M из структурной классификации де Бреверна [17]. При этом ошибочные ложноотрицательные предсказания (8%) относятся к классу неупорядоченных структур (обозначаемых “–“ в разметке 3 классов DSSP). Внутри этого класса, согласно структурной классификации DSSP, имеется класс T (turn), который структурно близок α-спирали. Этим фактором объясняют ложноположительные предсказания (8%) для класса H со стороны класса “–”. В нашей модели предсказания α-спирального класса и класса E (β-слой) практически не пересекаются. Это вполне ожидаемо, поскольку эти классы максимально структурно различаются (в смысле RMSD между пентапептидами), а предсказания DSSP-классов основаны на предсказании распределения по 16 структурным блокам де Бреверна. Отметим, что расстояние между структурными блоками M, соответствующими идеальной α-спирали, и D, близкими к антипараллельному β-слою, составляет 3.12 Å и близко к максимальному среди попарных расстояний для 16 структурных классов [14].
Точность предсказания DSSP класса E (β-слой) (76%) заметно ниже класса H (α-спираль) ввиду того, что среди класса неупорядоченных структур присутствуют конформации, близкие (в смысле RMSD) к предсказываемому классу E. Это приводит к значительным ложноположительным (23%) и ложноотрицательным (14%) ошибкам.
Структурную классификацию DSSP определяют преимущественно по водородным связям белковых цепей, поэтому такие DSSP-классы, как β-слои, имеют размытую структуру. Даже для случая 8 DSSP-классов эта проблема остается актуальной. Чтобы описать более детально локальную структуру, создали структурные классификации, основанные на других принципах. В частности, структурная классификация де Бреверна основана на фиксированных углах Phi и Psi для каждого из 16 структурных блоков. Точность предсказания, основанного на физико-химических свойствах аминокислот, в этом случае составила Q16 = 67.9% [14], при этом для α-спирального структурного блока “М” точность предсказания превысила 97%. Поскольку для предсказания мы не использовали множественные выравнивания последовательностей, эта точность, по-видимому, определена тем, что удалось найти существенные физико-химические параметры, определяющие структуру этого структурного блока. Эти параметры заданы в виде предикторов, созданных и отобранных с помощью предлагаемых нами алгоритмов создания предикторов и отладки предсказывающей модели. Многие предикторы удалось интерпретировать (например, предикторы, образованные по формуле (1)).
Как видно из табл. 4, на выборке CB513 [23] наш метод в его текущей реализации существенно лучше предсказывает α-спирали (превосходя на 8% даже “генетические” алгоритмы и на 9% и более другие state-of-the-art методы) и проявил себя несколько хуже в предсказании β-слоев (уступая 9% “генетическому” алгоритму и отставая от наиболее передовых state-of-the-art методов до 7%) и неупорядоченных структур (отставание от “генетического” алгоритма – 9%, от наиболее передовых state-of-the-art методов – 4%). Это может быть связано с тем, что наше предсказание α-спирали проще связать именно со структурными параметрами, поскольку структура α-спирали задана более формализовано, чем структура β‑слоев и тем более неупорядоченных структур. Кроме того, β-слои структурно близки к некоторым участкам, относимым к неупорядоченным (например, левая спираль типа полипролин II). Практически одинаковые пространственные структуры, близкие к конформации β-слоя, могут быть отнесены к разным DSSP-классам только по наличию/отсутствию характерных водородных связей, некоторые из которых вообще могут быть сформированы разными белковыми цепями. Информацию для отнесения таких спорных участков к одному из классов, по-видимому, можно получить исключительно из известной структуры гомологичных белков после их выравнивания по последовательности. В предлагаемом методе мы не использовали выравнивание последовательностей, поскольку занимались иллюстрацией возможностей метода генерации и отладка набора предикторов.
