

Молекулярная биология, 2023, T. 57, № 2, стр. 155-165
Молекулярные механизмы оптимизации элонгации трансляции существенно различаются у бактерий, имеющих и не имеющих кластеры генов биосинтеза нерибосомных пептидов
А. И. Клименко a, С. А. Лашин a, Н. А. Колчанов a, Д. А. Афонников a, Ю. Г. Матушкин a, *
a Институт цитологии и генетики Сибирского отделения Российской академии наук
630090 Новосибирск, Россия
* E-mail: mat@bionet.nsc.ru
Поступила в редакцию 27.07.2022
После доработки 29.08.2022
Принята к публикации 31.08.2022
- EDN: EGGOIC
- DOI: 10.31857/S002689842302012X
Аннотация
Нерибосомные пептиды, обладающие широкой биологической активностью, играют важную роль в жизнедеятельности бактерий. В частности, они действуют как антибиотики, токсины, поверхностно-активные вещества, сидерофоры, а также выполняют ряд других специфических функций. Биосинтез этих молекул происходит не на рибосомах, а с помощью специальных ферментов, гены которых образуют кластеры в бактериальных геномах. Мы предположили, что синтез нерибосомных пептидов является специфической особенностью метаболизма бактерий, которая может затрагивать и другие жизненно важные процессы, в том числе и связанные с трансляцией. Нами впервые показана связь между механизмом регуляции трансляции белоккодирующих генов в бактериях, который в значительной степени определяется эффективностью элонгации трансляции, и наличием в геномах кластеров генов биосинтеза нерибосомных пептидов. Проведен биоинформатический анализ эффективности элонгации трансляции белоккодирующих генов 11679 геномов бактерий, часть из которых содержала кластеры генов биосинтеза нерибосомных пептидов, а другая часть – нет. Показано, что бактерии, геномы которых содержат кластеры биосинтетических генов нерибосомных пептидов, и бактерии, которые не содержат кластеры таких генов, имеют значимые различия в молекулярных механизмах, обеспечивающих эффективность трансляции. Так, существенно меньшая часть микроорганизмов, геномы которых содержат кластеры генов нерибосомных пептидных синтетаз, характеризуется оптимизированной регуляцией количества локальных инвертированных повторов, большая же часть имеет геномы, оптимизированные за счет усредненной энергии шпилек инвертированных повторов в мРНК и дополнительно за счет состава кодонов. Полученные нами результаты позволяют предположить, что присутствие путей биосинтеза нерибосомных пептидов может влиять на структуру общего метаболизма бактерий, что выражается и в специфике механизмов рибосомного биосинтеза белков.
ВВЕДЕНИЕ
Нерибосомные пептиды (НРП) составляют важную фракцию бактериальных пептидомов. Будучи классом вторичных пептидных метаболитов, НРП обладают чрезвычайно широким спектром биологической активности и фармакологических свойств. Согласно базе данных Norine [1], НРП действуют как антибиотики (61%), токсины (17%), поверхностно-активные вещества (16%), сидерофоры (11%), противоопухолевые агенты (4%) и модификаторы иммунного ответа (4%); при этом ~25% НРП, включенных в базу данных, обладают несколькими видами активности. Биосинтез НРП является модульным, в его основе лежит использование особых ферментов – синтетаз нерибосомных пептидов (НРПС), которые кодируются кластерами генов в бактериальных геномах [2]. Эти ферменты достаточно хорошо аннотированы, что позволяет выявлять присутствие их генов в геномах бактерий биоинформатическими методами на основе сравнения аминокислотных последовательностей с профилями скрытых цепей Маркова (Hidden Markov Models) [3]. Это позволило на основе биоинформатического анализа большого количества геномов провести аннотацию кластеров генов биосинтеза НРП и описать их функциональные особенности [4].
Эффективность экспрессии генов играет решающую роль в синтетической биологии и геномной инженерии. Хотя на экспрессию генов бактерий влияют несколько процессов (транскрипция, трансляция, посттрансляционная модификация и др.), уровень их экспрессии в основном определяется эффективностью элонгации трансляции [5, 6].
