

Нефтехимия, 2019, T. 59, № 1, стр. 14-27
Получение моторного топлива из лигноцеллюлозы в трехстадийном процессе (обзорно-экспериментальная статья)
А. И. Нетрусов 1, 2, *, В. В. Тепляков 1, 2, М. В. Цодиков 2, А. В. Чистяков 1, 2, П. А. Жарова 1, 2, М. Г. Шалыгин 1, 2
1 Московский Государственный университет имени М.В. Ломоносова,
Биологический факультет, кафедра микробиологии
Москва, Россия
2 Институт нефтехимического синтеза им. А.В. Топчиева РАН
Москва, Россия
* E-mail: anetrusov@mail.ru
Поступила в редакцию 03.07.2018
После доработки 26.07.2018
Принята к публикации 22.07.2018
Аннотация
Описан трехстадийный процесс получения компонентов моторного топлива (МТ) из лигноцеллюлозного сырья. На первой стадии предобработки лигноцеллюлозу гидролизовали целлюлазами с последующим сбраживанием образующихся сахаров в этанол; затем разбавленные растворы этанола концентрировали мембранами в парофазном процессе с получением 70–80%-ных растворов. На третьей стадии водные растворы этанола (содержание воды 0–50%) в присутствии Pd–Zn/Al2O3/MFI-катализатора при 350°C и объемной скорости 0.6 ч–1 превращали в алканы и олефины С3–С8 и ароматические соединения С6–С12 (компоненты МТ). Установлено, что содержание воды в растворах этанола до 30% эффективно подавляет негативные процессы расщепления углеводородов (УВ) и закоксовывание поверхности катализатора, что приводит к уменьшению образования нежелательных С1 и С2 продуктов, а также увеличению операционного времени работы катализатора до 100 ч при уменьшении целевой фракции лишь на 10–15%. Последующая обработка поверхности катализатора водяным паром и водородом полностью восстанавливает его каталитическую активность.
ВВЕДЕНИЕ
| Список сокращений: | |
|---|---|
| FPIU | международная единица активности целлюлаз по фильтровальной бумаге |
| MVP | мембранное пароразделение |
| PV | первапорация |
| VP | паропроницаемость |
| VLE | паро-жидкостное равновесие |
| PDMS | полидиметилсилоксан |
| PTMSP | политриметилсилилпропин |
| PMP | поли-4-метил-2-пентин |
| MDK-1 | силоксановая промышленная мембрана РФ |
| MFI, ZSM-5 | алюмосиликатный цеолит пентазилового семейства |
| VHSV | время удерживания на катализаторе |
Биотопливо, как топливо из возобновляемого растительного или животного сырья, предназначено для замещения ископаемых невозобновляемых источников энергии, таких как нефть, уголь и природный газ. Использование ископаемого топлива привело к повышению концентрации СО2 в атмосфере и вследствие этого к возникновению глобального потепления. Биотоплива же рассматриваются как наиболее углерод-нейтральные источники энергии и их широкое использование должно привести к снижению содержания СО2 в атмосфере, что должно уменьшить негативный тепличный эффект на Земле [1].
Разработки биотоплив, как возобновляемых источников энергии, наиболее продвинуты в получении этанола. Как субстрат для массового производства биоэтанола наиболее подходит лигноцеллюлозная биомасса, которая дешева, доступна и образуется на Земле в больших количествах ежегодно. Однако, для снижения концентрации СО2 в атмосфере ежегодно следует производить лигноцеллюлозный биоэтанол в количествах, эквивалентных 50 ЭДж [2], тогда как сейчас его производят из зерна, свеклы и сахарного тростника только около 3.1 ЭДж [3]. Следует также отметить, что выработка лигноцеллюлозного этанола приведет к созданию рабочих мест и улучшению экономической ситуации в отдаленных районах развивающихся и даже развитых стран [4–6].
Производство биоэтанола первого поколения было организовано в США и Бразилии с использованием в качестве субстратов кукурузного крахмала и сахарного тростника, а также в Европе из сахарной свеклы [7]. Исследования по использованию лигноцеллюлозного сырья для получения биоэтанола начались в 70-х гг. и это следует рассматривать как биотопливо второго поколения [8, 9]. Его производство не конкурировало с пищевыми субстратами и не занимало площади сельскохозяйственных угодий.
Третье поколение биотоплив (в основном, этанол и бутанол) также использует для производства непищевые субстраты, такие как синтез-газ (СО-Н2) и, в меньшей степени, биомассу водорослей для выработки биодизеля [8–10].
С получением политической и финансовой поддержки несколько компаний на Западе уже начали промышленное производство лигноцеллюлозного биоэтанола, выработка которого достигла 45 млн л в год [11].
Однако, вследствие наличия значительного количества лигнина в лигноцеллюлозном сырье, микробное превращение растительной целлюлозы в этанол не столь прямой и простой процесс, как в случае использования простых сахаров [12].
Слой растительного лигнина выработан природой для защиты внешних оболочек растений от поражения микроорганизмами и придания им устойчивости на ветру. Соответственно, первым шагом на пути превращения лигноцеллюлозы в биоэтанол является процесс удаления лигнина в результате предобработки субстрата, делая одновременно целлюлозу более доступной для ферментативного гидролиза.
Щелочная термическая предобработка лигноцеллюлозы обычно приводит к растворению лигнина и его удалению, а также не способствует растворению молекул целлюлозы и гемицеллюлозы, как это имеет место при кислотном гидролизе [13, 14]. Поэтому, гидроксид натрия рассматривают как наиболее подходящий реагент для первичной химической предобработки биомассы, при которой распадаются связи лигнина с остальным растительным материалом с частичным его растворением, увеличением набухания целлюлозных фибрилл с частичным их гидролизом, что приводит к увеличению внутренней поверхности фибрилл, понижению степени ее кристаллизации и, в конечном итоге, ведет к повышению степени ее гидролиза целлюлазами [15].
Перед щелочной обработкой лигноцеллюлозы необходимо ее механически измельчить, при этом, чем мельче полученные частицы – тем полнее извлекаемость лигнина (идеальный размер 0.5–1.0 мм) [16]. Кроме того, такая обработка с умеренным нагревом (100–120°C) или без него, наиболее экологична, так как приводит к переводу целлюлозных фибрилл из кристаллического в аморфное состояние, также способствующее лучшему перевариванию целлюлазами [17, 18].
Целлюлозный биоэтанол получают в результате сбраживания дрожжами раствора глюкозы после расщепления молекул целлюлозы ферментами; конечные концентрации спирта при этом редко достигают 10% (обычно – 4–5%). После брожения этанол извлекают дистилляцией, а затем безводный этанол получают мембранной первапорацией с содержанием остаточной воды ниже 0.4 об. %; в такой форме он может быть напрямую использован как МТ или смешан с бензином в различных пропорциях. Ежегодное производство этанола достигает сейчас 86 млн т, причем 82% этого объема используют как топливо [19] в таких странах как США, Бразилия, Китай, Канада и в Европе [20].
Биоэтанол в промышленности вырабатывают дрожжи Saccharomyces cerevisiae или бактерии Zymomonas mobilis. При этом дрожжи доминируют в промышленных процессах вследствие их продуктивности, высокой степени устойчивости к образованному этанолу, устойчивости к токсичным ингибиторам процесса, образованным в результате предобработки биомассы. Их физиология хорошо изучена и разработаны генетические системы для получения более продуктивных штаммов [21, 22].
При сбраживании лигноцеллюлозного сырья дрожжами конечная его концентрация редко превышает 5%. Для дистилляции таких разбавленных растворов традиционными методами требуются значительные затраты тепловой энергии, поэтому мембранные методы концентрирования разбавленных растворов получают приоритет.
Исходя из вышеизложенного, данная работа посвящена проверке возможности получения МТ из биоэтанола первого и второго поколения с помощью парофазного мембранного концентрирования с последующим каталитическим превращением водных растворов этанола в алканы согласно приведенной схеме:
Брожение – Мембранное концентрирование – Катализ : схема процесса
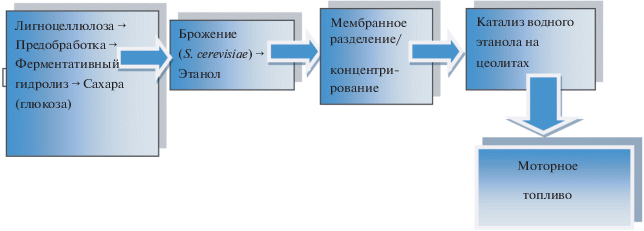
Схематическая диаграмма процесса получения возобновляемого моторного топлива из лигноцеллюлозы.
В настоящей работе изучен процесс получения моторных топлив на основе лигноцеллюлозного биоэтанола, как наиболее разработанного из целлюлозных топлив, для разработки научных основ создания новых технологий, которые обещают быть прорывными в этой области получения возобновляемой энергии.
Получение биоэтанола первого и второго поколения
Биоэтанол первого поколения для использования в качестве добавки к топливам получают с помощью дрожжей или бактерий и в таком процессе конечные концентрации спирта могут достигать 10–12 об. %. Поэтому для концентрирования спирта дистилляцией, требуются значительные затраты энергии. Этанол второго поколения, получаемый из лигноцеллюлозного сырья, после его предобработки даже более разбавлен – до 5 об. % вследствие плотности растительной пульпы, подвергаемой ферментативному гидролизу и ингибированию активности целлюлаз образуемым продуктом (глюкозой). Для сравнения эффективности последующего мембранного раз-деления и концентрирования спирта были ис-пытаны ферментационные образцы сброженной глюкозы первого и второго поколения биоэтанола.
Микроорганизмы и их культивирование. Штамм дрожжей Saccharomyces cerevisiae был получен из Коллекции микробных культур МГУ. Посевная среда содержала, г/л: глюкозу – 20; пептон – 5; дрожжевой экстракт – 3; рН – естественный (6.0). Культуры выращивали на орбитальной качалке при 180 об/мин при 30°C в течение 24 ч с последующим внесением клеток в ферментационную среду. Ферментационная среда для сбраживания этанола содержала, г/л: глюкозу – 100; пептон 0.5; дрожжевой экстракт – 0.3; рН – 6.8. Брожение проводили при 30°C в течение 48 ч в анаэробных условиях без перемешивания. Ферментационная среда для сбраживания целлюлозного этанола содержала, г/л: ферментированную целлюлозу (по глюкозному эквиваленту) – 50; пептон 0.5; дрожжевой экстракт – 0.3; рН – 6.8. Брожение вели в тех же условиях.
Предобработка лигноцеллюлозы. 100 г размолотой сухой лигноцеллюлозы (средний размер 0.5–1.0 мм) суспендировали в 1 л 5% (в/об.) NaOH (10% – конечная концентрация, в/об.) и смесь автоклавировали при 121°C (1 атм) в течение 30 мин. Осадок фильтровали под вакуумом и промывали 1% HCl до рН 5.0. Осадок биомассы без лигнина высушивали под вакуумом и хранили при комнатной температуре до обработки целлюлазами. Фильтрат, содержащий весь экстрагированный лигнин, нейтрализовали HCl (10%, об./об.), выдерживали при 4°С для формирования осадка, центрифугировали, высушивали под вакуумом и взвешивали.
Энзиматический гидролиз целлюлозы. После экстракции лигнина остаточную биомассу подвергали ферментативному гидролизу целлюлазами в 2 л колбах Эрленмейера в рабочем объеме 1 л, содержащем 50 мМ цитратный буфер (рН 4.8), 100 г/л субстрата (биомасса), 2500 FPIU ферментного коктейля (25 FPIU/г биомассы; СибФарм, Новосибирск, РФ). Колбы инкубировали при 50°С на орбитальной качалке при 50 об./мин в течение 48 ч. После проведения гидролиза рН смеси был доведен до 6.8 и к смеси были добавлена суспензия дрожжей (5 мас. %), после чего было проведено сбраживание сахаров в течение 36 ч при 30°C. Все эксперименты были проведены в трехкратных повторностях и их данные представлены средними значениями ± стандартные отклонения.
Аналитические измерения. Количество глюкозы, образованной в результате ферментативного гидролиза целлюлозы, измеряли спектрофотометрически с использованием реагентов теста “Фото-глюкоза” (Импакт, Москва, РФ) при 595 нм в глюкозо-оксидазной/пероксидазной реакции с использованием глюкозы (марка “ос. ч.”) в качестве стандарта. Концентрации этанола измеряли с помощью ГЖХ с использованием водно-этанольных смесей как стандарта.
Ферментационные процессы для получения этанола первого и второго поколений. Анаэробное сбраживание глюкозы дрожжами за 48 ч при 30°C (первое поколение) приводит к концентрация биоэтанола 10% (об./об.), в то время как при брожении лигноцеллюлозы (36 и 48 ч) достигается концентрация биоэтанола второго поколения лишь в количестве 2.5% (табл. 1).
Таблица 1.
Получение биоэтанола первого и второго поколения с помощью дрожжевого (штамм Saccharomyces cerevisiae) брожения
| Время | Биоэтанол первого поколения | Биоэтанол второго поколения | ||
|---|---|---|---|---|
| субстрат, г/л (глюкоза) | концентрация биоэтанола, % (об./об.) | субстрат, г/л (гидролизат целлюлозы) | концентрация биоэтанола, % (об./об.) | |
| 0 | 100 ± 0 | 0 ± 0 | 50 ± 3 | 0 ± 0 |
| 24 | 2 ± 1 (+100*) | 5 ± 0.2 | 10 ± 2 | 2 ± 0.2 |
| 36 | 50 ± 1 | 7.5 ± 0.2 | 1 ± 1 | 2.5 ± 0.1 |
| 48 | 0.5 ± 0.1 | 10 ± 0.2 | 0.1 ± 0.1 | 2.5 ± 0.2 |
Ферментационная среда после двух суток брожения с получением биоэтанола первого поколения содержала менее 0.05% остаточной глюкозы, что свидетельствует о полном потреблении дрожжами субстрата в количестве 20% за двое суток и способности переработать дополнительные 10% глюкозы за последующие сутки брожения (данные не приведены), образуя дополнительно 5% этанола. Изученный штамм дрожжей, таким образом, продемонстрировал исключительные возможности потребления глюкозы и накопления этанола до 15% за 72 ч брожения.
Ферментационная среда получения биоэтанола 2-го поколения после 36 ч брожения показала наличие очень небольшого количества остаточной глюкозы, образованной после гидролиза целлюлозы; при этом гемицеллюлозные молекулы остаются интактными, поскольку примененный штамм дрожжей не имеет гемицеллюлазных ферментов. Для гидролиза и последующего использования С5-сахаров гемицеллюлаз необходимы другие типы дрожжей, которые были бы способны сбраживать пентозы с получением биоэтанола 2-го поколения [23]. Получающиеся невысокие концентрации биоэтанола 2-го поколения (табл. 1) являются следствием в целом невысокой степени конверсии целлюлозы в биоэтанол. Прежде всего, концентрация сахаров в ферментационной среде зависит от количества гидролизуемой биомассы, которая в среде для гидролиза целлюлозы после экстракции лигнина не превышает 10% (в/об.) Соответственно этому, концентрация получающихся сахаров редко будет превышать 50 г/л при условии, что вся целлюлоза будет эффективно прогидролизована целлюлазными коктейлями, поскольку гемицеллюлазы находятся в лигноцеллюлозе в значительных количествах и не подвергаются гидролизу целлюлазами. В результате, даже при очень глубоком использовании глюкозы применяемыми штаммами дрожжей, конечная концентрация биоэтанола редко превышает 2.5%. Поэтому, для выделения и концентрирования таких разбавленных растворов биоэтанола применение обычных дистилляций будет чрезвычайно неэффективно с экономической точки зрения. Для создания экономичных технологий получения биоэтанола 2-го поколения представляется необходимым применять мембранные методы селекции, требующие невысоких энергетических затрат по сравнению с обычной дистилляцией. Для сравнения эффективностей выделения и концентрирования биоэтанола с помощью парофазного мембранного разделения были испытаны образцы биоэтанола 1-го и 2-го поколений, полученные после сбраживания глюкозы в первом случае и гидролизата сахаров после конверсии целлюлозы ферментами.
Мембранное выделение этанола из бродильной среды
Традиционные методы и мембранные процессы выделения спиртов. Традиционными методами разделения жидких смесей являются дистилляция, экстракция и адсорбция [24, 25]. Мембранные методы разделения жидких смесей появились сравнительно недавно, но уже рассматриваются как безреагентные или безадсорбентные технологии, обладающие более высокой энергетической эффективностью по сравнению с дистилляцией [26]. Мембранная первапорация является одним из наиболее подходящих мембранных методов извлечения и концентрирования этанола из ферментационного бульона. Тем не менее, она имеет такой существенный недостаток, как прямой контакт мембраны с биосредой, что приводит к загрязнению мембраны микроорганизмами и нелетучими органическими веществами [26]. Кроме того, первапорационные модули ограничивают площадь контакта мембраны с биосредой внутри биореактора, не позволяя обеспечить эффективное извлечение этанола как ингибитора. Для этих целей перспективен предложенный авторами новый, не имеющий аналогов, парофазный мембранный метод извлечения этанола из ферментационного бульона [27]. Метод основан на сочетании газовой сдувки (газового стриппинга) и мембранного разделения паров воды и этанола. Стриппинг используется для перевода этанола из жидкой фазы ферментационного раствора в паровую фазу в соответствии с положительным отклонением от закона Рауля для водно-этанольных растворов при пониженных концентрациях спирта в водной фазе [28, 29]. Собственно мембранный парофазный метод используется для извлечения этанола из ферментационного бульона сразу после газовой сдувки (стриппинга) [24, 27]. Такой подход позволяет осуществлять непрерывное извлечение этанола из биореактора, как это происходит при первапорации, и, в то же время, избежать недостатков первапорации. Кроме того, этот метод можно применять для выделения других летучих продуктов, если подобрана соответствующая мембрана. Следует отметить, что максимальная концентрация этанола, после которой биопроцесс ингибируется, обычно не превышает 10 мас. % [30]. В нашем случае концентрация целлюлозного этанола не более 2.5%. Извлечение этанола, являющегося незначительной примесью в воде, требует применения органофильных мембран, более проницаемых по отношению к органическим молекулам, чем к воде, что характерно для каучукоподобных мембранных материалов, например, для полидиметилсилоксана (ПДМС).
Мембраны на основе ПДМС детально исследованы в области выделения биоспиртов методом первапорации. Однако проницаемость ПДМС-мембран и их устойчивость к воздействию биосред недостаточны для ее промышленного применения [31]. Современные взгляды на явления парофазного мембранного разделения (ПМР) с фундаментальных позиций [32], а также с точки зрения потенциала их применения [33] демонстрируют важность и перспективность разработки неорганических и полимерных мембран для процессов разделения газообразных смесей, содержащих конденсируемые компоненты. Научная активность в области неорганических мембран представлена в ряде публикаций и ориентирована на условия синтеза для обеспечения бездефектного тонкого слоя на основе цеолитов или металлорганических каркасных структур (MOFs). В перспективе варьирование гидрофильно-гидрофобного баланса мембраны с эффектом “молекулярного просеивания” может обеспечить разделение различных органических (водно-органических) смесей (см., например [34, 35]).
К самым важным свойствам неорганических мембран можно отнести повышенную температуру применения и устойчивость к воздействию органических веществ. Сочетание неорганических и полимерных компонентов в качестве композитной мембраны также представляет определенный интерес [36]. Тем не менее, исторически для первапорации и пароразделения первыми были применены полимерные мембраны [33, 37]. Оба процесса протекают под градиентом парциального давления пара, которое при первапорации близко к давлению насыщения [32].
Характеристика парофазных мембранных процессов. Фактически, необходимость создания интегрированных систем с ПМР возникает, если учесть принципиальную особенность явлений стриппинга: положительное или отрицательное отклонение от закона Рауля для равновесия пар–жидкость. В целом положительное отклонение от закона Рауля для водно-органических систем (обогащение паровой фазы органикой) обеспечивает благоприятные условия для извлечения органических компонентов. В случае, когда вода является второстепенным компонентом (например, азеотроп этанол–вода), применение традиционной первапорации и гидрофильных мембран с будет предпочтительным. Гидрофильные мембраны с селективностью вода/органика $ \gg $1 более разработаны и используются в процессах обезвоживания водно-органических смесей [33–35, 37]. Например, мембраны PERVAP производства SULZER используются для непрерывной осушки растворителей, что позволяет экономично и эффективно обезвоживать органические компоненты до уровня ppm [38].
Другой класс паропроницаемых мембран основан на каучуко-подобных полимерах и высокопроницаемых гидрофобных стеклообразных полимерах с селективностью органика/вода >1 [32, 33]. В некоторой степени мембранное пароразделение на основе таких мембран может найти применение в производстве энергоносителей биопроисхождения. Практически же такая технология требует применения менее энергоемкой сепарационной системы для рекуперации и концентрирования биоспиртов [30, 37–39].
В настоящей работе описаны новые разработки для мембранного разделения парогазовых смесей, которые могут найти широкое применение, например для извлечения энергоносителей из продуктов переработки биомассы [40]. Ряд мембран, пригодных для этой цели, рассмотрен недавно в [41].
Основные тенденции мембранного пароразделения рассматривают на примере выделения биоспиртов из разбавленных водно-спиртовых растворов. Концентрирование биоспиртов можно реализовать с помощью органофильных мембран, которые обычно более проницаемы для органических молекул, чем для молекул воды. Это характерно для каучуков, например, ПДМС (см. выше).
В настоящее время стеклообразные органофильные полиацетилены также рассматривают как перспективные материалы для мембранного разделения водно-органических растворов, так как они могут обеспечить получение высокопроницаемых мембран. Например, наиболее проницаемым полимером в настоящее время является известный поли-1-триметилсилил-1-пропин (ПТМСП) [42, 43]. Он активно исследован в большом числе работ в качестве первапорационного мембранного материала для разделения водно-спиртовых смесей [43].
Поли(4-метил-2-пентин) (ПМП) – “углеродный” аналог ПТМСП и довольно новый мембранный материал. Он показывает высокие коэффициенты массопереноса для газов и паров и демонстрирует более высокую устойчивость к различным органическим растворителям по сравнению с ПТМСП [41]. Для понимания практического потенциала ПМР особое внимание уделяется фундаментальным аспектам сепарации: вкладу фазового перехода жидкость/пар и паро-селективным свойствам органофильных мембран. Фактически, эти две стадии реализуются в первапорации одновременно. Полученные к настоящему времени данные подробно рассмотрены для органофильных и гидрофобных мембран [41] и запатентованы [44].
В целом, лабораторные и коммерчески доступные мембраны для пароразделения могут быть классифицированы следующим образом: 1) высоко проницаемые эластомерные мембраны (например, на основе ПДМС) и гидрофобные стеклообразные полимерные мембраны с селективностью органика/вода >1 (например, на основе ПТМСП и ПМП); 2) средне-проницаемые гидрофобные стеклообразные полимерные мембраны с селективностью органика/вода <1 (например, на основе поливинилтриметилсилана (ПВТМС) и 3) гидрофильные мембраны с селективностью вода/органика $ \gg $1 (например, мембраны на основе поливинилового (ПВА) спирта, мембраны на основе целлюлозы). Практический интерес представляют коммерческие и лабораторные мембраны на основе гидрофобных и гидрофильных полимеров:
– плотная гидрофобная мембрана на основе поли-1-триметилсилил-1-пропина (ПТМСП);
– плотная гидрофобная мембрана на основе политриметилсилилпропина (ПМП);
– гидрофобная композитная мембрана МДК-1 на основе сополимера, содержащего ПДМС производства ЗАО НТЦ “Владипор”);
– гидрофобные асимметричные мембраны на основе поливинилтриметилсилана (ПВТМС), производимых Кусковским химическим заводом, РФ [45];
– плотная гидрофильная мембрана на основе промышленной целлофановой пленки;
– гидрофильная композитная мембрана НаРМ производства АО “РМ Нанотех”.
Следует отметить, что водоселективные мембраны обеспечивают концентрацию этанола в виде ретентата (см. ниже). Все исследованные мембраны демонстрируют высокую паропроницаемость, что представляет практический интерес. Экспериментальная установка и методика подробно описаны в [40]. Все основные технологические аспекты, включая роль мембранного блока, рассмотрены в [46].
Результаты исследования паропроницаемости. Движущей силой переноса паров через мембрану является перепад парциального давления пара через мембрану. Основной механизм переноса пара в обоих случаях – растворение–диффузия [32].
Проницаемость (Q) и коэффициент проницаемости (P) мембраны определяются следующим образом:
где Ji – поток пара, A – площадь мембраны, pi0 – парциальное давление компонента i над мембраной, pil – парциальное давление компонента i после мембраны, h – толщина мембраны.Идеальная селективность мембраны (αID) рассчитывается как отношение коэффициентов проницаемости, полученных для чистых компонентов:
Общий коэффициент разделения (α0) состоит из коэффициента разделения фазового перехода (αPh.T) из фазовой диаграммы “пар/жидкость” и коэффициента мембранного разделения (αM), которые рассчитываются следующим образом:
(5)
${{\alpha }_{0}} = \frac{{{{C_{{{\text{EtOH}}}}^{{\text{C}}}} \mathord{\left/ {\vphantom {{C_{{{\text{EtOH}}}}^{{\text{C}}}} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{C}}}}}} \right. \kern-0em} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{C}}}}}}}{{{{C_{{{\text{EtOH}}}}^{{\text{S}}}} \mathord{\left/ {\vphantom {{C_{{{\text{EtOH}}}}^{{\text{S}}}} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{S}}}}}} \right. \kern-0em} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{S}}}}}}},$(6)
${{\alpha }_{{{\text{Ph}}{\text{.T}}}}} = \frac{{{{C_{{{\text{EtOH}}}}^{{\text{V}}}} \mathord{\left/ {\vphantom {{C_{{{\text{EtOH}}}}^{{\text{V}}}} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{V}}}}}} \right. \kern-0em} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{V}}}}}}}{{{{C_{{{\text{EtOH}}}}^{{\text{S}}}} \mathord{\left/ {\vphantom {{C_{{{\text{EtOH}}}}^{{\text{S}}}} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{S}}}}}} \right. \kern-0em} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{S}}}}}}},$(7)
${{\alpha }_{{\text{M}}}} = \frac{{{{C_{{{\text{EtOH}}}}^{{\text{C}}}} \mathord{\left/ {\vphantom {{C_{{{\text{EtOH}}}}^{{\text{C}}}} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{C}}}}}} \right. \kern-0em} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{C}}}}}}}{{{{C_{{{\text{EtOH}}}}^{{\text{V}}}} \mathord{\left/ {\vphantom {{C_{{{\text{EtOH}}}}^{{\text{V}}}} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{V}}}}}} \right. \kern-0em} {C_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{\text{V}}}}}}},$Следует отметить, что коэффициент разделения αM дополнительно зависит от таких эксплуатационных параметров, как степень отбора, состав паровой фазы и др.
Некоторые опубликованные и полученные результаты переноса паров чистых компонентов через гидрофобные полимеры приведены в табл. 2. Как видно, большинство из исследованных мембран демонстрируют высокие потоки пара и низкую идеальную селективность вода/этанол (за исключением ПВТМС).
Таблица 2.
Проницаемость паров чистых компонентов через мембраны ПТМСП, ПМП, МДК-1* и ПВТМС при 50°C ([40], настоящая работа)
| Полимер | Компонент | Проницаемость паров, л (н.у.)/(м2 ч атм) | Идеальная селективность алкоголь/вода |
|---|---|---|---|
| ПТМСП | H2O | 2700 | – |
| C2H5OH | 2900 | 1.1 | |
| ПМП | H2O | 1000 | – |
| C2H5OH | 1000 | 1.0 | |
| МДК-1 | H2O | 11 500 | – |
| C2H5OH | 8000 | 0.7 | |
| ПВТМС | H2O | 10 400 | – |
| C2H5OH | 930 | 0.09 |
ПВТМС-мембраны демонстрируют водоселективные свойства (вода/этанол идеальная селективность (αID) > 1) и могут быть использованы для извлечения этанола по методу ПМР из ферментационной среды.
В соответствии с процессом паропереноса по механизму растворения–диффузии в полимере поток пара через мембрану пропорционален градиенту концентрации пара в мембране и уровню диффузии в полимерной среде (термодинамическая и кинетическая составляющие) [32, 33]. В случае водно-этанольных смесей для ПТМСП, ПМП и МДК-1 этот баланс приводит к селективности вода–этанол, близкой к единице. Для ПВТМС баланс в пользу водяного пара.
Исследованные мембраны являются одними из наиболее проницаемых, что подтверждается результатами поиска в функциональной базе данных ИНХС РАН [27]. Таким образом, концентрирование биоспиртов (биоэтанола) можно эффективно осуществлять комбинированными мембранными системами с гидрофобными мембранами (некоторые решения уже запатентованы [44]).
Однако исследование пароразделения за счет гидрофильных водо-селективных мембран показывает, что применение этих мембран является более перспективным. Как правило, такие мембраны с высокой селективностью к воде используются при обезвоживании органических растворителей (особенно в случае образования азеотропа) первапорацией. Примеры можно найти в [32, 33], но такой процесс неприемлем для разбавленных водных растворов из-за высокого энергопотребления, так как почти вся вода, представленная в растворе, должна испаряться.
В табл. 3 представлены результаты исследования переноса паров водно-этанольных смесей в мембране на основе целлофана и мембране НаРМ. Как видно из полученных данных, гидрофильные мембраны обладают значительной селективностью по отношению к водяному пару, что позволяет с высокой эффективностью концентрировать пары этанола в ретентате, где пермеат содержит преимущественно водяной пар, который может быть возвращен в биореактор.
Таблица 3.
Разделение водно-этанольной смеси методом парофазной мембраны с использованием гидрофильных мембран
| Мембрана | Температура, °P | Содержание этанола в жидкой фазе, мас. % | Содержание этанола в пермеате мас. % | Производительность по пермеату кг/(м2 ч) | Коэффициент мембранного разделения вода/этанол (αM) |
|---|---|---|---|---|---|
| Целлофан | 50–60 | 10–15 | 0.21–0.76 | 0.048–0.270 | 180–480 |
| НаРМ | 50–60 | 15 | 1.70–6.20 | 0.460–1.600 | 20–90 |
Полученные экспериментальные данные по паропроницаемости использованы нами для математического моделирования процесса парофазного мембранного разделения в применении к разбавленным водным растворам этанола (10 мас. %). Соответствующая схема процесса показана на рис. 1. Расчетные зависимости характеристик процесса при температуре 90°С для водно-этанольных смесей показаны на рис. 2, 3.
Рис. 1.
Схема одноступенчатого парофазного процесса мембранного извлечения этилового спирта из разбавленного водного раствора (ВН – вакуумный насос; Г – газодувка; К – конденсатор; MM – мембранный модуль).
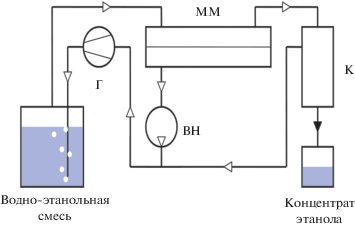
Рис. 2.
Зависимости степени извлечения этанола от концентрации этанола в продукте (ретентате) при использовании процесса парофазного мембранного разделения.
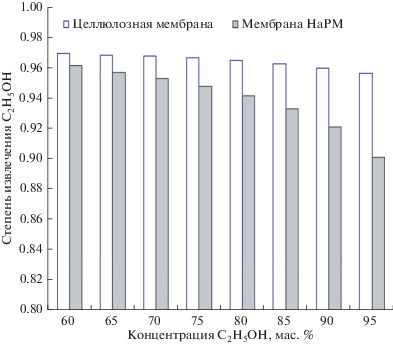
Рис. 3.