ЗАКЛЮЧЕНИЕ
Предложена методика формирования и усовершенствования наборов предикторов для последующего использования в задачах предсказания структуры и функций белка методами машинного обучения. Представлены примеры формирования наборов предикторов разных типов. Приведены примеры, иллюстрирующие работу пакета программ. Описана процедура задания пользователем собственных предикторов по аналогии с уже присутствующими в библиотеке. Представленная процедура усовершенствования предсказательной модели с помощью регрессионного и дискриминантного анализов – мощный инструмент поиска и тестирования наличия скрытых закономерностей, заложенных в исходных данных. Полученные с помощью машинного обучения результаты зачастую весьма сложно интерпретировать, в то время как предложенный метод позволяет выявить физико-химические факторы, определяющие структуру белка.
Генерацию предикторов и улучшение модели с применением классических статистических методов мы рассматриваем как подготовительную процедуру для современных методов машинного обучения, хотя при удачном выборе и последующей оптимизации набора входных предикторов использование классических методов построения предсказательных моделей обеспечивает вполне конкурентоспособные результаты.
Применение предложенной методики создания предикторов для предсказания структуры и функций белка методами машинного обучения будет продолжено нами в последующих работах. Кроме применения методов машинного обучения мы планируем подключить информацию о структуре гомологичных белков (template-based method) и создать алгоритм переключения между template-based method и non-template-based method в зависимости от предсказываемого белка. Показано, что подобный подход приводит к повышению точности на 3‒6% [33]. Весьма перспективным представляется применение изложенных в работе методов для предсказания более детальных, чем вторичная структура, структурных классификаций, например по структурным блокам де Бреверна [17] или еще более детальных классификаций с различным числом элементов и различной длиной фрагментов, которые мы специально разрабатываем [14].
В текущем виде предложенный метод и программное обеспечение могут быть использованы в составе комбинированного подхода, сочетающего использование различных методов/программ предсказания для первичного предсказания координат α-спиральных участков в предсказываемых пептидах, поскольку по точности предсказания таких участков предложенный нами метод и его программная реализация существенно превосходят все существующие аналоги.
Авторы благодарны Центру высокоточного редактирования и генетических технологий для биомедицины за вычислительные мощности, предоставленные для выполнения этой работы.
Работа выполнена при поддержке Российского научного фонда (22-24-01088).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Реализация предложенного метода в виде пакета кроссплатформенных программ на языке С++ доступна по адресу https://github.com/Milchevskiy/ protein-encoding-projects.
Список литературы
Anfinsen C.B. (1973) Principles that govern the folding of protein chains. Science. 181, 223‒230.
Yang Y., Gao J., Wang J., Heffernan R., Hanson J., Paliwal K., Zhou Y. (2018) Sixty-five years of the long march in protein secondary structure prediction: the final stretch? Brief. Bioinform. 19, 482‒494.
Zimmermann O., Hansmann U.H. (2008) LOCUSTRA: accurate prediction of local protein structure using a two-layer support vector machine approach. J. Chem. Inf. Model. 48, 1903‒1908.
Wuyun Q., Zheng W., Peng Z., Yang J. (2018) A large-scale comparative assessment of methods for residue-residue contact prediction. Brief. Bioinform. 19, 219‒230.
Zhang J., Kurgan L. (2018) Review and comparative assessment of sequence-based predictors of protein-binding residues. Brief Bioinform. 19, 821‒837.
Min S., Lee B., Yoon S. (2017) Deep learning in bioinformatics. Brief. Bioinform. 18, 851‒869.
Hu H.J., Pan Y., Harrison R., Tai P.C. (2004) Improved protein secondary structure prediction using support vector machine with a new encoding scheme and an advanced tertiary classifier. IEEE Trans Nanobioscience. 3, 265‒271.
Yoo P.D., Sikder A.R., Zhou B.B., Zomaya A.Y. (2008) Improved general regression network for protein domain boundary prediction. BMC Bioinformatics. 9(Suppl. 1), S12.
Miyazawa S., Jernigan R.L. (1999) Self-consistent estimation of inter-residue protein contact energies based on an equilibrium mixture approximation of residues. Proteins. 34, 49‒68.
Lin K., May A.C., Taylor W.R. (2002) Amino acid encoding schemes from protein structure alignments: multi-dimensional vectors to describe residue types. J. Theor. Biol. 216, 361‒365.
Asgari E., Mofrad M.R. (2015) Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One. 10, e0141287.
Jing X., Dong Q., Hong D., Lu R. (2020) Amino acid encoding methods for protein sequences: a comprehensive review and assessment. IEEE/ACM Trans. Comput. Biol. Bioinform. 17, 1918‒1931.
Kawashima S., Pokarowski P., Pokarowska M., Kolinski A., Katayama T., Kanehisa M. (2008) AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 36, D202‒205.
Milchevskaya V., Nikitin A.M., Lukshin S.A., Filatov I.V., Kravatsky Y.V., Tumanyan V.G., Esipova N.G., Milchevskiy Y.V. (2021) Structural coordinates: a novel approach to predict protein backbone conformation. PLoS One. 16, e0239793.
Taha K., Yoo P.D. (2015) Predicting protein function from biomedical text. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2015, 3275‒3278.
Dayhoff M.O. (1972) Atlas of protein sequence and structure. Silver Spring, Md.: National Biomedical Research Foundation.
de Brevern A.G., Etchebest C., Hazout S. (2000) Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins. 41, 271‒287.
Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22, 2577‒2637.
Hocking R.R. (1983) Developments in linear regression methodology: 1959‒1982. Technometrics. 25, 219‒223.
Ralston A., Wilf H.S., Enslein K. (1960) Mathematical methods for digital computers. New York: Wiley.
Wertz D.H., Scheraga H.A. (1978) Influence of water on protein structure. An analysis of the preferences of amino acid residues for the inside or outside and for specific conformations in a protein molecule. Macromolecules. 11, 9‒15.
Wang G., Dunbrack R.L., Jr. (2005) PISCES: recent improvements to a PDB sequence culling server. Nucleic Acids Res. 33, W94‒98.
Cuff J.A., Barton G.J. (1999) Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins. 34, 508‒519.
Rout S.B., Mishra S., Sahoo S.K. (2021) Q3 Accuracy and SOV measure analysis of application of GA in protein secondary structure prediction. Revue d’Intelligence Artificielle. 35, 403‒408.
Yang Y., Heffernan R., Paliwal K., Lyons J., Dehzangi A., Sharma A., Wang J., Sattar A., Zhou Y. (2017) SPIDER2: a package to predict secondary structure, accessible surface area, and main-chain torsional angles by deep neural networks. Methods Mol. Biol. 1484, 55‒63.
Drozdetskiy A., Cole C., Procter J., Barton G.J. (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389‒394.
Xie S., Li Z., Hu H. (2018) Protein secondary structure prediction based on the fuzzy support vector machine with the hyperplane optimization. Gene. 642, 74‒83.
Magnan C.N., Baldi P. (2014) SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics. 30, 2592‒2597.
Ma Y., Liu Y., Cheng J. (2018) Protein secondary structure prediction based on data partition and semi-random subspace method. Sci. Rep. 8, 9856.
Guo Z., Hou J., Cheng J. (2021) DNSS2: improved ab initio protein secondary structure prediction using advanced deep learning architectures. Proteins. 89, 207‒217.
Wang S., Peng J., Ma J., Xu J. (2016) Protein secondary structure prediction using deep convolutional neural fields. Sci. Rep. 6, 18962.
Zhang B., Li J., Lu Q. (2018) Prediction of 8-state protein secondary structures by a novel deep learning architecture. BMC Bioinformatics. 19, 293.
Krieger S., Kececioglu J. (2020) Boosting the accuracy of protein secondary structure prediction through nearest neighbor search and method hybridization. Bioinformatics. 36, i317‒i325.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология