

Российские нанотехнологии, 2020, T. 15, № 3, стр. 352-361
СТРУКТУРНЫЕ И ХИМИЧЕСКИЕ ПРЕВРАЩЕНИЯ ЭЛЕКТРОКАТАЛИЗАТОРОВ ПОЛУЧЕНИЯ ВОДОРОДА НА ОСНОВЕ КЛАТРОХЕЛАТОВ РУТЕНИЯ, КОБАЛЬТА И ЖЕЛЕЗА В ЭЛЕКТРОЛИЗЕРЕ ВОДЫ
Я. В. Зубавичус 1, *, С. А. Григорьев 2, 3, 4, А. С. Пушкарев 2, М. М. Борисов 2, М. Г. Бугаенко 5, Я. З. Волошин 5, 6, **, А. Г. Дедов 5, 7
1 Федеральный исследовательский центр “Институт катализа им. Г.К. Борескова СО РАН”
Новосибирск, Россия
2 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
3 Национальный исследовательский университет “МЭИ”
Москва, Россия
4 Центр водородной инфраструктуры, Северо-западный университет
Потчефструм, Южная Африка
5 Российский государственный университет нефти и газа им. И.М. Губкина
Москва, Россия
6 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
7 Институт нефтехимического синтеза им. А.В. Топчиева РАН
Москва, Россия
* E-mail: yzubav@gmail.com
** E-mail: voloshin@ineos.ac.ru
Поступила в редакцию 18.06.2020
После доработки 18.06.2020
Принята к публикации 21.07.2020
Аннотация
С использованием рентгеноабсорбционной спектроскопии XANES/EXAFS (X-ray Absorption Near-edge Structure/Extended X-ray Absorption Fine Structure) изучена структурная эволюция трех электрокаталитических систем получения водорода в электролизере воды с использованием клатрохелатных комплексов железа, кобальта и рутения(II) в качестве катализаторов. Показано, что комплексы характеризуются принципиально различной устойчивостью в условиях проведения электролиза воды в полупромышленных генераторах водорода. В случае клатрохелатов железа и кобальта(II) обнаружены сохранение их клеточной макробициклической структуры и восстановление инкапсулированного иона металла(II) до катиона металла(I), тогда как в случае клатрохелата рутения(II) наблюдается частичное разложение клеточного комплекса с образованием серосодержащих продуктов разложения инкапсулирующего гексасульфидного макробициклического лиганда, что приводит к накоплению дисульфида рутения RuS2 в отработанном клатрохелатсодержащем катодном материале. С использованием полученных экспериментальных данных о химической трансформации клатрохелатных электрокатализаторов в условиях протекания реакции 2Н+/Н2 обсуждаются пути увеличения эффективности электрокаталитических систем на основе этого класса координационных соединений.
ВВЕДЕНИЕ
Значительной технологической задачей для глобального будущего человечества является поиск безопасного, экологически чистого и возобновляемого источника энергии [1]. Если он не будет найден, увеличение спроса на энергию, обусловленное экономическим ростом и ростом населения, вскоре приведет к еще более высоким выбросам углекислого газа. Это воздействие на изменение климата может иметь драматические последствия для всей цивилизации. Водород является потенциальной альтернативой углеродсодержащему ископаемому топливу, если он получен из соответствующего источника. Выброс углеродсодержащих газов может быть значительно снижен, если вода как сырье и солнечный свет как источник энергии будут являться первичными углерод-нейтральными источниками H2 [1]. Однако широкое использование различных электрохимических систем (топливных элементов, электролизеров и т.д.) в водородной энергетике, высокотехнологичных отраслях промышленности, гибридном и водородном транспорте и других областях современной социальной и экономической жизни вызывает существенное увеличение потребления редких и драгоценных металлов, которые в настоящее время используются в качестве электрокатализаторов получения водорода. Их высокая стоимость значительно ограничивает широкое использование указанных выше электрокаталитических систем в водородной энергетике. Использование дешевых и распространенных соединений неплатиновых d-металлов в топливных элементах, электролизерах и электрохимических водородных компрессорах/концентраторах в ядерной энергетике сделает водородные энергетические устройства более доступными, чем когда-либо. Таким образом, одной из важнейших задач современной водородной энергетики является поиск высокоэффективных неплатиновых каталитических систем, позволяющих получать водород из водных растворов. В течение последних десятилетий электрокаталитическое восстановление ионов H+ до молекулярного водорода с использованием молекулярных соединений 3d-переходных металлов (прежде всего комплексов кобальта) в качестве электрокатализаторов широко использовалось [2–12] для получения H2 как экологически чистого топлива. Квазиароматические клатрохелаты d-металлов [13, 14] являются дешевыми, синтетически-доступными и экологически-чистыми соединениями, обладающими как химической, так и термодинамической устойчивостью как минимум в двух степенях окисления их инкапсулированного иона металла, а также в широком диапазоне окислительно-восстановительных потенциалов, pH и температуры. Они продемонстрировали высокую скорость переноса электронов, и их окислительно-восстановительные потенциалы могут быть тонко настроены с использованием функционализирующих заместителей в их инкапсулирющих лигандах; рациональный дизайн клатрохелатных электрокатализаторов позволяет получать комплексы, которые имеют наиболее низкое перенапряжение окислительно-восстановительной реакции 2H+/H2. Использование функционализирующих заместителей с терминальными полиароматическими группами позволяет эффективно иммобилизовать соответствующие клатрохелатные молекулы как электрокатализаторы посредством их сильной физической адсорбции на поверхность углеродных материалов или рабочих электродов, позволяя получать долгоживущие каталитические системы, обладающие высокой эффективностью в реакции получения водорода 2H+/H2. Однако механизм такой каталитический реакции в случае клеточных комплексов кобальта и железа остается до сих пор дискуссионным [15–17]. В случае клатрохелатов кобальта предложенный в [15] механизм их электрокаталитической активности включает либо чисто металл-центрированную активацию ионов H+, либо непосредственное взаимодействие макробициклического лиганда и инкапсулированного иона кобальта при образовании H2. С другой стороны, было однозначно доказано [15, 16], что электрохимическое восстановление этих клатрохелатов кобальта в присутствии Н-кислот приводит к электроосаждению на поверхности электрода кобальтсодержащих наночастиц, которые являются очень активными катализаторами получения H2 из воды. В случае клеточных комплексов железа механизм электрокаталитической реакции 2Н+/Н2 (молекулярный гомогенный или гетерогенный с образованием наночастиц) также неизвестен, но недавние результаты [18] продемонстрировали высокую устойчивость клатрохелата железа(I), полученного химическим восстановлением его железо(II)-содержащего предшественника, в сильнокислых условиях. Такое же химическое поведение обнаружено [18] в случае кобальт(I)-содержащего клатрохелатного аналога [19].
Недавние результаты тестирования показанных на схеме 1 фенилборатных клеточных комплексов железа, кобальта и рутения 1–3, дизайн молекул которых предназначался для их эффективной физической адсорбции на углеродные материалы (прежде всего на углеродную бумагу (УБ)), в электролизерах воды показали [20] их хорошие электрокаталитические характеристики в реакции получения водорода 2H+/H2. Использование подходящих углеродных материалов с большой площадью поверхности, таких как активированный уголь и восстановленный оксид графена, в качестве подложек для их эффективной иммобилизации привело к значительному увеличению электрокаталитической активности соответствующих клатрохелатсодержащих катодов на основе УБ, использованных в электролизере воды [20].

В настоящей работе с использованием синхротронного излучения изучили химический состав как исходных клатрохелатов железа, кобальта и рутения(II), так и катодных наноматериалов с иммобилизированными на УБ комплексами 1–3 как электрокатализаторами после их работы в электролизере воды с целью установить природу их каталитически-активных центров.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и методы. Показанные на схеме 1 клатрохелатные комплексы железа, кобальта и рутения(II) Fe((Pntr(CH2)3S)2Gm)3(BC6H5)2 (1), Co((Pntr(CH2)3S)2Gm)3(BC6H5)2 (2) и Ru((Pntr(CH2)3S)2Gm)3(BC6H5)2 (3) (где Gm – остаток глиоксима) получены с высокими выходами по описанным ранее методикам [21].
Для приготовления катодов использовали УБ марки Sigracet® GDL 39 BC (SGL Group, Германия). Иммобилизация упомянутых выше клатрохелатов путем физической адсорбции на полученные из УБ диски была осуществлена в закрытых стеклянных бюксах без какого-либо перемешивания или встряхивания. УБ-диск диаметром 30 мм погружали в 0.5 мМ раствор данного клатрохелатного комплекса в смеси ацетонитрил–хлористый метилен 2:1 на 24 ч. Затем записывали электронные абсорбционные спектры полученного раствора и вычисляли равновесную концентрацию клатрохелата, используя классическое уравнение A = εlcClt. Это позволило получить величины удельной адсорбции G, равные 1.454 × 10–5, 1.370 × 10–5 и 1.180 × 10–5 моль г–1 для комплексов 1–3 соответственно. Затем полученные модифицированные клатрохелатами УБ-диски были высушены на воздухе до достижения ими постоянной массы и были использованы в качестве катодов электролизера воды [22].
В качестве анодного катализатора использовали наноструктурную иридиевую чернь, полученную методом химического восстановления H2IrCl6 ⋅ 6H2O [23]. Суспензию катализатора в изопропиловом спирте с добавкой протонообменного полимера Nafion® (Ion Power, США) наносили на поверхность газодиффузионного электрода (пористый титан ВТ-1-0 толщиной 0.9 мм с пористостью 28% [24]) методом распыления в потоке воздуха [25]. Плотность нанесения анодного катализатора составила 2 мг см−2.
Используя мембрану Nafion® 117 (Chemours Company, США) в качестве твердого полимерного электролита и предварительно изготовленные, как описано выше, электроды, осуществляли сборку мембранно-электродного блока (МЭБ). Катод, анод и разделяющую их мембрану закрепляли в лабораторной электролизной титановой ячейке, состоящей из двух термостатируемых и стянутых болтами полуячеек с интегрированной системой каналов для подвода и отвода реагентов и продуктов [26]. При этом в случае клатрохелатсодержащих катодов дополнительно между электродом и полуячейкой помещали титановый газодиффузионный электрод для того, чтобы предотвратить повреждение электрода из УБ при затягивании болтов и сжатии МЭБ [22]. Спекание МЭБ осуществляли в рабочих условиях при температуре 80°C в течение 1 ч без подачи тока, а затем при постоянной плотности тока 0.5–1.0 А/см2 и периодическом контроле вплоть до достижения стабильной поляризационной характеристики. Все эксперименты проводили при температуре 80°C и анодной подаче воды по принципу “газ–лифт”.
Поляризационную характеристику каждого из собранных МЭБ регистрировали периодически в потенциостатическом ступенчатом режиме в диапазоне напряжений от 1.5 до 2.2 В с шагом 0.05–0.10 В. Переход к следующей величине напряжения осуществляли только после установления стационарной величины тока при данной величине напряжения (в течение 3–10 мин). После завершения каждого из экспериментов отработавшую ячейку разбирали, а составляющие МЭБ (анодный и катодный газодиффузионные электроды и мембрану с частично-связанными с ней каталитическими слоями) отделяли друг от друга и изучали различными физическими методами (прежде всего с использованием синхротронного излучения).
Спектры EXAFS и XANES на соответствующих К-краях поглощения для исходных клатрохелатов 1–3 и таковые УБ-катодов с иммобилизированными комплексами после их электрохимического тестирования 1'–3' были измерены на Курчатовском источнике синхротронного излучения. Спектры исходных комплексов 1–3 и рутенийсодержащего образца 3' были измерены на станции “Структурное материаловедение” [27] в геометрии пропускания с использованием ионизационных камер, заполненных смесями азот–аргон, обеспечивающими 20- и 80%-ное поглощение для I0 и It соответственно (камеры, расположенные перед и за образцом). Спектры образцов 1' и 2' ввиду низкой концентрации анализируемого металла были измерены в режиме регистрации выхода рентгеновской флуоресценции на станции “ФАЗА” [28]. Для регистрации интенсивности рентгеновской флуоресценции линий FeKα и CoKα использовали четырехэлементный полупроводниковый детектор XFlash (Bruker). Для обработки экспериментальных спектров использовали пакеты программ IFEFFIT [29] и FEFF [30]. Путем многосферной нелинейной подгонки уточняли величины межатомных расстояний R, таковые параметров Дебая–Валлера (σ2) и третьих кумулянт (о физическом смысле третьей кумулянты см. ниже). Величины шкальных множителей S02, учитывающих ненулевую вероятность многоэлектронных процессов, уточняли для спектров реперных соединений с известной структурой (соответствующие металлические фольги) и фиксировали в дальнейших уточнениях.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Экспериментальные спектры XANES на К‑краях Fe и Co для исходных клатрохелатных комплексов 1 и 2, а также отработанных УБ-катодов на их основе 1' и 2' приведены на рис. 1. В обоих случаях наблюдались однотипные изменения спектров, выражающиеся в незначительном сдвиге спектрального максимума в сторону меньших энергий. Как правило, такой сдвиг указывает на химическое восстановление, т.е. уменьшение степени окисления инкапсулированного иона металла. Отметим, что продуктом восстановления не может являться нульвалентный металл, поскольку в таком случае ожидаемый энергетический сдвиг должен быть более значительным: в случае Fe(0) интенсивный рост коэффициента рентгеновского поглощения должен наблюдаться начиная с энергии 7110–7115 эВ, а в случае Co(0) – с энергии 7707–7712 эВ. В экспериментальных спектрах в этих энергетических интервалах наблюдается лишь малоинтенсивный предпик, соответствующий дипольно-запрещенному электронному переходу 1s → 3d. Ранее путем химического восстановления были получены и структурно охарактеризованы методом рентгеноструктурного анализа (РСА) клатрохелаты кобальта(I) [19, 31, 32] и железа(I) [18].
Рис. 1.
Экспериментальные спектры XANES для исходных клатрохелатных комплексов 1 и 2, а также электрокаталитических материалов 1' и 2' на их основе после испытаний в электролизере воды.
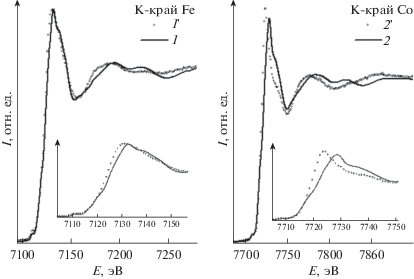
Кроме того, в полученных экспериментально спектрах наблюдается некоторое уменьшение энергетического зазора между спектральным максимумом и максимумом первой осцилляции коэффициента поглощения, которое может указывать на увеличение межатомного расстояния от инкапсулированного иона металла до легкого атома азота в его первой координационной сфере. Более детальная информация об изменении локального окружения инкапсулированных ионов железа и кобальта в клатрохелатсодержащих электрокаталитических наноматериалах в процессе электролиза воды в электрохимической ячейке былa полученa из количественного анализа спектров EXAFS.
Фурье-трансформанты спектров EXAFS для пар образцов 1–1' и 2–2' приведены на рис. 2 (левая и правая панели соответственно). Из приведенных изображений видно, что в отработанных железо- и кобальтсодержащих УБ-катодах сохраняется координационное окружение инкапсулированного иона металла легкими атомами, хотя и наблюдаются существенные изменения параметров локальной структуры. Отметим, что для целого ряда клатрохелатных комплексов железа и кобальта(II), аналогичных 1 и 2, по данным РСА характерен достаточно широкий разброс межатомных расстояний [33–35]. Другими словами, координационный полиэдр инкапсулированного иона металла достаточно далек от идеальной октаэдрической Oh-конфигурации. Его идеальный координационный полиэдр должен соответствовать тригональной призме с симметрией D3h или тригональной антипризме с симметрией D3, характеризующимся шестью равными расстояниями M–N (M = Fe, Co; рис. 3); также должен наблюдаться ряд более дальних межатомных расстояний M…C, М…O, M…S и M…B. Однако экспериментальные кристаллографические данные указывают на значительный разброс длин связей металл–донорный атом азота лиганда. Характер этого разброса (два коротких расстояния–четыре длинных, четыре коротких–два длинных, два коротких–два средних–два длинных и пр.) зависит от температуры и спинового состояния этого инкапсулированного катиона металла. Кроме того, в случае катионов металлов с ян–теллеровским структурным искажением реальный разброс межатомных расстояний в первой координационной сфере может достигать 0.2–0.3 Å. Эта структурная особенность изучаемых клатрохелатов и систем на их основе представляет методическую сложность для количественной обработки соответствующих спектров EXAFS, поскольку в классической теории EXAFS предполагается, что распределение межатомных расстояний является нормальным (гауссовым), и его ширина определяется лишь амплитудой тепловых колебаний атомов. Попытки описать полученные экспериментально спектры EXAFS для исходных комплексов 1 и 2 (в случае которых доступны результаты низкотемпературного РСА монокристаллов аналогичных клатрохелатов железа и кобальта(II)) не увенчались успехом. Относительно качественного описания экспериментальной кривой удалось добиться с использованием так называемого кумулянтного разложения, разработанного для систем с сильным ангармонизмом тепловых колебаний [36–38]. Суть подхода заключается в том, что в дополнение к стандартным структурным параметрам R (межатомное расстояние) и σ2 (параметр Дебая–Валлера, квадрат среднеквадратичного отклонения межатомного расстояния) для уточнения в такой модели используются более высокие моменты функции распределения межатомных расстояний (в частности, третья кумулянта, описывающая степень асимметрии (skewness)). Положительная величина третьей кумулянты соответствует асимметрии распределения межатомных расстояний с “хвостом” в сторону больших расстояний и наоборот.
Рис. 2.
Фурье-трансформанты спектров EXAFS для пар образцов 1–1' и 2–2': экспериментальные (сплошные линии) и оптимизированные теоретические кривые (кружочки). Наборы параметров локального окружения ионов железа и кобальта, соответствующие этим моделям, приведены в табл. 1 и 2.

Рис. 3.
Ожидаемое локальное окружение ионов d-металлов в клатрохелатных комплексах 1–3 (по данным РСА).
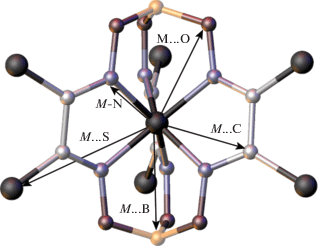
Параметры, полученные в ходе оптимизации модели, включающей в себя вклады координационных сфер M–N, M…C и M…O (M = Fe, Co; рис. 3), для исходных комплексов 1 и 2, а также электрокаталитических материалов 1' и 2' на их основе после электрохимических испытаний в электролизере воды приведены в табл. 1 и 2. Результаты уточнения свидетельствуют о том, что характерный набор межатомных расстояний, соответствующий клатрохелатному остову, сохраняется в ходе электрохимических превращений в электролизере воды, однако все эти расстояния систематически увеличиваются. Кроме того, в случае клеточного комплекса железа ширина распределения межатомных расстояний скачкообразно увеличивается при переходе от 1 к 1', а в случае клатрохелата кобальта, наоборот, уменьшается при переходе от 2 к 2'. Эти изменения, а также обсуждаемые выше изменения в спектрах XANES позволяют с высокой долей вероятности утверждать, что в ходе электрохимических испытаний в электролизере воды общая макробициклическая структура клатрохелатных комплексов железа и кобальта сохраняется, но наблюдается полное (или практически полное в пределах чувствительности метода) восстановление их инкапсулированных ионов металл(II) до катионов металл(I). Именно для клатрохелатных комплексов Fe и Co хорошо известен [19, 31, 32, 18 ] эффект необычной стабилизации их восстановленных металл(I)-содержащих клеточных комплексов, допускающий достаточно простые химические подходы к изменению степени окисления (II) ↔ (I) инкапсулированного иона металла. Увеличение ширины распределения межатомных расстояний указывает на переход иона 3d-металла в ян–теллеровски-искаженное состояние: в случае комплекса железа это продукт восстановления 1' с инкапсулированным ионом железа(I); а в случае клатрохелата кобальта – инкапсулированный ион кобальта(II) в исходном комплексе 2. Ранее наблюдались аналогичные эффекты в спектрах XANES и EXAFS клатрохелатных комплексов кобальта и железа(I), в случае которых степень окисления инкапсулированного иона металла была надежно установлена (в том числе с использованием магнитных измерений и данных РСА [32]).
Таблица 1.
Параметры локального окружения ионов кобальта в исходном комплексе 2 и электрокаталитическом материале 2' на его основе после электрохимических испытаний (по данным EXAFS)
| Образец | $S_{0}^{2}$ | Координацонная сфера | N | R, Å | σ2, Å2 | Третья кумулянта | ΔE, эВ |
Диапазон по R, Å | Диапазон по k, Å–1 | Rf, % |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.72 | Fe–N | 6 | 1.89 | 0.0018 | –0.0005 | – | 1.1–3.1 | 2.0–12.0 | 2.4 |
| Fe…C | 6 | 2.83 | 0.0035 | –0.0012 | 1.5 | |||||
| Fe…O | 6 | 2.98 | 0.0109 | 0.0044 | ||||||
| 1' | 0.72 | Fe–N | 6 | 2.03 | 0.0085 | 0.0005 | 4.6 | 1.1–3.1 | 2.0–12.0 | 1.4 |
| Fe…C | 6 | 3.14 | 0.0058 | 0.0061 | ||||||
| Fe…O | 6 | 3.45 | 0.0147 | 0.0027 |
Таблица 2.
