

Нефтехимия, 2019, T. 59, № 1, стр. 3-13
Парциальное каталитическое окисление спиртов 2. Катализаторы на основе соединений металлов переменной валентности (обзор)
П. Г. Мингалев 1, *, А. Ю. Оленин 1, Г. В. Лисичкин 1
1 Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
* E-mail: glo@petrol.chem.msu.ru
Поступила в редакцию 25.01.2018
После доработки 26.07.2018
Принята к публикации 21.03.2018
Аннотация
В обзоре рассмотрены методы селективного каталитического окисления первичных и вторичных спиртов в соответствующие альдегиды и кетоны. Данная часть обзора посвящена катализаторам на основе солей и оксидов переходных металлов. Наибольшее внимание уделяется реакциям окисления с помощью воздуха или молекулярного кислорода. Обсуждается влияние различных факторов (природа спирта, окислителя, катализатора и растворителя) на скорость реакции окисления и направление ее протекания.
СПИСОК СОКРАЩЕНИЙ
Cu-FMOF–Cu(II) {[Cu2(1,2-BDC)2(Fbtx)] · 3H2O}n; (1,2-BDC = 1,2-бензолдикарбоксилат)
Cys – цистеин
DDQ – 2,3-дихлор-5,6-дициано-1,4-бензохинон
Fbtx – 1,4-бис-(1,2,4-триазол-1-илметил)-2,3,5,6-тетрафторбензол
NHPI – N-гидроксифталимид
TBHP – трет-бутилгидропероксид
ВВЕДЕНИЕ
В первой части нашего обзора были рассмотрены моно- и биметаллические, а также металлокомплексные катализаторы парциального окисления спиртов (Нефтехимия. 2018. Т. 58. № 4). Однако, не меньший, а возможно и больший, интерес представляют катализаторы на основе соединений переходных металлов, способных обратимо изменять степень окисления. Этой теме и посвящена предлагаемая читателю вторая часть обзора.
ПАРЦИАЛЬНОЕ ОКИСЛЕНИЕ СПИРТОВ НА КАТАЛИЗАТОРАХ, СОДЕРЖАЩИХ СОЕДИНЕНИЯ ПОЛИВАЛЕНТНЫХ ЭЛЕМЕНТОВ
Известно, что многие переходные металлы образуют соединения в разных степенях окисления, обратимо переходящие друг в друга. Это явление положено в основу создания катализаторов окислительно-восстановительных процессов, в частности парциального окисления спиртов. Активный компонент катализатора может существовать в двух формах: окисленной (Кат[о]) и восстановленной (Кат[в]). Окисление исходного соединения в продукт происходит с участием окисленной формы, при этом она восстанавливается. Замыкание цикла реализуется за счет взаимодействия восстановленной формы с окислителем. Необходимым условием для возникновения каталитического эффекта являются более низкие энергии активации реакции исходного соединения с окисленной формой и восстановленной формы с окислителем по сравнению с аналогичным параметром прямого превращения исходного соединения в продукт под действием окислителя.
Каталитическая активность в реакции парциального окисления спиртов зафиксирована для соединений меди [1–3], железа [4, 5], марганца [6, 7], ванадия [8], палладия [4, 5, 9, 10], бинарных систем, содержащих оксиды железа и марганца [11], марганца и циркония [12], церия и марганца [13], вольфрама и алюминия [14] в сочетании с оксидами d- и f-элементов [15–20].
Авторами работы [1] в качестве модельного субстрата был выбран бензиловый спирт. Его окисление молекулярным кислородом (0.1 МПа) возможно в каталитической системе NHPI/CuBr2/HNO3 при 25°C. NHPI, HNO3 и CuBr2 являются обязательными компонентами для окисления, без любого из них реакция практически не идет. Каталитическая система NHPI/HNO3/CuBr2 проявляет высокую активность в окислении первичных бензильных спиртов, которые окисляются до соответствующих альдегидов с выходом до 51% за 4 ч. Электроноакцепторные группы, такие как нитро- и хлор-, обеспечивают селективность не менее 95%, тогда как электронодонорные группы, включая метоксильные и метильные группы, – только 80%. Это связано, по-видимому, с протеканием конкурирующих реакций окисления по электронодонорной ароматической системе. В тех же условиях первичные алифатические спирты в реакцию не вступают. В работе предложен возможный механизм реакции окисления.
В работе [6] был изучен процесс аэробного окисления спиртов в присутствии различных количеств MnO2. При использовании одного эквивалента MnO2 через 7 ч при комнатной температуре наблюдался выход продукта окисления в 84%. Количество MnO2 может быть ограничено 20 мол. %, что подтверждает его каталитическую роль в реакции. Это количество можно снизить даже до 5 мол. %, однако скорость реакции в этом случае очень низка даже при 40°С. При 80°С можно достичь высоких конверсий и выходов при количестве MnO2 до 10 мол. % и продолжительности реакции до 7 ч. В отсутствие диоксида марганца реакция практически не идет. В то же время MnO2 способен окислять 1-фенилэтанол как стехиометрический окислитель. При этом с высокой селективностью образуется ацетофенон. Использование водного раствора TBHP в качестве окислителя также дало хорошие результаты. Существенно, что присутствие воды в системе не оказывает отрицательного влияния на реакцию. Наконец, была предпринята попытка использовать пероксид водорода как более экологичный окислитель вместо TBHP, но реакция не пошла. Авторы справедливо предполагают, что реакция разложения пероксида водорода на воду и кислород, катализируемая MnO2, происходит быстрее, чем окисление спирта. Область применения реакции была исследована на спиртах бензильного, алифатического и аллильного типа. Во всех рассмотренных случаях присутствие MnO2 сильно увеличивает скорость окисления.
Описана методика окисления спиртов с применением DDQ. Она дает желаемый продукт с выходом 95% менее чем за 10 мин. Однако высокая стоимость DDQ затрудняет его использование в крупномасштабных синтезах. Принимая во внимание этот факт, авторы [7] предложили методику, которая позволила использовать DDQ как катализатор в присутствии дешевого стехиометрического соокислителя Mn(OAc)3. Обнаружено, что уменьшение количества DDQ приводит к резкому падению выходов продуктов. Процесс наиболее селективен для аллиловых спиртов, которые эффективно превращаются в соответствующие ненасыщенные карбонильные соединения. Бензильные спирты с электронодонорными заместителями давали более высокие выходы продуктов окисления, чем спирты с электроноакцепторными заместителями. Учитывая селективный характер этого окисления, быстроту протекания реакции и то, что реакция протекает в мягких условиях, можно полагать, что рассмотренная методика имеет хорошие перспективы.
В работе [8] сообщается о простой и эффективной каталитической системе на основе ванадия для окисления первичных и вторичных спиртов до альдегидов или кетонов. В качестве окислителя используют гидропероксид трет-бутила, а в качестве катализатора – ванадилсульфат. Реакция идет при комнатной температуре. В той же статье сравнили эффективность VOSO4 с другими ванадийсодержащими катализаторами и TBHP с пероксидом водорода в качестве окислителей. Было найдено, что ни V2O5, ни VO(acac)2 не столь эффективны, как VOSO4. Аналогичную реакцию проводили с H2O2 как окислителем. В качестве катализатора использовали VOSO4. Выход составил всего 35%. В отсутствие катализатора реакция не идет вообще. Вышеуказанные эксперименты убедительно продемонстрировали эффективность VOSO4 как катализатора для окисления спиртов с помощью TBHP. В воде реакция протекала очень медленно. В то же время при использовании метанола был получен неплохой выход (63%). Аналогичные результаты получены для дихлорметана, ТГФ и ДМФ. Для ДМСО и уксусной кислоты выходы ацетофенона составили 81 и 84% соответственно. Высокая конверсия наблюдалась также в ацетонитриле, где выход составил 90%. Проведение окисления фенилэтанола в смеси вода−ацетонитрил (1 : 1) дает ожидаемый ацетофенон с выходом 96% в течение очень короткого времени реакции. Это можно объяснить разницей в растворимости субстрата и VOSO4 в воде, ацетонитриле и спиртах.
Изменение природы заместителей в молекуле спирта не оказывает существенного влияния на выход реакции. Разработанная каталитическая система эффективна также для 1,2- и 1,4-диолов. Она применима и для окисления холестерина до соответствующего карбонильного соединения с хорошим выходом. С успехом эта каталитическая система была использована и для окисления гекс-5-ен-1-ола с образованием альдегида; двойная связь в этом случае сохраняет свое положение.
При использовании системы Pd(OAc)2 – оксалат натрия в отсутствие растворителя [9] вторичные алифатические спирты окислялись до соответствующих кетонов с выходами не менее 99%. Первичные алифатические спирты также окислялись в этой системе с высокими выходами (около 90%), однако скорость их окисления весьма низка. Как сообщается в этой работе, O2 или H2O2 играют роль окислителя при использовании комплексов нуль- и двухвалентного палладия в качестве катализаторов. Добавление уксусной кислоты может повысить эффективность окисления. Механизм реакции окисления на почти всех исследованных в работе каталитических системах практически одинаков. Ацетатные лиганды несут двоякую функцию: в качестве лиганда иона Pd и в качестве основания для депротонирования Pd-связанного спирта. Однако системы с бидентатными лигандами функционируют по различным механизмам, причем роль уксусной кислоты остается не ясной; добавление ее уменьшает выход продуктов окисления. Принципиальным является наличие в системе ацетата палладия, поскольку в его отсутствие реакция не идет. Было показано, что пероксид водорода просто источник молекулярного кислорода, который и является реальным окислителем в данной реакции. Об этом свидетельствует тот факт, что если реакцию прекратить немедленно после полного разложения пероксида водорода (примерно через 1 ч), выход не превышает 10%. С другой стороны, если сразу окислять спирт молекулярным кислородом в исследованной системе, то выход также превышает 90%. Однако следует отметить, что пероксид водорода более удобен для использования, так как он жидкий.
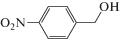
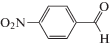
Работа [4] посвящена исследованию окисления различных спиртов в соответствующие альдегиды молекулярным кислородом при температуре 50°С и давлении в 1 атм. В качестве катализатора выступала система Fe3O4/Cys/Pd. Субстратами были первичные алифатические спирты (бутанол, гексанол), а также бензиловый спирт и его производные, содержащие в ядре различные заместители. Было показано, что селективность реакции составляет во всех случаях более 99%, т. е. побочные продукты практически не образуются. Алифатические спирты окисля ются труднее спиртов бензильного ряда, для осуществления реакции требуется 4 ч, а выходы не превышают 75%. Для сравнения, бензиловый спирт дает бензальдегид с 85%-ным выходом уже через полтора часа. Донорные заместители облегчают реакцию; так, п-метоксибензиловый спирт дает соответствующий альдегид с 87% выходом уже через час. Введение в пара-положение бензилового спирта галогенов (хлор, бром) немного снижает выход (до 82–83%), однако эти заместители слабо влияют на протекание реакции. м-Гидроксибензиловый спирт также окисляется с несколько худшим выходом (82%). По-видимому, в случае галогенов положительный мезомерный эффект заместителя практически компенсируется его отрицательным индуктивным эффектом; в случае же, когда мезомерная стабилизация невозможна (м-гидроксибензиловый спирт), определяющим становится индуктивный эффект заместителя. Введение в пара-положение сильной электроноакцепторной группы (п-нитробензиловый спирт) ведет к резкому падению активности и выхода – выход соответствующего альдегида даже через 3 ч после начала реакции составляет всего 58%.
Авторы [5] исследовали реакцию окисления различных спиртов до альдегидов и кетонов с помощью молекулярного кислорода. В качестве катализатора выступали этилендиаминовые комплексы палладия, иммобилизованные на магнетитсодержащих частицах кремнезема. Было показано, что наилучшие результаты могут быть достигнуты для бензилового спирта, в этом случае получен практически количественный выход. Катализатор, по всей видимости, чувствителен к пространственным факторам, так как введение любых заместителей в бензольное ядро ведет к снижению выхода. Электронные факторы, по-видимому, также важны, так как в случае акцепторных заместителей снижение выхода более заметное. Катализатор активен также при окислении спиртов аллильного типа и вторичных алифатических спиртов, однако его активность в этих случаях заметно ниже.