Для того чтобы провести необходимые эксперименты по определению уровня экспрессии генов в интересующем организме, нужно затратить немало финансовых и временных ресурсов. Методы биоинформатики позволяют в первом приближении решить эту проблему, оценив уровень экспрессии гена в данном организме на основе анализа его нуклеотидного состава.
Эффективность элонгации трансляции – это характеристика “оптимальности” нуклеотидной последовательности генов: чем активнее происходит экспрессия указанных генов, тем выше индекс эффективности элонгации (EEI – Elongation Efficiency Index) [5, 6]. Оптимизация может проходить по частотам используемых кодонов, минимизации количества и “прочности” потенциальных шпилек на мРНК и комбинации этих параметров. Программа EloE [7, 8] – это инструмент ранжирования генов на основе предполагаемой эффективности элонгации трансляции их мРНК, определяемой по нуклеотидным последовательностям, с учетом таких факторов, как состав кодонов, наличие и стабильность вторичных структур в мРНК [5, 6]. Полученные предсказанные значения коррелируют с экспериментальными данными по экспрессии генов у различных микроорганизмов [7, 8]. Таким образом, EloE – это биоинформатический инструмент для аннотации генома, позволяющий исследователю на основе только нуклеотидных последовательностей всего генома выводить априорные оценки эффективности экспрессии генов. В настоящей работе мы провели оценку значений EEI для генов, кодирующих НРПС.
Анализ эффективности элонгации трансляции мРНК генов НРПС является важным шагом на пути к получению знаний о свойствах бактериального биосинтеза НРП, а также эволюции этих белков в различных организмах.
В нашей работе впервые проведен поиск связи между механизмом регуляции трансляции мРНК белоккодирующих последовательностей в бактериях, которая в значительной степени определяется эффективностью стадии элонгации и присутствием в геномах кластеров генов биосинтеза НРП.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Мы провели биоинформатический анализ кластеров биосинтетических генов (BGCs) НРП, полученных из ANTISMASH-DB [9], используя полногеномные последовательности бактериальных геномов, доступные в NCBI GenВank. В основе анализа лежит метод прогнозирования эффективности элонгации трансляции, реализованный в программе EloE [7, 8]. Скрипты статистического и биоинформатического анализа разработаны на языках Python и R с использованием программной библиотеки Biopython и пакета robCompositions [10].
Данные о геномах бактерий, содержащих кластеры генов биосинтеза НРП, взяты из ANTISMASH-DB [9]. В качестве общей выборки бактериальных геномов использовали данные о номерах доступа геномных проектов, содержащихся в базе данных геномов Joint Genome Institute (JGI GOLD) [11] со статусом ‘Complete and Published’. На основе номеров доступа из NCBI GenВank получены полногеномные последовательности геномов бактерий для проведения дальнейшего биоинформатического анализа.
ИНДЕКС ЭФФЕКТИВНОСТИ ЭЛОНГАЦИИ ТРАНСЛЯЦИИ EEI
В основе работы лежит алгоритм расчета индекса эффективности элонгации трансляции EEI, разработанный В.А. Лихошваем и Ю.Г. Матушкиным [5, 6]. Этот индекс рассчитывается для каждого гена организма и имеет смысл средней скорости прохождения стадии элонгации трансляции.
Индекс EEI рассчитывается по следующей формуле:
(1)
${\text{EEI}}\left( i \right) = {K \mathord{\left/ {\vphantom {K {\left( {{{w}_{1}}{{T}_{a}}\left( i \right) + {{w}_{2}}{{T}_{e}}\left( i \right)} \right)}}} \right. \kern-0em} {\left( {{{w}_{1}}{{T}_{a}}\left( i \right) + {{w}_{2}}{{T}_{e}}\left( i \right)} \right)}},$а) w1 = 1, w2 = 0 – учитывается только слагаемое Ta(i), которое имеет смысл среднего времени размещения в A-сайте рибосомы изоакцепторной аминоацил-тРНК;
б) w1 = 0, w2 = 1 – учитывается только слагаемое Te(i), которое имеет смысл среднего времени, затрачиваемого рибосомой на стадию транслокации;
в) w1 = 1, w2 = 1 – учитываются оба слагаемых Ta(i) и Te(i).
УЧЕТ КОДОННОГО СОСТАВА ГЕНА ПРИ РАСЧЕТЕ ИНДЕКСА EEI
Первое слагаемое Ta имеет смысл среднего времени размещения в A-сайте рибосомы изоакцепторной аминоацил-тРНК. Чем выше концентрация изоакцепторной аминоацил-тРНК, тем быстрее комплементарная тРНК попадает в A-сайт рибосомы. Концентрации тРНК пропорциональны концентрациям соответствующих кодонов в выборке высокоэкспрессирующихся генов. Параметр Ta вычисляют на основе анализа состава кодонов гена по следующей формуле:
(2)
${{T}_{a}}\left( i \right) = \sum\limits_{j = 1}^{{{n}_{i}}} {{{{{\beta }_{{\delta \left( {i,~j} \right)}}}} \mathord{\left/ {\vphantom {{{{\beta }_{{\delta \left( {i,~j} \right)}}}} {{{n}_{i}}}}} \right. \kern-0em} {{{n}_{i}}}}} ,$(3)
${{\beta }_{{\delta \left( {i,~j} \right)}}} = \frac{{\sum\limits_{m = 1}^C {\sqrt {{{\alpha }_{m}}} } }}{{\sqrt {{{\alpha }_{{\delta \left( {i,~j} \right)}}}} }},$ВТОРИЧНЫЕ СТРУКТУРЫ В мРНК
Второе слагаемое Te(i) имеет смысл среднего времени, затрачиваемого рибосомой на стадию транслокации. Этот параметр вычисляется на основе оценки самокомплементарности i-ой мРНК по следующей формуле:
(4)
${{T}_{e}}\left( i \right) = {{t}_{{{\text{min}}}}}(1--p\left( i \right)) + {{t}_{{{\text{max}}}}}p\left( i \right),$(5)
$p\left( i \right) = ~\mathop \smallint \limits_0^{{\text{LCI}}\left( i \right)} \frac{{{{k}^{{n + 1}}}{{x}^{n}}}}{{G\left( {n + 1} \right)}}{{e}^{{ - kx}}}dx,$ИНДЕКС ЛОКАЛЬНОЙ КОМПЛЕМЕНТАРНОСТИ
Индекс локальной комплементарности (LCI) отражает насыщенность нуклеотидной последовательности мРНК вторичными структурами. При расчетах используют два типа индекса LCI: без энергии (LCI1) и с энергией (LCI2). Первый тип основан на предположении, что рибосома последовательно расплетает вторичную структуру независимо от ее свободной энергии. Второй тип предполагает, что время задержки рибосомы перед стабильной вторичной структурой может определяться свободной энергией этой структуры.
Индекс LCI1 (без энергии) рассчитывается по следующей формуле:
(8)
${\text{LCI}}1\left( i \right) = \frac{{\sum\limits_{m = 1}^{{{m}_{i}} - {{s}_{{\max }}} - {{l}_{{\max }}}} {\left\{ {\sum\limits_{s = {{s}_{{\min }}}}^{{{s}_{{\max }}}} {\left[ {\sum\limits_{l = {{l}_{{\min }}}}^{{{l}_{{\max }}}} {\zeta \left( {con\left( {m,~m + s + 1} \right),~\overline {con\left( {m + s + l - 1,~m + 2s + l - 2} \right)} } \right)} } \right]} } \right\}} }}{{{{m}_{i}} - 2{{s}_{{\max }}} - {{l}_{{\max }}} + 1}},$Индекс LCI2 (с энергией) рассчитывается по формуле:
(9)
${\text{LCI}}2\left( i \right) = \frac{{\sum\limits_{m = 1}^{{{m}_{i}} - {{s}_{{\max }}} - {{l}_{{\max }}}} {\left\{ {\sum\limits_{s = {{s}_{{\min }}}}^{{{s}_{{\max }}}} {\left[ {\sum\limits_{l = {{l}_{{\min }}}}^{{{l}_{{\max }}}} {\psi \left( {con\left( {m,~m + s + 1} \right),~\overline {con\left( {m + s + l - 1,~m + 2s + l - 2} \right)} } \right)} } \right]} } \right\}} }}{{{{m}_{i}} - 2{{s}_{{\max }}} - {{l}_{{\max }}} + 1}},$ПЯТЬ ТИПОВ EEI
Как отмечено выше, всего имеются три нетривиальные комбинации индикаторных коэффициентов w1 и w2 в формуле расчета EEI. Также в расчетах используются два типа индекса LCI. В итоге получаются пять типов индекса EEI:
а) EEI1 = K/Ta – учитывается только кодонный состав гена;
б) EEI2 = K/Te(LCI1) – учитывается только количество вторичных структур в мРНК;
в) EEI3 = K/Te(LCI2) – учитывается только энергетическая прочность вторичных структур в мРНК;
г) EEI4 = K/(Ta + Te(LCI1)) – учитываются и кодонный состав, и количество вторичных структур в мРНК;
д) EEI5 = K/(Ta+Te(LCI2)) – учитываются и кодонный состав, и энергетическая прочность вторичных структур в мРНК.
ОПРЕДЕЛЕНИЕ РАБОЧЕГО ТИПА ИНДЕКСА EEI В ОРГАНИЗМЕ
Для определения типа индекса, лучше всего оценивающего эффективность элонгации трансляции алгоритмом EloE [7, 8], в отсортированных списках значений каждого из пяти индексов выделяют гены рибосомных белков и рассчитывают их среднее положение (M) и стандартное отклонение от среднего (R) по формулам:
(11)
$R = ~\sqrt {\frac{1}{{{{N}_{{rib}}}}}\mathop \sum \limits_{i = 1}^{{{N}_{{rib}}}} {{{\left( {M - {{x}_{i}}} \right)}}^{2}}} ,$Гены рибосомных белков выбраны в качестве маркеров высокоэкспрессирующихся генов, так как известно, что рибосомные гены большинства одноклеточных организмов характеризуются высоким уровнем экспрессии. В частности, в работе [13], где введены индексы: RSCU (Relative Synonymous Codon Usage), отражающий частоту использования синонимичных кодонов в эталонной выборке, и CAI (Codon Adaptation Index), отражающий степень неравномерности кодонного состава гена. Эталонная выборка включала специально выбранные гены с заведомо высоким уровнем экспрессии (рибосомные белки, белки внешнего мембранного слоя и др.).
Основным для каждого организма считается тот тип индекса EEI, у которого параметр M принимает наибольшее значение, а параметр R – наименьшее, т.е. рибосомные гены больше смещены в сторону высокоэкспрессирующихся генов и расположены плотнее. Например, у Herpetosiphon aurantiacus DSM 785 основным является 1-ый тип индекса EEI, так как он показывает более высокий уровень экспрессии рибосомных генов, чем остальные четыре (рис. 1).

РАСЧЕТЫ EEI ДЛЯ ГЕНОВ СИНТЕТАЗ НЕРИБОСОМНЫХ ПЕПТИДОВ
Нами изучены особенности трансляции генов, кодирующих НРПС. Мы оценивали эффективность элонгации трансляции мРНК этих генов на основе индекса EEI. Величины EEI позволяют оценить важность экспрессии белоккодирующих генов для жизнедеятельности микроорганизма: чем выше этот индекс, тем активнее происходит экспрессия гена [5, 6]. Для расчета EEI по нуклеотидной последовательности в геноме используется комплекс программ EloE [7, 8]. Индекс рассчитывают на основе частот встречаемости кодонов в гене и локальной вторичной структуры мРНК. Однако не выявлено никаких характерных особенностей значений EEI для этих генов на фоне остальных генов микроорганизмов.
Мы провели анализ типов индексов EEI и оценили закономерности их распределения в бактериях, которые содержат/не содержат кластеры генов НРПС. Программа EloE [7, 8] на основе анализа полного набора генов в геноме бактерии определяет индекс, оптимальный для этого организма, один из пяти возможных (EEI1, EEI2, EEI3, EEI4, EEI5). Каждый из этих типов характеризует связанные с трансляцией процессы в геноме: EEI1 учитывает только кодонный состав рамки трансляции, влияние локальной вторичной структуры мало; EEI2 – эффективность трансляции определяется присутствием локальных инвертированных повторов; EEI3 – учитывает усредненную энергию повторов, в которых энергетически возможно образование шпильки; EEI4 – комбинация вкладов EEI1, EEI2; EEI5 – комбинация вкладов EEI1, EEI3.
Для оценки связи между кластерами генов, кодирующих НРПС, в геноме бактерий и типом оптимизации элонгации трансляции использовали 2191 геном бактерий, содержащих НРП (5676 кластеров). Предварительный статистический анализ показал, что большое число (>90) генов в кластерах часто встречается в геномах представителей рода Streptomyces (шесть кластеров), большое число кластеров (>2) найдено в геномах представителей родов Mycobacterium (12), Streptomyces (12), Myxococcus (5), Paenibacillus (5), Xenorhabdus (4), Nocardia (3) и Rhodococcus (3).
В процессе анализа оказалось, что у ряда геномов один из определяющих параметров алгоритма выбора оптимального индекса эффективности элонгации трансляции – средний ранг генов рибосомных белков (M-значение) – оказался низким, т.е. смещение генов рибосомных белков в сторону высоких значений EEI относительно других генов было незначительным. Это не позволяет с уверенностью отнести данные геномы к одному из пяти типов и может сказываться на значимости результатов поиска связи между EEI и наличием кластеров генов НРПС. Поэтому, чтобы исключить влияние таких геномов на оценку EEI и обеспечить устойчивость проведенного анализа, мы пересчитали результаты, полученные на предыдущем этапе, отфильтровав геномы с низким индексом, т.е. эти геномы не брали в дальнейшие расчеты. Были рассмотрены два порога качества – базовый (M ≥ 50) и более строгий (M ≥ 75), для которых произвели пересчет полученных результатов. После фильтрации по базовому порогу качества (M ≥ 50) было взято 1855 геномов, содержащих гены НРПС, из 10111, прошедших фильтрацию и проанализированных EloE. Также взято 8256 геномов бактерий без генов НРПС. После фильтрации по строгому порогу качества (M ≥ 75) в анализ взяли 1473 генома с генами НРПС. Геномов бактерий без генов НРПС взяли 5960.
Выборки организмов с генами синтетаз НРП и без НРП анализировали методом главных компонент для композиционных данных [14], полученных в ходе процедуры генерации повторных выборок (ресэмплинга) организмов из групп двух типов – содержащих НРП и не содержащих НРП – и представляющих собой композицию частот оптимальных типов индексов элонгации трансляции в этих выборках. Результаты отображаются в виде диаграммы распределения выборок в пространстве главных компонент.
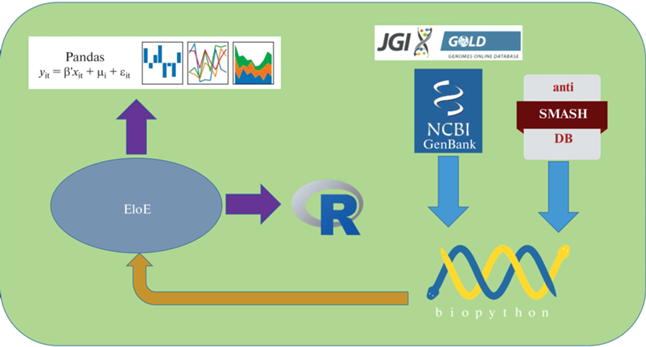
Общая схема обработки данных представлена на рис. 2.
Рис. 2.
Общая схема биоинформатического анализа. По данным из ANTISMASH-DB о кластерах НРПС из NCBI GenBank с использованием библиотеки Biopython были загружены последовательности кластеров и геномы соответствующих видов бактерий, а также проведен предварительный статистический анализ. Далее с помощью EloE теоретически оценена эффективность элонгации трансляции всех генов из геномов бактерий как содержащих кластеры НРПС, так и без этих кластеров, и проведен статистический анализ с использованием библиотеки Pandas языка программирования Python и библиотеки robCompositions статистического пакета R.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
На основе данных об аннотированных геномах мы провели анализ оптимальных индексов эффективности элонгации трансляции (EEI) и оценили распределение геномов по типам оптимального индекса в группах бактерий, которые содержат/не содержат кластеры генов НРПC. Полученные результаты свидетельствуют, что организмы, геномы которых кодируют/не кодируют НРПC, имеют существенно различные частоты встречаемости оптимальных типов элонгации трансляции EEI2, EEI5 и EEI3 (см. рис. 3).
Рис. 3.
Сравнение разных групп геномов бактерий по композициям типов EEI. а – Геномы, содержащие кластеры генов нерибосомных пептидсинтетаз (НРПС), согласно информации из ANTISMASH-DB; б – геномы, не содержащие кластеры генов НРПС, согласно информации из ANTISMASH-DB. в – Все геномы бактерий, геномные проекты которых имеют статус “Complete and Published” согласно JGI GOLD.

Как видно из диаграмм, выборка организмов, которые содержат кластеры генов НРПС, существенно отличается от выборки без кластеров НРПС по EEI2 (5% с НРПС против 12% без НРП), EEI3 (14% с НРПС против 10% без НРПС) и EEI5 (17% с НРПС и 13% без НРП). Таким образом, можно сделать вывод, что среди геномов, содержащих кластеры генов НРПС, эффективность трансляции в меньшей степени обуславливается процессами, связанными с ролью инвертированных повторов (EEI2), в то же время большую роль в контроле эффективности трансляции играют процессы, связанные со стабильностью локальной вторичной структуры мРНК (EEI3 и EEI5).
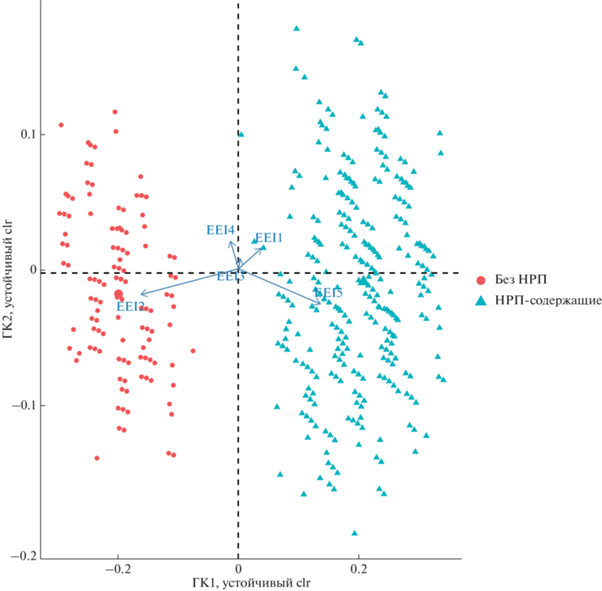
Эти результаты подтверждаются анализом выборок организмов с/без НРП методом главных компонент для композиционных данных [14]. Этот метод основан на генерации повторных выборок из организмов двух типов – содержащих НРП и не содержащих НРП – и анализу частот индексов в этих выборках. Результаты представлены на рис. 4, где каждый круг – выборка геномов, не содержащих НРП, треугольник – выборка геномов, содержащих НРП (выборки сгенерированы случайным независимым образом из исходной выборки геномов бактерий). Видно, что выборки с кластерами генов НРПС и без них существенно расходятся по компонентам, связанным с индексами EEI2 и EEI5.
Рис. 4.
Результаты анализа методом главных компонент (ГК) для композиционных данных (устойчивый, с обратным преобразованием счетов в пространство clr – центрированных отношений логарифмов) [14] множества композиций по типам EEI, полученного в результате ресэмплинга. Кружочками отмечены случайные выборки геномов из родов, принадлежащих к группе геномов, содержащих кластеры генов НРПС, а треугольниками – случайные выборки геномов из родов, принадлежащих к группе геномов, не содержащих НРП.
Мы проверили устойчивость полученных ранее результатов с использованием фильтрации геномов, для которых оптимальный индекс EEI не может быть установлен с заданным порогом качества. Как указано в разделе “Расчеты EEI для генов синтетаз нерибосомных пептидов”, средний ранг генов рибосомных белков (M-значение) оказался низким. Поэтому мы рассмотрели два пороговых значения этого параметра, $M \geqslant 50$ и более строгий $--{\text{\;}}M \geqslant 75$. Геномы, не удовлетворяющие этому критерию, были исключены из анализа.
Результаты анализа для двух порогов фильтрации представлены на рис. 5а, б. Каждый кружок – это выборка геномов, не содержащих НРП, а каждый треугольник – выборка геномов, содержащих НРП. Видно, что эти выборки существенно расходятся по компонентам, связанным с индексами EEI2, EEI5 и EEI3.
Рис. 5.
Результаты анализа методом главных компонент (ГК) для композиционных данных (устойчивый, с обратным преобразованием счетов в пространство clr – центрированных отношений логарифмов) множества композиций по типам EEI, полученного путем ресэмплинга. Кружками отмечены случайные выборки геномов из родов, принадлежащих к группе геномов, НЕ содержащих кластеров генов НРПС, а треугольниками отмечены случайные выборки геномов из родов, принадлежащих к группе геномов, содержащих кластеры генов НРПС. а – Фильтрация по базовому порогу ($M \geqslant 50$); б – фильтрация по строгому порогу ($M \geqslant 75$).

Рис. 5.
Окончание.
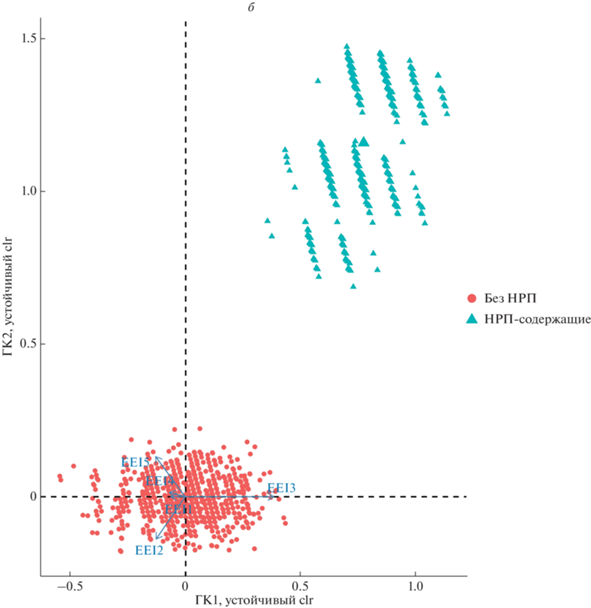
В пространстве первых двух главных компонент группы так же четко кластеризуются (см. рис. 4), как и до фильтрации. Однако стоит отметить, что при фильтрации по базовому ($M \geqslant 50$) и строгому порогу ($M \geqslant 75$) изменяются сами главные компоненты. Если без фильтрации львиная доля (94%) объясненной дисперсии приходилась на первую главную компоненту, складывающуюся преимущественно из долей EEI2 и EEI5, то после фильтрации по строгому порогу ($M \geqslant 75$) на главную компоненту 1 приходится 76% объясненной дисперсии, а на главную компоненту 2, которая преимущественно и различает наши кластеры (и все так же объясняется соотношениями между EEI2 и EEI5) – 14%. Т.е. возросла роль доли геномов с оптимальным типом индекса EEI3 в дисперсии общей выборки.
Наши результаты показывают, что в группах бактерий, различающихся по физиологическому типу (в нашем случае это наличие или отсутствие кластеров генов НРПС), распределение типов EEI может существенно различаться. Эти результаты получены впервые и, по-видимому, свидетельствуют о различиях в метаболизме бактерий, геномы которых кодируют НРПС, и бактерий, которые их не кодируют. Эти различия могут быть связаны с энергетическими процессами (оптимизацией метаболизма), лежащими в основе трансляции РНК и биосинтеза белков.
Гены синтетаз, которые участвуют в синтезе НРП, могут быть связаны с рядом специфических молекулярных процессов, которые отсутствуют у бактерий, не способных синтезировать НРП. Т.е. способность синтезировать НРП сопровождается увеличением доли геномов, оптимизирующих метаболизм с учетом как частот кодонов, так и минимизации количества и прочности шпилек. Поскольку синтез НРП несет энергетические издержки для бактерий, это приводит к необходимости оптимизировать процесс трансляции на уровне всего генома бактерий данной группы.
Проведенный биоинформатический анализ предоставил информацию о распределении кластеров генов биосинтеза НРПС бактерий (а также самих генов), полученном на основе предсказания эффективности элонгации трансляции. Эти распределения могут существенно различаться у разных таксонов. Выделенные кластеры могут служить объектом дальнейшего изучения функциональной роли НРПС, экспрессия которых обеспечивается данными кластерами.
Работа поддержана грантами Российского фонда фундаментальных исследований (17-00-00470 (K) и 17-00-00462). Данные обрабатывали с использованием вычислительных ресурсов ЦКП “Биоинформатика” при поддержке бюджетного проекта № FWNR-2022-0020.
Настоящая статья не содержит каких-либо исследований с использованием животных в качестве объектов.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Caboche S., Pupin M., Leclère V., Fontaine A., Jacques P., Kucherov G. (2008) NORINE: a database of nonribosomal peptides. Nucl. Acids Res. 36, 326–331. https://doi.org/10.1093/nar/gkm792
Süssmuth R.D., Mainz A. (2017) Nonribosomal peptide synthesis – principles and prospects. Angew. Chemie – Int. Ed. 56, 3770–3821.
Kim H.U., Blin K., Lee S.Y., Weber T. (2017) Recent development of computational resources for new antibiotics discovery. Curr. Opin. Microbiol. 39, 113–120.
Blin K., Shaw S., Kautsar S.A., Medema M.H., Weber T. (2021) The antiSMASH database version 3: increased taxonomic coverage and new query features for modular enzymes. Nucl. Acids Res. 49(D1), D639‒D643.
Лихошвай В.А., Матушкин Ю.Г. (2000) Предсказание эффективности экспрессии генов по их нуклеотидному составу. Молекуляр. биология. 34, 406–412.
Likhoshvai V.A., Matushkin Yu.G. (2002) Differentiation of single-cell organisms according to elongation stages crucial for gene expression efficacy. FEBS Lett. 516. 87–92.
Соколов В.С., Зураев Б.С., Лашин С.А., Матушкин Ю.Г. (2014) EloE – веб-приложение для оценки эффективности элонгации трансляции генов. Вавиловский журн. генетики и селекции. 18, 904–909.
Korenskaia A.E., Matushkin Y.G., Lashin S.A., Klimenko A.I. (2022). Bioinformatic assessment of factors affecting the correlation between protein abundance and elongation efficiency in Prokaryotes. Internat. J. Mol. Sci. 23(19), 11996. https://doi.org/10.3390/ijms231911996
Blin K., Medema M.H., Kottmann R., Lee S.Y., Weber T. (2017) The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucl. Acids Res. 45, D555–D559. https://doi.org/10.1093/nar/gkw9601
Filzmoser P., Hron K., Templ M. (2018) Applied compositional data analysis. with worked examples. In: Statistics. Springer Ser., Nature Switzerland AG, Cham, Switzerland. ISBN 978-3-319-96420-1
Mukherjee S., Stamatis D., Bertsch J., Ovchinnikova G., Katta H.Y., Mojica A., Chen I.M.A., Kyrpides N.C., Reddy T.B.K. (2019) Genomes OnLine database (GOLD) v.7: Updates and new features. Nucl. Acids Res. 47(D1), D649–D659. https://doi.org/10.1093/nar/gky977
Turner D.H., Sugimoto N. (1988) RNA structure prediction. Annu. Rev. Biophys. Biophys. Chem. 17, 167–192.
Sharp P.M., Li W.H. (1987) The codon adaptation index – a measure of directional synonymous codon usage bias, and its potential applications. Nucl. Acids Res. 15, 1281–1295.
Filzmoser P., Hron K., Reimann C. (2007) Principal component analysis for compositional data with outliers. Environmetrics. 20, 621–632.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология