

Российские нанотехнологии, 2020, T. 15, № 6, стр. 733-740
КАТОДНЫЕ ЭЛЕКТРОКАТАЛИЗАТОРЫ НА ОСНОВЕ Pt/Ti1 – xRuxO2 – δ ДЛЯ ТОПЛИВНЫХ ЭЛЕМЕНТОВ
А. А. Бельмесов 1, *, А. В. Левченко 1, А. А. Баранов 1, 2, С. Е. Надхина 1, А. П. Мельников 1, 2, 3
1 Институт проблем химической физики РАН
Черноголовка, Россия
2 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
3 Московский физико-технический институт
Долгопрудный, Россия
* E-mail: belmesovaa@mail.ru
Поступила в редакцию 16.06.2020
После доработки 26.08.2020
Принята к публикации 31.08.2020
Аннотация
Проведены исследования электрокаталитической активности и стабильности материалов в системе Pt/Ti1 –xRuxO2 – δ, в реакциях электровосстановления кислорода, в том числе в условиях работы в топливном элементе (ТЭ). Показано, что все полученные электрокаталитические материалы стабильны в диапазоне электродных потенциалов работы водородно-воздушного ТЭ, а электровосстановление кислорода протекает преимущественно по четырехэлектронному механизму. Мощностные характеристики ТЭ с полученным электрокатализатором сопоставимы с характеристиками ТЭ с коммерческим электрокатализатором, при этом стабильность полученных электрокатализаторов в 5–6 раз выше, чем Pt/C-катализаторов.
ВВЕДЕНИЕ
Электровосстановление кислорода на катоде топливного элемента (ТЭ), как правило, более медленный процесс по сравнению с большинством анодных реакций. Например, кажущаяся плотность тока обмена для реакции восстановления кислорода (РВК) iокатод ~ 6 мкA/cм2 [1], что на несколько порядков меньше, чем для реакции окисления водорода iо анод ~ 0.1 A/cм2. Кроме того, на катоде образуются промежуточные продукты, активирующие коррозионные процессы на электродных материалах. В связи с этим срок службы и рабочие характеристики ТЭ в значительной степени определяются электрохимическими параметрами катодного каталитического слоя [2]. Основным катодным процессом, протекающим в водородно-воздушном ТЭ, является РВК:
(1)
$2{{{\text{Н}}}^{ + }} + 2{{е}^{ - }} + 0.5{{{\text{О}}}_{2}} = {{{\text{Н}}}_{2}}{{{\text{О}}}_{{({\text{ж}})}}}.$В основном эффективность преобразования энергии в ТЭ определяют кинетические затруднения этого процесса [3]. В соответствии с [3] (рис. 1) механизмы РВК на Pt включают в себя несколько отдельных реакций. Во-первых, кислород может непосредственно восстанавливаться до Н2О в кислом электролите с присоединением четырех электронов (прямая РВК). Другой путь восстановления О2 включает в себя промежуточное образование H2O2 без разрыва связи О–О (по двухэлектронному механизму) и последующее превращение перекиси водорода. Для применения в ТЭ наиболее эффективным катализом РВК является восстановление кислорода до молекул воды по четырехэлектронному пути.

Неполное восстановление О2 до Н2О2 не только ведет к низкой эффективности конверсии энергии, но и может продуцировать свободные радикалы, активирующие протекание процессов окисления материалов электрода, в первую очередь углеродного носителя, и мембраны [4].
Добиться увеличения каталитической активности в РВК можно с помощью платиновых сплавов, поскольку на их поверхности может ингибироваться формирование OHадс на атомах Pt за счет совместного влияния геометрии поверхности и электронной структуры [5]. Согласно [6] потенциал начала адсорбции OHадс на сплавах может смещаться к более положительным потенциалам (для Pt/C это значение, как правило, выше 0.8 В) [7].
Для повышения каталитической активности катодных катализаторов важно ориентироваться на создание материалов, активирующих протекание РВК по четырехэлектронному пути, а также обладающих стабильностью к окислению. Носитель электрокатализатора должен соответствовать следующим требованиям: быть однородным по всему объему (с узким распределением частиц по размерам) и иметь высокую электропроводность [8].