Широкий круг первичных и вторичных спиртов алифатического и ароматического ряда может быть селективно окислен кислородом воздуха до соответствующих альдегидов или кетонов с применением системы PhSeOOH – Fe(NO3)2 – MnSO4. Отмечено, что окисление идет в мягких условиях и не дает вредных побочных продуктов [11].
Идея использования смешанных оксидов лежит в основе работы [12], в которой катализаторы состава ZrOx–MnCO3 и ZrOx–Mn2O3 использованы для парциального окисления спиртов бензильного типа. Эти катализаторы получали соосаждением солей циркония и марганца основанием с последующим отжигом полученных осадков. Спирты бензильного типа количественно переходят в альдегиды уже через 5–8 ч. Наличие заместителей в молекуле, как правило, не оказывает сильного слияния на активность системы. При переходе от спиртов бензильного типа к алифатическим скорость реакции падает на порядок. Правда, и в этом случае при достаточно большой длительности реакции может быть получен количественный выход альдегида. Двойные C=C-связи в процессе окисления не затрагиваются.
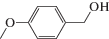
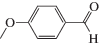
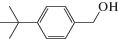
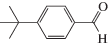
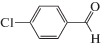
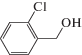
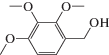
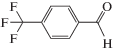
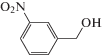
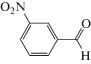
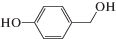
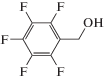
Каталитические характеристики описываемой системы (табл. 1) имеют ярко выраженную зависимость от содержания оксида циркония в катализаторе. Так, при жидкофазном окислении кислородом бензилового спирта в толуоле в области 1 мас. % ZrOx при 100% степени конверсии и более чем 99% селективности наблюдаются максимальные значения специфической активности и числа оборотов катализатора. Эти же параметры существенным образом зависят от температуры и достигают максимальных значений при 100°С.
Таблица 1.
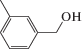
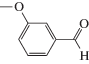
Характеристики реакций селективного окисления спиртов до альдегидов на катализаторе 1% ZrOx–MnCO3 [12]. Конверсия во всех случаях 100%, селективность >99%
| Субстрат | Продукт | Продолжительность реакции, мин |
|---|---|---|
 |
 |
5 |
 |
 |
5 |
 |
 |
5 |
 |
 |
5 |
 |
 |
5 |
 |
 |
6 |
 |
 |
6 |
 |
 |
6 |
 |
 |
6 |
 |
 |
6 |
 |
 |
7 |
 |
 |
7 |
 |
 |
7 |
 |
 |
8 |
 |
 |
8 |
 |
 |
8 |
 |
 |
9 |
 |
 |
13 |
 |
 |
14 |
 |
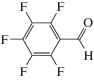 |
17 |
 |
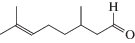 |
90 |
Авторы [13] исследовали каталитическую активность мезопористых композитов из оксидов церия, марганца, меди, кобальта и ванадия. В качестве модельной реакции было выбрано окисление бензилового спирта в бензальдегид молекулярным кислородом. Было обнаружено, что наилучшими характеристиками обладают церий-марганцевые композиты, в то время как чистые CeO2 и Mn3O4 малоактивны. Однако активность этих композитов также не особенно высока, конверсия бензилового спирта даже в лучшем случае лишь немного превосходит 60%.
В работе [14] предложен новый перспективный катализатор для окисления спиртов молекулярным кислородом в соответствующие карбонильные соединения на основе композиции WO3–Al2O3. Катализатор проявляет активность в отношении чрезвычайно широкого ряда первичных и вторичных спиртов, содержащих как алкильные, так и ароматические (включая гетероциклические) заместители. Наиболее подробно свойства этого катализатора изучены на реакции окисления бензилового спирта в бензальдегид. Показано, что оксид алюминия каталитической активностью в этой реакции не обладает, а активность чистого триоксида вольфрама низка. Предложенный катализатор обладает существенно более высокими активностью и селективностью по сравнению с исследованными в работе соединениями Ti, Nb, Mo, V, Fe и Ru. Замена оксида алюминия в катализаторе на диоксид титана ведет к улучшению каталитических свойств. Еще более активным оказался композит на основе триоксида вольфрама с диоксидом циркония. Обнаружено, что скорость окисления вторичных спиртов в обсуждаемой системе в 2–3 раза ниже, чем первичных; природа радикалов существенного влияния на ход реакции не оказывает. В то же время можно добиться увеличения активности и селективности этой системы с помощью небольших добавок стабильных свободных радикалов, таких, как дифенилпикрилгидразил или тетраметилпиперидиноксил. Замена кислорода на воздух во всех случаях ведет к существенному снижению конверсии и, как следствие, выхода. Наилучшими растворителями для этой системы являются толуол и ацетон. Применение менее полярного циклогексана приводит к резкому снижению селективности реакции. Использование более полярных растворителей также ведет к снижению выхода – либо за счет снижения селективности (диметилацетамид), либо за счет снижения конверсии (вода, ацетонитрил).
Возможность повторного использования катализатора была исследована в работе [17] для окисления бензилового спирта кислородом в присутствии композита золото – оксид меди(II) – гидроксилапатит (Au/CuO/HAP). Были исследованы свойства катализатора Au/CuO/HAP при окислении бензилового спирта в условиях рециркуляции. Было найдено, что активность катализатора в циклах со второго по четвертый включительно практически постоянна. Методом ПЭМ было обнаружено, что размер частиц в ходе рециркуляции существенно не изменяется. Результаты РФЭС показывают, что химическое состояние Au не меняется, однако около 50% поверхности CuO превратилось в Cu2O. Изменение поверхностной структуры CuO не оказало значительного влияния на каталитическую активность повторно используемого Au/CuO–HAP. Это свидетельствует о том, что в обеспечении активности катализатора главную роль играет золото при незначительном вкладе CuO. Роль наночастиц CuO, по видимому, сводится к защите наночастиц Au от агрегации.
Селективное жидкофазное окисление бензилового спирта до бензальдегида гидропероксидом трет-бутила на γ-Al2O3 с нанесенными наночастицами меди и золота проводили с использованием различных концентраций TBHP для определения оптимального отношения бензилового спирта/TBHP [19]. Окисление бензилового спирта исследовали для мольных соотношений бензилового спирта/TBHP 1 : 0.5; 1 : 1; 1 : 1.5; 1 : 2; 1 : 2.5. Показано, что конверсия бензилового спирта растет с увеличением количества TBHP. Чтобы исследовать влияние наличия в системе свободного кислорода, эксперименты также проводили в отсутствие TBHP. Было обнаружено, что конверсия бензилового спирта в этом случае составляет всего 10 и 12% для катализаторов на основе меди и золота соответственно. Однако в присутствии небольших количеств TBHP конверсия для тех же катализаторов составляла 61 и 63% соответственно. При высоких мольных соотношениях TBHP : спирт селективность по бензальдегиду заметно снижается. Это связано с дальнейшим окислением бензальдегида до бензойной кислоты и образованием бензилбензоата. Поэтому, принимая во внимание как конверсию бензилового спирта, так и селективность по бензальдегиду, было найдено, что оптимально для этой реакции соотношение бензиловый спирт : TBHP = 1 : 1.5 [19].
В [20] описан катализатор окисления спиртов на основе золота, нанесенного на цеолитоподобный носитель. В качестве окислителя выступает молекулярный кислород при 80°С и атмосферном давлении. Продуктом реакции являются соответствующие альдегиды или кетоны. Показано, что катализатор активен как по отношению к алифатическим спиртам, так и к спиртам аллильного и бензильного типа. Окисление вторичных спиртов почти во всех случаях протекает количественно вне зависимости от природы заместителей, единственный продукт – соответствующий кетон. Первичные алифатические спирты окисляются хуже, хотя селективность реакции также весьма высока. Спирты аллильного и бензильного типа количественно окисляются до альдегидов. Введение в бензольное ядро донорных заместителей (в том числе гидроксигруппы) не влияет на скорость реакции, введение акцепторных заместителей снижает скорость реакции (но не выход). Так, п-хлорбензиловый спирт окисляется в 2.5 раза медленнее бензилового, а п-нитробензиловый – в 5 раз. Замена бензольного ядра на пиридиновое, являющееся электронодефицитной ароматической системой, также ведет к снижению скорости реакции в 5 раз. Любопытно, что замена бензольного ядра на электронодонорное тиофеновое также ведет к существенному замедлению реакции (в 6 раз). Это, по-видимому, можно объяснить тем, что сера образует с поверхностью золота донорно-акцепторные связи, в результате чего электронная плотность в ароматической системе снижается.
ВЛИЯНИЕ ПРИРОДЫ ОКИСЛИТЕЛЯ НА РЕАКЦИЮ ПАРЦИАЛЬНОГО ОКИСЛЕНИЯ СПИРТОВ
Специфика окисления первичных спиртов состоит в том, что реакция окисления спирта в альдегид может сопровождаться окислением альдегида до карбоновой кислоты. Стандартный электродный потенциал второй реакции, как правило, ниже такового для реакции спирт–альдегид. Поэтому практически всегда в продуктах окисления спиртов, в том числе и каталитическом, присутствуют карбоновые кислоты и/или сложные эфиры.
Традиционные неорганические окислители, такие, как соединения марганца, хрома, железа, в высших степенях окисления обладают достаточными значениями стандартных потенциалов полуреакций восстановления. Этим обусловлено их применение в реакциях парциального окисления спиртов [21–23]. Вместе с тем, зачастую реакция окисления спиртов не останавливается на карбонильных соединениях, а в конечной реакционной массе присутствуют значительные количества продуктов дальнейшего окисления. К тому же восстановленные формы соединений марганца, хрома и железа существенным образом загрязняют конечные продукты реакции, что неизбежно требует использования специальных приемов очистки и выделения целевых продуктов.
Альтернативой традиционным неорганическим окислителям служат пероксид водорода и молекулярный кислород. Но и они не лишены недостатков. Оба окислителя в большинстве случаев образуют гетерофазную систему, что требует использования специальных приемов для повышения площади контакта реагентов. Кроме того, концентрированные растворы пероксида водорода нестабильны и взрывоопасны. Использование кислорода в качестве окислителя также сопряжено с определенными трудностями – так, повышение давления кислорода при наличии значительных количеств органических веществ, особенно летучих, может привести к лавинообразному ускорению экзотермической реакции окисления, сопровождаемой взрывом.
Тем не менее, в настоящее время эти два окислителя наиболее часто используются для окисления. Авторами [24] исследована модельная реакция окисления бензилового спирта пероксидом водорода в присутствии различных гетерогенных катализаторов. В зависимости от состава катализатора в одних и тех же условиях конечный продукт может содержать до 100% бензилового спирта, 72% бензальдегида, 34% бензойной кислоты. Молекулярный кислород [25] и воздух [26–33]. используются для парциального окисления спиртов гораздо чаще, чем пероксид водорода.
Авторами [34] было исследовано окисление бензилового спирта пероксидом водорода в присутствии соединений хрома(III). Конверсия бензилового спирта росла с увеличением количества пероксида водорода в реакционной смеси. Максимальная конверсия (76%) была достигнута при мольном соотношении H2O2 : субстрат = 3 : 1.
В работе [35] исследовано влияние различных количеств DDQ на реакцию окисления бензилового спирта. Обнаружено, что как конверсия, так и выход увеличиваются вместе с увеличением количества DDQ от 1.0 до 7.0 мол. %; между тем селективность по бензальдегиду также постепенно увеличивается. Максимальная конверсия и выход бензальдегида составляют 99 и 94% в присутствии 7.0 мол. % DDQ соответственно.
Весьма активным окислителем для получения альдегидов является закись азота; ее применение описано в патенте [36]. В качестве катализаторов выступает широкий ряд комбинаций свободных переходных металлов или их оксидов с алюмо- или ферросиликатами. Система проявляет высокую активность и окисляет до альдегидов не только спирты, но и алканы, однако для окисления алканов требуются более высокие температуры. При этом селективность катализатора падает.
В работе [35] окисление бензилового спирта молекулярным кислородом было выбрано в качестве модели для исследования каталитических свойств различных систем. В результате было установлено, что в отсутствие катализатора в течение 2 ч при 90°С и 0.3 МПа O2 в дихлорметане конверсия бензилового спирта (1) составляет всего 2.7%. Конверсия повышается до 4.5% при добавлении 3 мол. % N-бромсукцинимида (NBS). Более того, конверсия (1) составляет 14.3 или 23.1%, когда в качестве катализатора используют NBS-DDQ или NBS-NaNO2 соответственно. Однако конверсия (1) и селективность по бензальдегиду может быть повышена до 99 и 94.5% соответственно в аналогичных условиях, когда используют каталитические количества NBS-DDQ-NaNO2. Кроме того, была исследована активность комбинации DDQ-NaNO2 и DDQ без добавок, однако выходы альдегида в этих условиях невысоки (около 10%). Также было исследовано влияние количества NBS на протекание реакции. Обнаружено, что выход бензальдегида растет с увеличением количества NBS, а самая высокая селективность по бензальдегиду составляет 86.2% в присутствии 50 мол. % NBS.
Помимо этого, для окисления бензилового спирта были изучены каталитические системы, включающие аналоги NBS и NaNO2-DDQ. Обнаружено, что конверсии бензилового спирта в 37.6, 73.8 и 21.0% получают при 90°С в течение 2 ч, когда NBS заменяют на N-иодсукцинимид, N-хлорсукцинимид и сукцинимид, соответственно. Эти результаты показывают, что NBS является оптимальным компонентом каталитической системы по сравнению со своими аналогами.
В качестве катализатора аэробного окисления бензилового спирта была предложена нитрозилсерная кислота. Отмечена высокая активность этого катализатора и высокая селективность по бензальдегиду [37].
В работе [38] представлен новый, эффективный и экологически чистый метод селективного аэробного окисления спиртов до альдегидов и кетонов в присутствии нитрата индия In(NO3)3. В качестве растворителя использовали ионную жидкость – соль 1-додецил-3-метилимидазолия [C12mim][FeBr4]. Помимо In(NO3)3 были использованы и другие нитраты металлов, такие как Cu(NO3)2, Fe(NO3)3, AgNO3, Ba(NO3)2. Результаты показали, что In(NO3)3 демонстрирует наиболее высокую активность. Кроме того, во всех случаях не было обнаружено продуктов дальнейшего окисления.
Несмотря на наличие работ, направленных на поиск новых окислителей, которые могут быть использованы в реакции селективного окисления спиртов, в настоящее время наиболее эффективными из них являются газообразный кислород, воздух и водные растворы пероксида водорода.
ВЛИЯНИЕ ПРИРОДЫ СПИРТА И РАСТВОРИТЕЛЯ НА ПРОЦЕСС ПАРЦИАЛЬНОГО ОКИСЛЕНИЯ
Способность спиртов вступать в реакции парциального каталитического окисления существенным образом зависит от природы углеводородного радикала. Во-первых, в зависимости от природы углеродного атома, непосредственного связанного с функциональной гидроксильной группой, возможно образование альдегида (для первичного атома) или кетона (для вторичного атома). В случае третичного атома углерода парциальное окисление в мягких условиях не идет, а их ужесточение приводит к разрушению основной цепи, приводящей, как правило, к простейшим продуктам, таким, как оксиды углерода и вода. Во-вторых, наличие в углеводородном радикале бензольных колец, связанных с фрагментом С–ОН, способствует его преобразованию в карбонильную группу.
Разнообразные спирты (алифатические, аллильные, бензильные и др.) могут быть окислены до альдегидов с использованием в качестве катализатора рутения, нанесенного на оксид алюминия [39]. Наличие в молекуле двойных С=С-связей или аминогрупп не мешает протеканию реакции, а сами эти группы в реакцию не вступают. На Ru/Al2O3 легко окисляются спирты, содержащие в молекуле гетероциклические фрагменты – пиридиновый, тиофеновый, 1,3-диоксолановый. Спирты бензильного и аллильного типов окисляются лучше других, однако более четкую корреляцию между строением молекулы окисляемого спирта и легкостью его окисления в этом случае установить сложно. Высокую активность по отношению к первичным спиртам проявляют также гидроксиды рутения, нанесенные на композит магнетит/кремнезем [40], однако она ниже, чем у вышеописанной системы. Таким же путем могут быть окислены как алифатические спирты, так и спирты бензильного типа. Наличие двойной связи не мешает осуществлению реакции. Следует также отметить, что эта система показала очень хорошие качества при окислении вторичных спиртов до соответствующих кетонов – почти во всех случаях, вне зависимости от природы заместителей, выходы близки к количественным.
В [41] в качестве катализатора окисления спиртов в альдегиды и кетоны был выбран никельсодержащий гидроталькит. Было обнаружено, что катализатор активен в окислении как алифатических спиртов, так и спиртов аллильного и бензильного типов, однако в окислении алифатических спиртов активность его низка. Для спиртов бензильного типа показано, что введение в бензольное ядро донорных заместителей, как правило, повышает скорость реакции окисления. Для акцепторных заместителей, судя по всему, важна возможность резонансной стабилизации промежуточной частицы на заместителе. Так, введение слабоакцепторного заместителя (п-хлорбензиловый спирт) несколько снижает скорость реакции и конверсию спирта, а введение сильноакцепторного заместителя (п-нитробензиловый спирт) ее ускоряет. Замена бензольного ядра на электронодефицитное пиридиновое существенно снижает скорость реакции, а замена на электроноизбыточное тиофеновое – заметно повышает. К сожалению, авторы приводят только данные о конверсии соответствующих спиртов, но не значения селективности. Как интересный факт авторы отмечают низкие значения конверсии п-гидроксиметилбензилового спирта и п-гидроксибензилового спирта (в последнем случае реакция вообще не идет), однако объяснения этому авторы не дают.