Параметры локального окружения ионов кобальта в исходном комплексе 2 и электрокаталитическом материале 2' на его основе после электрохимических испытаний (по данным EXAFS)
| Образец | $S_{0}^{2}$ | Координационная сфера | N | R, Å | σ2, Å2 | Третья кумулянта | ΔE, эВ |
Диапазон по R, Å | Диапазон по k, Å–1 | Rf, % |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 0.72 | Co–N | 6 | 2.07 | 0.0066 | 0.0027 | 7.5 | 1.1–3.1 | 2.0–12.0 | 0.3 |
| Co…C | 6 | 2.74 | 0.0122 | 0.0193 | ||||||
| Co…O | 6 | 2.94 | 0.0096 | 0.0007 | ||||||
| 2' | 0.72 | Co–N | 6 | 2.10 | 0.0088 | –0.0008 | 4.4 | 1.1–3.1 | 2.0–12.0 | 2.8 |
| Co…C | 6 | 2.80 | 0.0009 | 0.0015 | ||||||
| Co…O | 6 | 3.15 | 0.0005 | 0.0063 |
Спектры XANES и фурье-трансформанты EXAFS на К-крае рутения для пары образцов 3 и 3' приведены на рис. 4. Изменения в спектре XANES (левая панель) предполагают, что в отработанном электрокаталитическом материале 3' наряду с частичным сохранением исходного состояния инкапсулированных ионов рутения появляется дополнительная компонента, максимум поглощения которой сдвинут вправо, что соответствует формальному окислению этого иона (т.е. увеличению степени окисления иона рутения, несмотря на протекание восстановительного процесса в электролитической ячейке). Значительные изменения, затрагивающие даже первую координационную сферу, наблюдаются и в EXAFS этого образца. При этом в ближайшем окружении иона рутения появляются атомы более тяжелые, чем атомы азота. Вероятно, это атомы серы, содержавшиеся изначально в инкапсулирующем клатрохелатном лиганде комплекса 3. Как результат, уточняемая модель для интерпретации этого EXAFS была составлена из четырех координационных сфер, характерных для исходного комплекса 3 (Ru–N, Ru…C, Ru…O и Ru…S, рис. 3), а также включала в себя координационные сферы Ru–S и Ru...Ru, характерные для сульфида рутения RuS2. Соответствующие пути рассеяния были добавлены в модель с фиксированными координационными числами; варьировался только параметр, соответствующий мольной доле исходного комплекса 3. Результаты анализа приведены в табл. 3. Высокая стабильность при уточнении, а также близкое соответствие уточненных величин межатомных расстояний ожидаемым, подтвердили такую интерпретацию. Полученные экспериментальные данные свидетельствуют о том, что в ходе электрохимических испытаний электрокаталитического наноматериала на основе клатрохелатного комплекса рутения 3 претерпевает редокс-реакции с частичным химическим превращением его гексасульфидного макробициклического лиганда, которое приводит к образованию RuS2. По результатам уточнения соотношение исходный комплекс 3/RuS2 в образце 3' составляет приблизительно 2.6 : 1 (72 : 28%, табл. 3). Отметим, что, как и в случаях с клатрохелатными комплексами железа и кобальта, ожидаемого катодного восстановления инкапсулированного иона рутения до металлического состояния Ru(0) не наблюдается.
Рис. 4.
Экспериментальные спектры XANES (слева) и фурье-трансформанты спектров EXAFS для пары образцов 3–3': экспериментальные (сплошные линии) и оптимизированные теоретические кривые (кружочки). Наборы параметров локального окружения ионов рутения, соответствующие этим моделям, приведены в табл. 3.

Таблица 3.
Параметры локального окружения ионов рутения в исходном комплексе 3 и электрокаталитического материала 3' на его основе после электрохимических испытаний (по данным EXAFS)
| Образец | $S_{0}^{2}$ | Координационная сфера | N | R, Å | σ2,Å2 | ΔE, эВ | Диапазон по R, Å | Диапазон по k, Å–1 | Rf, % |
|---|---|---|---|---|---|---|---|---|---|
| 3 | 0.96 | Ru–N | 6 | 1.98 | 0.0043 | –5.5 | 1.1–4.2 | 2.0–12.0 | 2.2 |
| Ru…C | 6 | 2.91 | 0.0015 | ||||||
| Ru…O | 6 | 2.98 | 0.0060 | ||||||
| Ru…S | 6 | 4.31 | 0.0034 | ||||||
| 3' | 0.96 | Ru–N | 6 · 0.72 | 1.98 | 0.0071 | –6.7 | 1.1–4.2 | 2.0–12.0 | 0.5 |
| Ru…C | 6 · 0.72 | 2.93 | 0.0032 | ||||||
| Ru…O | 6 · 0.72 | 3.17 | 0.0088 | ||||||
| Ru…S | 6 · 0.72 | 4.17 | 0.0015 | ||||||
| Ru–S | 6 · 0.28 | 2.32 | 0.0025 | ||||||
| Ru…S | 6 · 0.28 | 3.57 | 0.0022 | ||||||
| Ru…S | 2 · 0.28 | 3.82 | 0.0022 | ||||||
| Ru…Ru | 12 · 0.28 | 4.03 | 0.0017 |
ВЫВОДЫ
Для протестированных в качестве электрокатализаторов реакции восстановления ионов Н+ до молекулярного водорода в электролизере воды клатрохелатов железа и кобальта, производных широко распространенных в природе неплатиновых 3d-металлов, с использованием синхротронного излучения экспериментально установлено образование их восстановленных металл(I)-содержащих форм. Напротив, в случае клеточного комплекса рутения, металла платиновой группы, электрохимические превращения в электролизере воды вызвали частичную деструкцию его инкапсулирующего серосодержащего лиганда и химические превращения его синтонов с участием ионов рутения, приведшие к образованию дисульфида платинового металла RuS2. Это соответствует известному эффекту “отравления” катализаторов на основе металлов платиновой группы соединениями серы. Действительно, ранее была неожиданно обнаружена [22] более высокая электрокаталитическая активность клатрохелатного комплекса кобальта 2 (неплатинового электрокатализатора) по сравнению с его рутенийсодержащим аналогом 3 (производным металла платиновой группы). Этот результат может быть объяснен упомянутым выше “отравлением” металла серосодержащими производными клатрохелатного лиганда. Поэтому в случае инкапсулирования катионов металлов этой группы представляется целесообразным переход к использованию несеросодержащих макробициклических лигандов для последующего использования их клатрохелатных комплексов в качестве катодных электрокатализаторов получения высокочистого водорода в электролизерах воды.
Работа выполнена при поддержке Российского научного фонда (проект № 17-13-01468) в части синтеза клеточных комплексов, Российского фонда фундаментальных исследований (грант № 18-29-23007) в части электрокаталитических экспериментов (Я.З. Волошин), (грант № 19-58-26022) в части исследований МЭБ электролизера (С.А. Григорьев и А.С. Пушкарев); при частичной финансовой поддержке Министерства науки и высшего образования РФ (проект AAAA-A19-119020890025-3) в части количественного анализа спектров XANES и EXAFS (Я.В. Зубавичус). Адсорбционные исследования выполнены в рамках государственного задания ИОНХ РАН в области фундаментальных научных исследований.
Список литературы
Esswein A.J., Nocera D.G. // Chem. Rev. 2007. V. 107. P. 4022. https://doi.org/10.1021/cr050193e
Jacques P.A., Artero V., Pecaut J., Fontecave M. // Proc. Natl. Acad. Sci. 2009. V. 106. P. 20627. https://doi.org/10.1073/pnas.0907775106
Stubbert B.D., Peters J.C., Gray H.B. // J. Am. Chem. Soc. 2011. V. 133. P. 18070. https://doi.org/10.1021/ja2078015
Lakadamyali F., Kato M., Muresan N.M., Reisner E. // Angew. Chem. Int. Ed. 2012. V. 51. P. 9381. https://doi.org/10.1002/anie.201204180
McCrory C.C.L., Uyeda C., Peters J.C. // J. Am. Chem. Soc. 2012. V. 134. P. 3164. https://doi.org/10.1021/ja210661k
Hu X., Brunschwig B.S., Peters J.C. // J. Am. Chem. Soc. 2007. V. 129. P. 8988. https://doi.org/10.1021/ja067876b
Hu X., Cossairt B.M., Brunschwig B.S. et al. // Chem. Commun. 2005. P. 4723. https://doi.org/10.1039/b509188h
Razavet M., Artero V., Fontecave M. // Inorg. Chem. 2005. V. 44. P. 4786. https://doi.org/10.1021/ic050167z
Carole B., Artero V., Fontecave M. // Inorg. Chem. 2007. V. 46. P. 1817. https://doi.org/10.1021/ic061625m
Bigi J.P., Hanna T.E., Harman W.H. et al. // Chem. Commun. 2010. V. 46. P. 958. https://doi.org/10.1039/B915846D
McNamara W.R., Han Z., Yin C.-J. et al. // Proc. Natl. Acad. Sci. 2012. V. 109. P. 15594. https://doi.org/10.1073/pnas.1120757109
Leung C.-F., Chen Y.-Z., Yu H.-Q. et al. // Int. J. Hydrogen Energy. 2011. V. 36. P. 11640. https://doi.org/10.1016/j.ijhydene.2011.06.062
Voloshin Y.Z., Kostromina N.A., Kraemer R. Clathrochelates: Synthesis, Structure and Properties, Elsevier, 2002.
Voloshin Y.Z., Belaya I.G., Kraemer R. Cage metal complexes: clathrochelates revisited. Heidelberg: Springer, 2017.