Для повышения стабильности к окислительной деградации многие исследователи проводят модификацию углеродных носителей катализаторов или их замену на другие типы носителей, в частности различные оксиды металлов, например SnO2 [9], TiO2 [10], MoOx [11]. В настоящее время встречаются публикации, в которых в качестве материала-носителя для электрокатализаторов используют оксид титана [8, 12–14]. Такой выбор обусловлен высокой химической и электрохимической стабильностью в кислой среде, низкой стоимостью оксида титана, возможностью его применения в виде высокодисперсного порошка. Авторы [15, 16] отмечают, что при допировании оксида титана оксидом рутения или оксидом ниобия наблюдается значительный рост проводимости оксидного носителя примерно до 1 × × 10–4 См/см при содержании допанта 7–8 мол. %.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
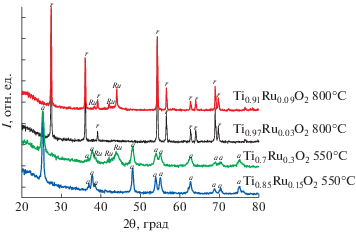
Синтез оксидных носителей состава Ti1 –xRuxO2 – δ, где x = 0–0.3 (0 ≤ Ru ≤ 30 мол. %), осуществляли путем осаждения из спиртового раствора Ti(OBu)4 (Acros Organics 99%) и RuCl3 (“х.ч.”) водным раствором азотной кислоты (“х.ч.”). Полученный гель оставляли созревать при комнатной температуре в течение недели, затем на стеклянном пористом фильтре удаляли остаток растворителя и промывали осадок [17, 18]. Полученный сухой осадок отжигали на воздухе при 300°С, затем в водороде при температуре 550°С для получения образцов со структурой анатаза и 800°С – со структурой рутила [19]. После отжига образцов при 800°С, содержащих 0–3 мол. % Ru, в водороде на рентгенограмме, полученной с помощью порошкового дифрактометра ДРОН-УМ2 (Россия), присутствуют только пики рутильной фазы, а на рентгенограммах образцов с содержанием Ru ≥ 4 мол. % появляются также пики, относящиеся к фазе металлического рутения (рис. 2). После отжига образцов при 550°С, содержащих 0–15 мол. % Ru, в водороде на рентгенограмме присутствуют пики фазы анатаза, а на рентгенограммах образцов с содержанием Ru 30 мол. %, появляются также пики, относящиеся к фазе металлического рутения (рис. 2). Все полученные образцы были использованы для дальнейших исследований.
Рис. 2.
Рентгенограммы образцов носителей катализаторов. Символом а отмечены рефлексы анатаза, r – рутила, Ru – металлического рутения.
Удельную площадь поверхности оксидных носителей определяли методом БЭТ по десорбции азота на приборе NOVA 3200 (Quantachrome Instruments, США). Измерение проводимости оксидных носителей проводили в четырехзондовой ячейке.
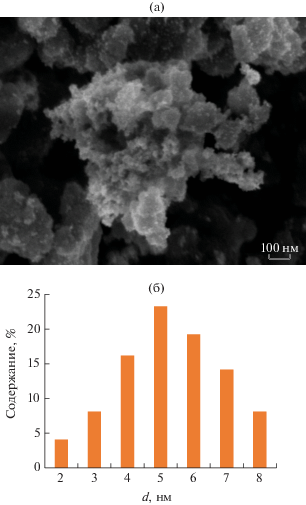
Осаждение платины на оксидный носитель проводили путем восстановления H2PtCl6 этиленгликолем (Sigma Aldrich), он выступал в роли растворителя и восстановителя [17]. Содержание платины в полученных образцах было определено рентгенофлуоресцентным анализом (“Х-Арт М” (КОМИТА, Россия)) и энергодисперсионным рентгеновским микроанализом (LEO SUPRA 25 (Carl Zeiss, Германия)). Микрофотографии (рис. 3а), полученные с помощью сканирующего автоэмиссионного электронного микроскопа LEO SUPRA 25 (Carl Zeiss, Германия), показали, что платина равномерно распределена по поверхности оксидов, средний размер частиц составляет 4–6 нм.
Рис. 3.
Микрофотография образца 10%Pt/TiO2 (а), распределение частиц платины по размерам, полученное по результатам анализа микрофотографий (б).
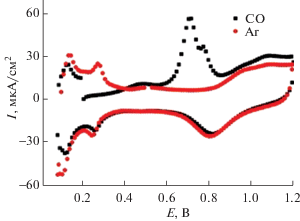
Электрохимически активную площадь поверхности платины, нанесенной на оксидные носители, определяли по десорбции водорода (Sуд Н Pt), монооксида углерода (Sуд СО Pt) и адатомов меди (Sуд Cu Pt) [17, 20] (рис. 4). Для увеличения проводимости (до σ = 3 × 10–2 См/см) в состав образцов добавили углеродные нанотрубки (УНТ) [21] ~5 мас. % (диаметр 1–5 нм, S = 280 м2/г), проводимость композитов описана в [22]. В работе использовали дисковый электрод из стеклоуглерода и изотропного пироуглерода (Sэ = 0.196 см2). Катализатор наносили на электрод в виде предельно тонкого слоя из “каталитических чернил”, которые готовили следующим образом: 10 мг исследуемого катализатора с 20 мкл 10%-ного раствора Nafion в 1 мл изопропилового спирта подвергали ультразвуковой обработке в течение 30 мин при t = 30–40°С. Из полученной суспензии брали аликвоту 6 мкл раствора (60 мкг катализатора), которую наносили на электрод. Электрод сушили в течение 30 мин на воздухе. После полного высыхания подготовленного слоя электрод помещали в трехэлектродную электрохимическую ячейку. Перед измерением фоновый электролит (0.5 М H2SO4) насыщали кислородом в течение 2 ч. Поляризационные кривые РВК записывали в интервале 1–0.2 В при скорости наложения потенциала 5 мВ/с в катодном и анодном направлении при скорости вращения электрода в интервале 600–3500 об./мин.
Рис. 4.
ЦВА фона образца 20%Pt/TiO2 анатаз (Sуд = = 216 м2/г) (атмосфера Ar), кривая десорбции монооксида углерода. 0.5 М H2SO4, трехэлектродная жидкостная ячейка, стеклоуглеродный электрод, 20 мВ/с.
Электрохимические исследования по десорбции водорода, монооксида углерода и адатомов меди проводили в стандартной трехэлектродной ячейке на потенциостате ПИ-50 Pro (Elins, Россия). В качестве рабочего электрода использовали стеклоуглеродный диск, в качестве электрода сравнения – нормальный водородный электрод (НВЭ), относительно которого приведены все потенциалы, в качестве вспомогательного электрода – платиновую фольгу. Электролит – 0.5 М H2SO4. Все измерения проводили при комнатной температуре.
Исследования в ТЭ проводили в ячейке ElectroChem 2.2 × 2.2 см. При сборке мембранно-электродных блоков использовали мембрану Nafion® 212 и газо-диффузионный слой Freudenberg C8. Состав каталитического слоя для Pt/C-материалов: 30% Nafion®, 70% электрокатализатора; для Pt/TiO2-материалов – 5% УНТ, 17% Nafion®, 78% электрокатализатора, в обоих случаях загрузка платины на электродах была около 0.5 мг/см2. Прессование проводили при 130°С при давлении 80 кг/см2 в течение 3 мин. Тестирование ТЭ проводили на станции G40 (Greenlight Innovation, США). Измерение вольтамперных характеристик ТЭ проводили на электронной нагрузке P – 400LX (Elins, Россия). Ускоренные деградационные тесты проводили с помощью циклической вольтамперометрии 0.6–1.1 В, 0.1 В/с, 30 000 циклов H2/Ar, Rh = 100%. Для контроля степени деградации после 0, 1000, 5000, 10 000, 20 000 и 30 000 проводили замер электрохимически активой площади поверхности платины с помощью циклической вольтамперометрии 0–1.2 В, 0.05 В/с, H2/Ar, Rh = 100%. Также измеряли мощностные характеристики ТЭ в этих точках путем снятия стационарных кривых в потенциостатическом режиме [23].
Для сравнения получаемых электрохимических характеристик материалов использовали коммерческий катализатор 20% Pt/C компании “Еtec” (США).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Стабильность платинированных оксидных систем исследовали методом циклической вольтамперометрии (ЦВА) в интервале 0.05–1.2 В. Полученные фоновые кривые (рис. 5) имеют характерный вид для ЦВА платины в фоновом электролите: пики на анодной ветви при 50–350 мВ – десорбция водорода, при 800–1200 мВ – адсорбция кислорода, на катодной ветви при 600–1200 мВ – десорбция кислорода, при 50–350 мВ – адсорбция водорода. Отсутствие явных различий ЦВА чистой платины говорит об устойчивости к электроокислению и электровосстановлению полученной системы в исследуемом диапазоне потенциалов.
Рис. 5.
Фоновые кривые 10%Pt/Ti0.92Ru0.08O2-δ (рутил) + 10% УНТ, Ti0.92Ru0.08O2-δ (рутил) + 10% УНТ. 0.5 М H2SO4, Ar, трехэлектродная жидкостная ячейка, стеклоуглеродный электрод, 20 мВ/с.
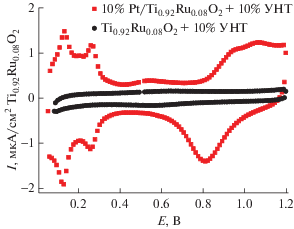
Рис. 6.
Поляризационные кривые РВК на катализаторе 20%Pt/TiO2 + 5% УНТ (а) и 20%Pt/C Vulcan XC72(б), 25°С, $v$ = 5 мВ/с, атмосфера кислорода, трехэлектродная жидкостная ячейка, НВЭ. Скорость вращения ВДЭ 1-6 соответствует 84, 105, 147, 190, 210, 263 рад/с.
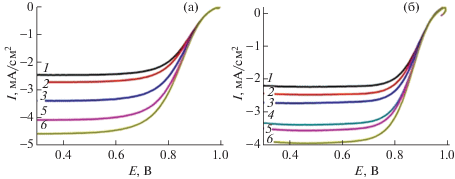
Наиболее интересными с точки зрения применения в ТЭ являются электрокатализаторы, на которых РВК протекает по четырехэлектронному механизму, в этом случае на катоде не образуется перекись водорода, являющаяся сильным окислителем для материалов каталитического слоя. Для исследования механизма РВК применяли метод вращающегося электрода.
Исследования в ячейке с вращающимся дисковом электродом (ВДЭ) показали, что величина предельного тока зависит от скорости вращения электрода. Учет влияния концентрационной поляризации проводили с использованием соотношения Коутецкого–Левича, экспериментальные данные сопоставляли с коммерчески производимым электрокатализатором – 20%Pt/C Vulcan XC72:
(2)
$1{\text{/}}i = 1{\text{/}}{{i}_{k}} + 1{\text{/}}Z{{\omega }^{{0.5}}} = 1{\text{/}}{{i}_{k}} + 1{\text{/}}{{i}_{D}},$Известно, что РВК на платине в кислой среде преимущественно протекает по четырехэлектронному механизму. Так как тангенсы угла наклона прямых на рис. 7 при 0.3 В, равные 1/Z, близки, это свидетельствует о сходном с Pt/C механизме РВК на Pt/TiO2.
Рис. 7.
Зависимости 1/i–1/ω–1/2, полученные при Е = 0.9–0.3 В для катализатора 20%Pt/C “Etek” (а) и 20%Pt/TiO2 + + 5% УНТ (б), на основе измеренных поляризационных кривых.
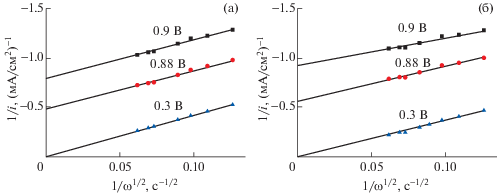
Каталитическая активность 20%Pt/TiO2 составляет 1.1, 1.8 мА/см2 (35 ± 4, 58 ± 6 мкА/см2Pt) при потенциале 0.9, 0.88 В соответственно, для 20%Pt/C “Etek” – 1.2, 2.1 мА/см2 (36 ± 4, 63 ± ± 6 мкА/см2Pt) при потенциале 0.9, 0.88 В соответственно. Площадь электрохимически активной Pt на исследуемых электродах была 31 ± ± 3 см2Pt/см2 электрода для 20%Pt/TiO2 и 33 ± ± 3 см2Pt/см2 электрода для 20%Pt/C “Etek”. Таким образом, 20%Pt/TiO2 имеет активность в РВК в пределах погрешности эксперимента, близкую с 20%Pt/C “Etek”, аналогичные данные получены в [12].
Исследования и сопоставление электрокаталитической активности 20%Pt/C “Etek” и 20%Pt/TiO2 + 5% УНТ, проведенные в жидкостной полуячейке, показали, что структура оксидного носителя влияет на электрокаталитическую активность платины в РВК. Так, при использовании носителя на основе TiO2 со структурой анатаза скорость РВК близка к скорости данной реакции на катализаторах 20%Pt/C “Etek”. В то время как на катализаторах, использующих носитель на основе TiO2 со структурой рутила, скорость РВК существенно меньше и увеличивается с увеличением степени допирования рутением (рис. 8). Причины изменения электрокаталитической активности при изменении структуры и состава оксидного носителя вероятнее всего связаны с изменением скорости отдельных стадий электровосстановления кислорода [24], но для подтверждения этого требуются дополнительные исследования. В связи с более высокой электрокаталитической активностью материала с носителем на основе TiO2 со структурой анатаза дальнейшие исследования эффективности работы в ТЭ и исследования стабильности проводили только с этим материалом.
Рис. 8.
Данные по исследованию электрокатализаторов 10%Pt/TiO2 рутил (1), 10%Pt/Ti0.98Ru0.02O2 (2), ETek 20%Pt/C (3), 10%Pt/Ti0.97Ru0.03O2 (4), 20%Pt/TiO2 (5) анатаз в трехэлектродной газожидкостной полуячейке.
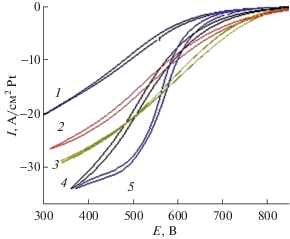
Исследования электрокатализаторов в модельных единичных ТЭ показали, что использование электрокаталитического материала на основе 20%Pt/TiO2 (анатаз) + 5% УНТ позволяет обеспечить эффективность работы ТЭ, сопоставимую с коммерческим катализатором 20%Pt/C “Etek”. Однако в случае катализаторов на основе 20%Pt/TiO2 (анатаз) потери, связанные с диффузионными затруднениями в каталитическом слое, начинают проявляться раньше, чем для катализаторов на основе 20%Pt/C “Etek” (рис. 9). Вероятнее всего это следствие недостаточной оптимизации состава каталитического слоя при использовании катализаторов на основе 20%Pt/TiO2 (анатаз). Плотность носителей на основе TiO2 существенно выше, а гидрофобность существенно ниже, чем у носителей на основе углеродных материалов. Это приводит к формированию более плотных каталитических слоев, более склонных к затоплению водой, образующейся в процессе работы ТЭ.
Рис. 9.
Вольтамперные и мощностные характеристики топливных элементов, использующих 20%Pt/C “Etek” и 20%Pt/TiO2 + 5% УНТ в качестве катализаторов.
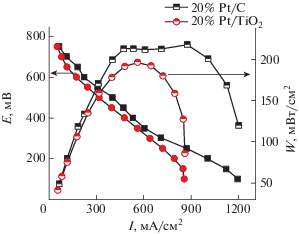
С помощью методики ускоренной деградации ТЭ [23, 25] было проведено сравнение стабильности катализаторов 20%Pt/C “Etek” и 20%Pt/TiO2 + + 5% УНТ. Использовали методику ускоренной деградации, аналогичную примененной в работе [25], в которой испытания коммерческого катализатора HiSPEC 13100 (70Pt/C) в диапазоне потенциалов 0.6–1.1 В 10 000 циклов со скоростью 100 мВ/с показали, что после ускоренного состаривания у ТЭ осталось только 20% от исходной мощности.
Исследования процессов деградации катализаторов 20%Pt/TiO2 (анатаз) + 5% УНТ показали, что разработанный катализатор обладает высокой стабильностью к окислительной деградации (рис. 10) и существенно (в 5–6 раз) стабильнее коммерческого катализатора 70%Pt/C HiSPEC 13100 (данные [25]) (табл. 1)
Рис. 10.
ЦВА(а) и мощностные характеристики топливного элемента (б), использующих катализатор 20%Pt/TiO2 + 5% УНТ после различного количества циклов деградации.
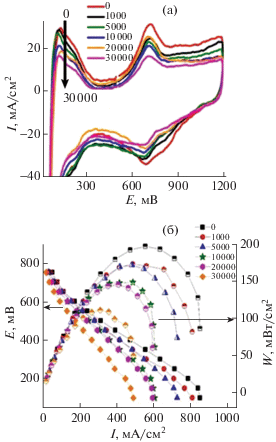
Таблица 1.
Сравнение остаточных токов в топливном элементе после различного количества циклов деградации для катализаторов 70%Pt/C HiSPEC 13100 и 20%Pt/TiO2 (анатаз) + 5% УНТ
| Количество деградацион-ных циклов | SH2 N/SH2 0 20%Pt/TiO2 | SH2 N/SH2 0 70%Pt/C HiSPEC 13100 [25] |
|---|---|---|
| 0 | 1 | 1 |
| 1000 | 0.96 | 0.89 |
| 5000 | 0.88 | 0.56 |
| 10 000 | 0.68 | 0.41 |
| 20 000 | 0.62 | |
| 30 000 | 0.52 |
ЗАКЛЮЧЕНИЕ
Проведены исследования катализаторов с оксидными носителями в системе Ti1 –xRuxO2 – δ. Показано, что механизм электровосстановления кислорода на катализаторах Pt/Ti1 –xRuxO2 – δ аналогичен механизму на Pt/C-катализаторах – восстановление кислорода протекает преимущественно по четырехэлектронному механизму. Однако состав и структура оксидного носителя влияют на скорости отдельных стадий электровосстановления кислорода, что приводит к изменению общей скорости РВК, но для подтверждения этого требуются дополнительные исследования.
Использование электрокаталитического материала на основе 20%Pt/TiO2(анатаз) + 5% УНТ позволяет обеспечить эффективность работы топливного элемента, сопоставимую с коммерческим катализатором 20%Pt/C “Etek”. Однако в случае катализаторов на основе 20%Pt/TiO2 (анатаз) потери, связанные с диффузионными затруднениями переноса кислорода в каталитическом слое, начинают проявляться раньше, чем для катализаторов на основе 20%Pt/C “Etek”, это связано с более высокой плотностью и гидрофильностью носителей на основе TiO2, чем у носителей на основе углеродных материалов. Это приводит к формированию более плотных каталитических слоев, более склонных к затоплению водой, образующейся в процессе работы ТЭ.
Исследование процессов деградации катализаторов 20%Pt/TiO2 (анатаз) + 5% УНТ показали, что разработанный катализатор обладает высокой стабильностью к окислительной деградации и в 5–6 раз стабильнее коммерческого катализатора 70%Pt/C HiSPEC 13100.
Работа частично выполнена по теме Государственного задания, № государственной регистрации АААА-А19-119061890019-5, темкарта 0089-2019-007.
Работа выполнена с использованием ресурсов Центра компетенций НТИ по технологиям новых и мобильных источников энергии при ИПХФ РАН и с использованием оборудования Аналитического центра коллективного пользования ИПХФ РАН.
Работа выполнена при поддержке Министерства образования и науки Российской Федерации (соглашение № 05.605.21.0188 от 3 декабря 2019 г. (RFMEFI60519X0188)).
Список литературы
Wagner N., Schnurnberger W., Mueller B., Lang M. // Electrochim Acta. 1998. V. 43. P. 3785. https://doi.org/10.1016/S0013-4686(98)00138-8
Stenina I.A., Safronova E.Y., Yaroslavtsev A.B. et al. // Thermal Eng. 2016. V. 63. № 6. P. 385. https://doi.org/10.1134/S0040601516060070
Wang B. // J. Power Sources. 2005. V. 152. P. 1. https://doi.org/10.1016/j.jpowsour.2005.05.098
Dobrovolsky Yu., Leonova L., Vakulenko A. // Solid State Ionics. 1996. V. 86. P. 1017. https://doi.org/10.1016/0167-2738(96)00244-5
Neyerlin K.C., Srivastava R., Yu C., Strasser P. // J. Power Sources. 2009. V. 186. № 2. P. 261. https://doi.org/10.1016/j.jpowsour.2008.10.062
Mukerjee S., Srinivasan S., Soriaga M.P. // J. Electrochem. Soc. 1995. V. 142. P. 1409.https://doi.org/10.1149/1.2048590
Koffi R.C., Coutanceau C., Garnier E. et al. // Electrochim. Acta. 2005. V. 50. № 20. P. 4117. https://doi.org/10.1016/j.electacta.2005.01.028
Mukerjee S., Lee S. J., Ticianelli E. A. et al. // Electrochem. Solid-State Lett. 1999. V. 2 (1). P. 12. https://doi.org/10.1149/1.1390718
Frolova L., Lyskov N., Dobrovolsky Yu. // Solid State Ionics. 2012. V. 225. P. 92. https://doi.org/10.1016/j.ssi.2012.02.013
Lo C.-P., Kumar A. // ECS Transactions. 2010. V. 33. № 1. P. 493. https://doi.org/10.1149/1.3484547
Gutz M., Wendt H. // Electrochim. Acta. 1998. V. 43. P. 3637. https://doi.org/10.1016/S0013-4686(98)00121-2
Liu X., Chena J., Liu G. et al. // J. Power Sources. 2010. V. 195. P. 4098. https://doi.org/10.1016/j.jpowsour.2010.01.077
Ruiz-Camacho B., Martínez-Álvarez O., Rodríguez-Santoyo H.H., Granados-Alejo V. // J. Electroanalytical Chem. 2014. V. 725. P. 19. https://doi.org/10.1016/j.jelechem.2014.04.019
Ruiz-Camacho B., Martı´nez-Gonza´lez H., Gonza´lez-Huerta R.G., Tufin˜o-Vela´zquez M. // Int. J. Hydrogen Eenergy. 2014. V. 39. P. 16731. https://doi.org/10.1016/j.ijhydene.2014.02.109
Фролова Л.А., Добровольский Ю.А. // Изв. РАН. Сер. хим. 2011. Т. 60. № 6. С. 1076.
Colomer M.T., Jurado J.R. // Chem. Mater. 2000. V. 12. P. 923. https://doi.org/10.1021/cm9903879
Belmesov A.A., Baranov A.A., Levchenko A.V. // Russian J. Electrochemistry. 2018. V. 54. № 6. P. 493. https://doi.org/10.1134/S1023193518060046
Bel'Mesov A.A., Levchenko A.V., Palankoev T.A. et al. // Russian J. Electrochemistry. 2013. V. 49. № 8. P. 831. https://doi.org/10.1134/S1023193513080053
Zyubin A.S., Zyubina T.S., Dobrovol’Skii Yu.A. et al. // Russian J. Inorganic Chemistry. 2014. V. 59. № 8. P. 816. https://doi.org/10.1134/S0036023614080221
Green C.L., Kucernak A. // J. Phys. Chem. B. 2002. V. 106. P. 1036. https://doi.org/10.1021/jp0131931
Tarasov B.P., Muradyan V.E., Volodin A.A. // Russ. Chem. Bull. 2011. V. 60. P. 1261. https://link.springer.com/content/pdf/10.1007/s11172-011-0194-8.pdf
Volodin A.A., Belmesov A.A., Murzin V.B. // Inorg. Mater. 2013. V. 49. № 7. P. 656. https://doi.org/10.1134/S0020168513060174
Аваков В.Б., Богдановская В.А., Василенко В.А. и др. // Электрохимия. 2015. Т. 51. № 8. С. 813.
Minhua Shao, Qiaowan Chang, Jean-Pol Dodelet, Regis Chenitz // Chem. Rev. 2016. V. 116. P. 3594. https://doi.org/10.1021/acs.chemrev.5b00462
U.S. Drive fuel cell tech team cell component accelerated stress test and polarization curve protocols for PEM fuel cells. January 2013. http://energy.gov/sites/prod/files/2015/08/f25/fcto_dwg_usdrive_fctt_accelerated_stress_tests_jan2013.pdf.
Дополнительные материалы отсутствуют.
Инструменты
Российские нанотехнологии