Система также чувствительна к пространственным факторам, так как в общем случае вторичные спирты бензильного ряда окисляются хуже, чем первичные. Например, если скорость окисления 1-фенилэтанола примерно соответствует таковой для бензилового спирта, то скорость окисления 1-фенилпропанола-1 в полтора раза ниже, а для 1-фенилбутанола-1 даже значение конверсии в полтора раза ниже.
Комбинация N-гидроксифталимида и трет-бутилнитрита в ацетонитриле является перспективной каталитической системой для получения альдегидов из спиртов бензильного типа при помощи молекулярного кислорода. Реакцию проводят при 80°С и атмосферном давлении в течение 3 часов [42]. Показано, что большинство исследованных заместителей (п-метил-, о-фенил-, о-хлор-, м-метокси-) слабо влияют на процесс. Бензаннелирование также слабо сказывается на протекании реакции (1-гидроксиметилнафталин окисляется примерно так же, как и бензиловый спирт). Введение в бензольное ядро электроноакцепторных заместителей реакцию затрудняет. Любопытен тот факт, что с п-нитробензиловым спиртом реакция протекает легче, чем с м-нитробензиловым. По-видимому, это объясняется возможностью резонансной стабилизации промежуточных частиц нитрогруппой. Система активна также в окислении вторичных спиртов, как алифатических, так и содержащих ароматические фрагменты (в том числе пиридиновые, фурановые и тиофеновые). Первичные алифатические спирты, такие, как н-пентанол или 2-фенилэтанол, в реакцию практически не вступают.
В работе [30] на примере аэробного окисления широкого ряда спиртов исследована активность и селективность привитого на поверхность мезопористого носителя МСМ-41 бипиридильного комплекса CuI. Показано, что в случае первичных спиртов окисление селективно дает соответствующие альдегиды, однако реакция идет только для спиртов аллильного и бензильного типа. Наличие и положение донорных (метил-, метокси-, диметиламино-) или слабоакцепторных (галогены) заместителей в бензольном ядре существенным образом не сказывается на активности и селективности катализатора. Введение в любое положение бензольного ядра сильноакцепторной нитрогруппы резко снижает выход продукта. Заметно снижается выход также в случае о-аминобензилового спирта. Очевидно, в последнем случае протекают конкурирующие реакции окисления аминогруппы и/или ароматического кольца.
Замена бензольного ядра на пиридиновое или фурановое заметно снижает выход альдегида. По-видимому, в случае пиридинового ядра, также, как и в случае нитробензиловых спиртов, акцепторная ароматическая система хуже стабилизирует переходное состояние реакции. Фурановое ядро обладает электронодонорными свойствами, однако стабильность его не слишком высока, и в этом случае могут протекать реакции окисления ядра (как и в случае о-аминобензилового спирта). Замена бензольного ядра на достаточно стабильное тиофеновое не приводит к существенному изменению выхода реакции. В то же время алифатические спирты в этой системе не окисляются. По отношению к 1-фенилэтанолу система также неактивна. Авторами проведена оптимизация условий окисления бензилового спирта.
В [43] проведено исследование окисления разнообразных спиртов молекулярным кислородом в присутствии интеркалированного в Zn, Al-гидроталькит порфиринового комплекса кобальта и N-гидроксифталимида. Найдено, что вторичные спирты в этой системе окисляются до соответствующих кетонов с почти количественным выходами, а природа заместителей в этом случае не играет роли. По отношению к первичным спиртам система также активна. Основным продуктом окисления является соответствующий альдегид, однако он содержит заметные количества продукта окисления – карбоновой кислоты. Найдено, что наихудшие выходы получаются при окислении бензилового спирта или β-гидроксиметилтиофена; окисление же алифатических спиртов, α-гидрокисметилпиридина или замещенных бензиловых спиртов протекает более гладко.
Каталитическая система на основе H2O2–Ru(bbp)(pydic) [44] весьма эффективна для окисления насыщенных первичных алифатических спиртов, таких как 2-фенилэтанол и 1-октанол. Эта система оказалась весьма эффективной и для окисления вторичных спиртов. Реакция хорошо проходит в случае спиртов, содержащих объемистые заместители, например, 2-адамантанола. Многие вторичные циклические спирты были эффективно окислены в этой каталитической системе. Каталитическое окисление также может быть успешно выполнено в случае алифатических вторичных спиртов. Эта каталитическая система эффективно окисляет спирты в отсутствие растворителя.
Различные растворители, такие как вода, СН2Сl2, ДМСО, ДМФА, СН3СN и ТГФ были использованы при окислении бензилового спирта молекулярным кислородом в присутствии N-бромсукцинимида. Высокие выходы продукта были получены только в случае хлористого метилена. Для других растворителей было найдено, что апротонные растворители (ДМСО, ТГФ, ДМФА и СН3СN) эффективнее, чем вода. Это, вероятно, связано с тем, что катализатор плохо растворим в воде [35].
В работе [18] показано влияние природы различных растворителей при окислении п-метоксибензилового спирта кислородом до п-метоксибензальдегида. В качестве катализатора использовали наночастицы золота на оксиде магния. Результаты свидетельствуют о том, что катализатор демонстрирует наилучшие характеристики в отсутствие какого-либо растворителя, хотя использование ДМСО вело лишь к незначительному уменьшению селективности. Наблюдаемый существенно более низкий выход альдегида в присутствии растворителей (кроме ДМСО) может быть следствием конкурентной адсорбции молекул растворителя на поверхности катализатора, что препятствует свободному доступу молекул реагентов к активным центрам.
В статье [2] проводили эксперименты по аэробному окислению 4-метоксибензилового спирта в среде различных растворителей, таких как ацетонитрил, 1,4-диоксан, ДМФА, N-метилпирролидон и толуол. В качестве окислителя использовали Cu-FMOF. Обнаружено, что лучшим растворителем является ацетонитрил, для которого как конверсия, так и селективность составляют 99%, в то время как наихудшим – толуол, в среде которого конверсия составляет 27%, а селективность – 88%.
Таким образом, можно сделать вывод о том, что в ряду ароматические спирты–аллиловые спирты–алифатические спирты реакционная способность субстратов падает. Кроме того, вторичные спирты, как правило, окисляются легче первичных. Это, по-видимому, связано со стабильностью образующихся промежуточных частиц (радикалов). Природа заместителей в производных бензилового спирта обычно оказывает влияние на скорость окисления. Как правило, электронодонорные заместители облегчают окисление, а электроноакцепторные – затрудняют. Однако известны многочисленные примеры, когда указанная закономерность не соблюдается, поэтому еe нельзя считать жeстким правилом.