Anxolabéhère-Mallart E., Costentin C., Fournier M. et al. // J. Am. Chem. Soc. 2012. V. 134. P. 6104. https://doi.org/10.1021/ja301134e
El Ghachtouli S., Fournier M., Cherdo S. et al. // J. Phys. Chem. C. 2013. V. 117. P. 17073. https://doi.org/10.1021/jp405134a
Lacy D.C., Roberts G.M., Peters J.C. // J. Am. Chem. Soc. 2015. V. 137. P. 4860. https://doi.org/10.1021/jacs.5b01838
Voloshin Y.Z., Novikov V.V., Nelyubina Y.V. et al. // Chem. Commun. 2018. V. 54. P. 3436. https://doi.org/10.1039/c7cc09611a
Voloshin Y.Z., Varzatskii O.A., Vorontsov I.I., Anti-pin M.Yu. // Angew. Chem. Int. Ed. 2005. V. 44. P. 3400. https://doi.org/10.1002/anie.200463070
Voloshin Y.Z., Buznik V.M., Dedov A.G. // Abst. Book of 2a XXI Mendeleev Congress on General and Applied Chemistry, Saint Petersburg. 2019. P. 43.
Voloshin Y.Z., Chornenka N.V., Belov A.S. et al. // J. Electrochem. Soc. 2019. V. 166. P. H598. https://doi.org/10.1149/2.0391913jes
Pushkarev A.S., Solovyev M.A., Grigoriev S.A. et al. // Int. J. Hydrogen Eenergy. 2020. V. 45. P. 26206.https://doi.org/10.1016/j.ijhydene.2020.02.098
Pushkareva I.V., Pushkarev A.S., Grigoriev S.A. et al. // Russ. J. Appl. Chem. 2016. V. 89. № 12. P. 2109. https://doi.org/10.1134/S1070427216120260
Kalinnikov A.A., Ostrovskii S.V., Porembskii V.I. et al. // Russ. J. Appl. Chem. 2018. V. 91. № 6. P. 927. https://doi.org/10.1134/S1070427218060071
Grigoriev S.A., Pushkarev A.S., Pushkareva I.V. et al. // Int. J. Hydrogen Energy. 2017. V. 42. P. 27845. https://doi.org/10.1016/j.ijhydene.2017.05.048
Valiollahi R., Vagin M., Gueskine V. et al. // Sustain. Energy Fuels. 2019. V. 3. № 12. P. 3387. https://doi.org/10.1039/C9SE00687G
Chernyshov A.A., Veligzhanin A.A., Zubavichus Y.V. // Nucl. Instrum. Methods. Phys. Res. A. 2009. V. 603. № 1–2. P. 95. https://doi.org/10.1016/j.nima.2008.12.167
Senin R., Borisov M., Mukhamedzhanov E., Kovalchuk M. // Acta Cryst. A. 2017. V. 73. P. C862.
Ravel B., Newville M. // J. Synchr. Rad. 2005. V. 12 (4). P. 537. https://doi.org/10.1107/S0909049505012719
Newville M. // J. Synchr. Rad. 2001. V. 8(2). P. 322. https://doi.org/10.1107/S0909049500016964
Voloshin Y.Z., Varzatskii O.A., Novikov V.V. et al. // Eur. J. Inorg. Chem. 2010. P. 5401. https://doi.org/10.1002/ejic.201000444
Zelinskii G.E., Pavlov A.A., Belov A.S. et al. // ACS Omega. 2017. V. 2. P. 6852. https://doi.org/10.1021/acsomega.7b01088
Zelinskii G.E., Dudkin S.V., Chuprin A.S. et al. // Inorg. Chim. Acta. 2017. V. 463. P. 29. https://doi.org/10.1016/j.ica.2017.04.011
Belov A.S., Voloshin Y.Z., Pavlov A.A. et al. // Inorg. Chem. 2020. V. 59 (9). P. 5845. https://doi.org/10.1021/acs.inorgchem.9b03335
Zelinskii G.E., Belov A.S., Vologzhanina A.V. et al. // Polyhedron. 2019. V. 160. P. 108. https://doi.org/10.1016/j.poly.2018.12.031
Bus E., Miller J.T., Kropf A.J. et al. // Phys. Chem. Chem. Phys. 2006. V. 8. P. 3248. https://doi.org/10.1039/b605248g
Fornasini P., Monti F., Sanson A. // J. Synchr. Rad. 2001. V. 8. P. 1214. https://doi.org/10.1107/S0909049501014923
Edwards A.B., Tildesley D.J., Binsted N. // Mol. Phys. 1997. V. 91 (2). P. 357. https://doi.org/10.1080/002689797171643
Дополнительные материалы отсутствуют.
Инструменты
Российские нанотехнологии