Обобщая результаты работ, направленных на изучение влияния растворителя на процесс каталитического окисления спиртов, можно заключить, что по экономическим соображениям предпочтительна вода, однако ее использование наиболее уместно при получении водных растворов карбонильных соединений. Для синтеза чистых высококонцентрированных продуктов имеет смысл работать с легко отгоняемым растворителем, либо вообще в его отсутствие. Применение неводных растворителей оправдано только в тех случаях, когда реакция по каким-либо причинам затруднена и в отсутствие растворителя и в водном растворе исходного спирта.
ФОТОКАТАЛИТИЧЕСКИЕ РЕАКЦИИ СЕЛЕКТИВНОГО ОКИСЛЕНИЯ СПИРТОВ
В некоторых случаях введение катализатора в реакционную среду, содержащую спирт и окислитель, не приводит к существенному ускорению парциального окисления, но процесс может быть интенсифицирован путем облучения системы видимым светом [25, 45].
В работе [45] была изучена фотокаталитическая активность композита нитрид бора/сульфид индия (BN/In2S3) в реакции окисления бензилового спирта до бензальдегида молекулярным кислородом в бензотрифториде при облучении видимым светом в течение 3 ч. Сначала конверсия бензилового спирта растет с увеличением содержания BN. Однако при увеличении содержания BN выше 7% конверсия бензилового спирта начинает снижаться. Это может быть связано с тем, что избыток BN экранирует активные центры на поверхности сульфида индия, а также препятствует поглощению последним света.
Когда содержание нитрида бора составляет 7 мас. %, образец демонстрирует оптимальную фотокаталитическую активность, а конверсия бензилового спирта достигает 60%. Однако при использовании чистого сульфида индия в качестве фотокатализатора конверсия бензилового спирта составляет лишь 25%. Контрольный эксперимент с попыткой использования BN в качестве катализатора показывает, что бензальдегид вообще не образуется. Это указывает на то, что основным фотокаталитически активным компонентом системы является именно композит BN/In2S3. Фотокаталитическая активность таких композитов для селективного окисления бензилового спирта до бензальдегида значительно выше, чем у механической смеси нитрида бора (7%) и сульфида индия (93%). Кроме того, эта смесь демонстрирует более низкую фотокаталитическую активность, чем чистый сульфид индия.
Авторами [25] описан процесс селективного фотокаталитического окисления спиртов, содержащих алифатические углеводородные радикалы, молекулярным кислородом на ванадийоксидных катализаторах. При использовании интенсивного излучения при 450 нм степени конверсии первичных спиртов, содержащих от 5 до 10 атомов углерода, достигают 90% при 70% селективности образования соответствующих альдегидов. В случае вторичных спиртов, таких как гексанол-3 конверсия составляет 100% при 96% селективности по гексанону-3.
Рассматривая перспективы промышленного использования фотокаталитического окисления спиртов, логично предположить, что такой метод может быть с успехом реализован для получения продуктов тонкого органического синтеза.
ЗАКЛЮЧЕНИЕ
Приведенный в статье материал свидетельствует об интересе исследователей к проблеме парциального каталитического окисления спиртов. Этот интерес стимулирован, главным образом, запросами компаний, связанных с тонким органическим синтезом и фармацевтикой. Перспективы широкого вовлечения биомассы в качестве источника органического сырья также стимулируют развитие исследований, целью которых является разработка эффективных технологий окисления этилового и н-бутилового спирта в соответствующие альдегиды. Этанол и н-бутанол – крупнотоннажные продукты переработки биомассы растений.
Важно отметить, что накопленные в литературе сведения позволяют сделать, хотя и неоднозначные, но все же достаточно определенные выводы о влиянии на конверсию сырья, выход продуктов и селективность процесса таких факторов, как строение исходного спирта, природа окислителя и растворителя, продолжительность реакции.
В литературе пока отсутствуют полновесные данные о попытках использования сларри-реактора для реализации процесса парциального окисления спиртов [46–49]. Вместе с тем, такой подход может оказаться продуктивным, особенно в случае использования гомогенных металлокомплексных катализаторов, поскольку возникает возможность организовать непрерывный технологический процесс, минуя стадию гетерогенизации металлокомплкса. Важно, что температура кипения продуктов парциального окисления ниже, чем у сырья и за счет этого, удаляя продукт из сферы реакции, можно повысить выход.
Список литературы
Chen B., Li J., Yang G., Gao S. // Res. Chem. Intermed. 2015. V. 41. № 6. P. 3929.
Chen S.-C., Lu S.-N., Tian F., Li N., Qian H.-Y., Cui A.-J., He M.-Y., Chen Q. // Catal. Commun. 2017. V. 95. P. 6.
Kheirjou S., Kheirjou R., Rezayan A. H., Shakourian-Fard M., Hashemi M. M. // Compt. Rend. Chim. 2016. V. 19. № 3. P. 314.
Zamani F., Hosseini S. M. // Catal. Commun. 2014. V. 43. P. 164.
Sun X., Zheng Y., Sun L., Lin Q., Su H., Qi C. // NanoStruct. NanoObj. 2016. V. 5. P. 7.
Bhaumik C., Stein D., Vincendeau S., Poli R., Manoury É. // Compt. Rend. Chim. 2016. V. 19. № 5. P. 566.
Cosner C. C., Cabrera P. J., Byrd K. M., Thomas A., Helquist P. // Org. Lett. 2011. V. 13. № 8. P. 2071.
Sarmah G., Bharadwaj S. K., Dewan A., Gogoi A., Bora U. // Tetrahedron Lett. 2014. V. 55. № 36. P. 5029.
Liu J., Wang F., Sun K., Xu X. // Catal. Commun. 2008. V. 9. № 3. P. 386.
Mondal A., Das A., Adhikary B., Mukherjee D.K. // J. Nanopart. Res. 2014. V. 16. № 4. Art. 2366.
Yu Lei, Wu Yubing, Zhang Xu, Fan Xin, CN Patent № 106673974 (A) (2017).
Assal M.E., Kuniyil M., Khan M., Al-Warthan A., Siddiqui M.R.H., Tremel W., Tahir M.N., Adil S.F. // ChemistryOpen. 2017. V. 6. № 1. P. 112.
Xu J., Shang J.-K., Wang Y., Chen Y., Li Y.-X. // Catal. Lett. 2017. V. 147. № 2. P. 328.
Zhu Y., Xu J., Lu M. // Catal. Commun. 2014. V. 48. P. 78.
Choudhary V. R., Dumbre D.K. // Appl. Catal. A. 2010. V. 375. № 2. P. 252.
Patil N., Uphade B., Jana P., Bhargava S., Choudhary V. // Chem. Lett. 2004. V. 33. № 4. P. 400.
Tian T., Liu Y., Zhang X. // Chin. J. Catal. 2015. V. 36. № 8. P. 1358.
Choudhary V.R., Dumbre D.K. // Catal. Commun. 2011. V. 13. № 1. P. 82.
Ndolomingo M.J., Meijboom R. // Appl. Surf. Sci. 2017. V. 398. P. 19.
Liu H., Liu Y., Li Y., Tang Z., Jiang H. // J. Phys. Chem. C. 2010. V. 114. № 31. P. 13362.
Wang Y.-Y., Song H., Song H.-L., Jin Z.-S. // Rus. J. Phys. Chem. A. 2016. V. 90. № 10. P. 1931.
Sun H.-Y., Hua Q., Guo F.-F., Wang Z.-Y., Huang W.-X. // Adv. Synth. Catal. 2012. V. 354. № 4. P. 569.
Keypour H., Saremi S.G., Veisi H., Azadbakht R. // RSC Adv. 2016. V. 6. № 80. P. 77020.
Kawamura K., Yasuda T., Hatanaka T., Hamahiga K., Matsuda N., Ueshima M., Nakai K. // Chem. Eng. J. 2016. V. 285. P. 49.
Zavahir S., Xiao Q., Sarina S., Zhao J., Bottle S., Wellard M., Jia J., Jing L., Huang Y., Blinco J.P., Wu H., Zhu H.-Y. // ACS Catal. 2016. V. 6. № 6. P. 3580.
Allen S.E., Walvoord R.R., Padilla-Salinas R., Kozlow-ski M.C. // Chem. Rev. 2013. V. 113. № 8. P. 6234.
Albadi J., Alihoseinzadeh A., Razeghi A. // Catal. Commun. 2014. V. 49. P. 1.
Zhang P., Deng J., Mao J., Li H., Wang Y. // Chin. J. Catal. 2015. V. 36. № 9. P. 1580.
Shaabani A., Shaabani S., Afaridoun H. // RSC Adv. 2016. V. 6. № 54. P. 48396.
Zhao H., Chen Q., Wei L., Jiang Y., Cai M. // Tetrahedron. 2015. V. 71. № 46. P. 8725.
Zhang P., Gong Y., Li H., Chen Z., Wang Y. // Nature Commun. 2013. V. 4. P. 1593.
Ando K., Nakazawa J., Hikichi S. // Eur. J. Inorg. Chem. 2016. V. 2016. № 15–16. P. 2603.
Wolfson A., Wuyts S., De Vos D.E., Vankelecom I.F.J., Jacobs P.A. // Tetrahedron Lett. 2002. V. 43. № 45. P. 8107.
Noshiranzadeh N., Bikas R., Ślepokura K., Mayeli M., Lis T. // Inorg. Chim. Acta. 2014. V. 421. P. 176.
Tong X., Sun Y., Yan Y., Luo X., Liu J., Wu Z. // J. Molec. Catal. A. 2014. V. 391. P. 1.
Харитонов А.С., Чернявский В.С., Панов Г.И., Пирютко Л.В. // Патент РФ № 2212396. Дата приор. 18.03.2002. Б.И. 2003. № 26.
Sheng Xuebin, Zhang Lu, Li Shenghui, Zheng Changyong // Patent CN №106565440 (A). 2017.
Hu Y. L., Fang D. // Compt. Rend. Chim. 2015. V. 18. № 6. P. 614.
Zotova N., Hellgardt K., Kelsall G.H., Jessiman A.S., Hii K.K. (Mimi) // Green Chem. 2010. № 12. P. 2157.
Costa V.V., Jacinto M.J., Rossi L.M., Landers R., Gusevskaya E.V. // J. Catal. 2011. V. 282. № 1. P. 209.
Zhou W., Tao Q., Pan J., Liu J., Qian J., He M., Chen Q. // J. Molec. Catal. A. 2016. V. 425. P. 255.
Hu Y., Chen L., Li B. // Catal. Commun. 2016. V. 83. P. 82.
Zhou W., Chen D., Cui A., Qian J., He M., Chen Q. // J. Chem. Sci. 2017. V. 129. № 3. P. 295.
Borthakur R., Asthana M., Kumar A., Koch A., Lal R.A. // RSC Adv. 2013. V. 3. № 45. P. 22957.
Meng S., Ye X., Ning X., Xie M., Fu X., Chen S. // Appl. Catal. B. 2016. V. 182. P. 356.
Хаджиев С.Н., Колесниченко Н.В., Ежова Н.Н. // Нефтехимия. 2016. Т. 56. № 2. С. 95 [Petrol. Chem. 2016. V. 56. № 2. P. 93.]
Хаджиев С.Н., Ежова Н.Н., Яшина О.В. // Наногетерогенный катализ. 2017. Т. 2. № 1. С. 3.
Иванцов М.И., Куликова М.В., Губанов М.А., Дементьева О.С., Чудакова М.В., Бондаренко Г.Н., Хаджиев С.Н. // Наногетерогенный катализ. 2017. Т. 2. № 1. С. 23.
Кононов С.В., Павлов В.С., Иванова И.И., Хаджиев С.Н. // Наногетерогенный катализ. 2016. Т. 1. № 2. С. 151.
Дополнительные материалы отсутствуют.
Инструменты
Нефтехимия