Зависимости относительной производительности процесса от концентрации этанола в продукте (ретентате) при использовании процесса парофазного мембранного разделения.
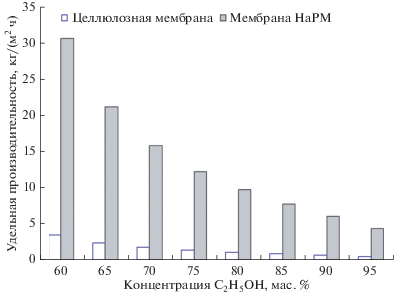
Как видно из гистограмм на рис. 2–3, высокая степень извлечения этанола достигается с помощью целлофановой мембраны, которая, в то же время, демонстрирует низкую производительность. Оптимальные результаты демонстрирует мембрана НаРМ, обеспечивающая одновременно высокую степень извлечения этанола и высокую производительность (около 3.5 кг/(м2 ч) при концентрировании этанола в диапазоне от 10% исходных до 95 мас. %. (на рис. 3 показан диапазон 60–95% этанола). Следует отметить, что ПМР позволяет оценить те же параметры, когда концентрация этанола будет ниже (например, 2.5 мас. %, как мы и ожидали). В последнем случае мы увидим снижение производительности соответственно меньшей движущей силой.
Процесс может быть дополнительно интенсифицирован, например, сжатием пара перед мембранным пароотделением, как это рассмотрено в [28, 29]. Представленная информация показывает, что применение метода парофазной мембранной сепарации для извлечения спиртов и их концентрации из разбавленных водных растворов дает возможность достичь в одну стадию требуемого высокого уровня концентрации спирта, что зачастую является достаточным для его дальнейшей утилизации в каталитических технология получения компонентов моторных топлив.
Каталитический синтез компонентов топлива из водных растворов биоэтанола
Продукты биомассы – сырье для каталитического образования моторных топлив. В последнее время большое внимание уделяется разработке эффективных подходов к использованию возобновляемого сырья в качестве сырьевого источника для получения энергоносителей, компонентов МТ и важных мономеров [46–50]. Эти задачи решаются так называемой “зеленой химией”, базирующейся на использовании возобновляемой биомассы и ее продуктов в качестве исходного сырья [47, 51, 52]. К перспективным источникам получения углеводородов, как компонентов топлив и ряда важных мономеров можно отнести, в первую очередь, этанол, бутанол, ацетон, глицерин, растительные масла. В [46] представлены перспективные схемы включения продуктов биомассы в промышленные процессы получения мономеров и топлив.
В зависимости от типа используемого катализатора этанол превращается в алкан-олефиновую или ароматическую фракции С3–С12 [47, 51, 52, 53, 54]. В этих работах было показано, что в присутствии металлсодержащих катализаторов на основе оксида алюминия этанол вступает в реакцию кросс-конденсации углеродного скелета с сореагентами, такими, как пропанол, глицерин, ацетон, приводя к образованию алкан-олефиновых УВ.
В присутствии Pd–Zn- и Au-содержащих цеолитных катализаторов типа ZSM-5 этанол практически с исчерпывающим выходом превращается в алкан-ароматические УВ бензиновой и керосиновой фракций [52, 55]. Образование ароматических УВ сопровождается выделением в реакционную среду водорода, который расходуется на гидрирование первоначально образуемых из этанола олефинов в алканы. Протекание сопряженных реакций позволило продемонстрировать возможность в присутствии опытного образца Pd−Zn/Al2O3/MFI-катализатора реализовать совместное превращение этанола и рапсового масла в алкан-ароматические УВ С4–С18, без использования в процессе молекулярного водорода [55]. Недостатком цеолитсодержащих катализаторов является быстрая дезактивация в течение 3 ч непрерывной работы в результате конденсации на поверхности катализатора ароматических УВ и последующего закоксовывания [46, 56, 57]. Продукты микробиологического синтеза содержат разбавленные водные растворы органических субстратов, в которых содержание этанола не превышает 10–15% [58, 59]. Вследствие этого в технологии их выделения используется энергозатратная стадия ректификации. При этом энергозатратность процессов ректификации относится, главным образом, к получению высококонцентрированных субстратов. В этой связи c использованием H-ZSM катализаторов на основе цеолита MFI проведены исследования по превращению обводненного этанола [46, 60–62]. В этих работах было показано, что присутствие воды при температурах, не превышающих 400°С, обеспечивает более высокую стабильность цеолитного катализатора к коксообразованию и обеспечивает более высокую селективность в образовании ароматических углеводородов бензиновой фракции [46, 62]. При более высоких температурах присутствие воды приводит к необратимомой потере активности в результате деалюминирования цеолитного катализатора.
Нанесение Pd–Zn-металлсодержащих компонентов на поверхность MFI позволило повысить устойчивость в реакции превращения этанола до 20–25 ч непрерывной работы [46, 52, 62]. Модифицированный Pd–Zn-содержащий катализатор проявляет более высокую селективность в превращении этанола в алкан-ароматическую фракцию С5–С12 по сравнению с немодифицированным катализатором. При этом [47, 50–52], было показано, что в зависимости от соотношения активных компонентов возможно можно изменять селективность в образовании алканов до С8 или ароматических углеводородов.
С целью разработки наиболее эффективного получения углеводородных компонентов бензиновой и керосиновой фракций по технологической схеме, представляющей: микробиологическую ферменатацию – мембранное выделение этанола – превращение этанола в углеводородные компоненты топлив, становится важной задачей изучения каталитических превращений обводненного этанола в углеводородные компоненты топлив и мономеров в присутствии опытного образца Pd–Zn/[ZSM-5 + Al2O3] [63–65].
МЕТОДИКА КАТАЛИТИЧЕСКИХ ЭКСПЕРИМЕНТОВ
Каталитические эксперименты проводили на микрокаталитической установке PID Eng&Tech при температуре 350°C, давлении Ar (5 атм) и объемной скоростью подачи этанола и его водных смесей 0.6 ч–1. Эти условия являются оптимальными для превращения этанола на цеолитсодержащем катализаторе [52].
Анализ продуктов превращения этанола и его обводненных растворов. Газообразные продукты реакции анализировали методом газовой хроматографии в режиме “on line”. Анализ углеводородных газов С1–С5 проводили на превращения этанола и его водных растворов в углеводородную фракцию. В работе использовали опытно-промышленные образцы катализатора Pd–Zn/ЦВМ, производства ангарского завода с содержанием активных компонентов 0.6 мас. % Pd и 1 мас. % Zn, отношение Al/Si = 30. В качестве реагента использовали этанол марки “ч. д. а.” ГОСТ 5964-93 без предварительной очистки, а также его водные растворы.
Анализ продуктов реакции осуществляли на хроматографе Кристалл-4000 (ПИД, He 70 см3/мин; 120°С; P 1.65 МПа, HP-PLOT/Al2O3, 50 м × 0.32 мм). Анализ СО, СО2 и Н2 проводили на хроматографе Кристалл-4000 (ДТП, Ar (ос. ч.) колонка СКТ, 150 × 0.4 см, 130°С, 30 мл/мин). Низкие концентрации СО (<0.4 об. %) определяли с помощью газового анализатора Riken Keiki c ИК-ячейкой (модель RI-550A).
Жидкие органические продукты реакции в водной и органической фазах идентифицировали хромато-масс-спектрометрически на MSD 6973 (Agelent) и Automass-150 (Delsi Nermag), EI = = 70 эВ, объем пробы 1 мкл, колонки: HP-5MS, 0.32 × 50, Df = 0.52, 50°С (5 мин), 10°С/мин, 270°С, Тинж = 250°С, постоянный поток 1 мл/мин, деление потока 1/(100–200); CPSil-5, 0.15 × 25, Df = 1.2, 50°С (8 мин), 10°С/мин, 270°С, Тинж = = 250°С, Ринж = 2.2 бар, деление потока 1/300.
Количественная идентификация оценка состава жидких продуктов проводилась методом ГЖХ на Varian 3600, колонка: Хромтэк SE-30, 0.25 × 25, Df = 0.3, 50°С (5 мин), 10 °С/мин, 280°С, Тинж = 250°С, Ринж = 1 бар, деление потока 1/200, ДИП, внутренний стандарт для органического слоя – трифтометилбензол. Содержание этилового спирта в водной фазе определяли ГХ-МС по соотношению интегральных сигналов спирта и воды методом абсолютной градуировки, внутренний стандарт для водного слоя – внутренняя нормализация.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В табл. 4 представлены результаты по выходу и составу продуктов превращения лигноцеллюлозного этанола, при различном количестве добавленной воды.
Таблица 4.
Выход и состав углеводородных продуктов в процессе превращения обводненного этанола, мас. %
| Содержание воды, об. % | 5 | 20 | 30 | 40 | 50 |
| Конверсия этанола, % | 100 | 100 | 100 | 100 | 100 |
| Выход продуктов | |||||
| Углеводороды | 57.83 | 48.70 | 42.61 | 36.52 | 30.43 |
| Вода | 42.17 | 51.30 | 57.39 | 63.48 | 69.57 |
| Выход углеводородных продуктов, мас. % | |||||
| С1 | 0.83 | 0.51 | 0.27 | 0.32 | 0.10 |
| С2 | 12.02 | 7.68 | 5.79 | 6.89 | 3.20 |
| С2= | 4.82 | 8.73 | 12.33 | 6.71 | 11.87 |
| алифатические УВ С3–С8 | 48.53 | 48.44 | 47.53 | 46.54 | 47.05 |
| ароматические УВ С6–С12 | 33.80 | 34.64 | 34.08 | 39.45 | 37.69 |
| Состав алифатической фракции С3–С8, мас. % | |||||
| С3 | 15.71 | 13.35 | 12.44 | 13.34 | 9.81 |
| С3= | 3.06 | 4.29 | 5.37 | 4.40 | 6.07 |
| С4 | 30.30 | 28.53 | 27.40 | 10.98 | 24.71 |
| С4= | 4.34 | 6.19 | 5.60 | 5.34 | 7.65 |
| С5 | 20.56 | 17.67 | 17.29 | 21.49 | 14.92 |
| С5= | 2.17 | 2.70 | 2.74 | 3.39 | 2.16 |
| С6 | 12.33 | 12.05 | 11.51 | 18.00 | 10.31 |
| С6= | 1.48 | 1.51 | 1.47 | 1.93 | 1.36 |
| С7 | 7.91 | 8.57 | 9.81 | 13.75 | 10.72 |
| С7= | 0.56 | 0.61 | 1.19 | 0.77 | 4.14 |
| С8 | 1.60 | 4.53 | 5.18 | 6.61 | 8.16 |
| Состав ароматической фракции С6–С12, мас. % | |||||
| бензол | 0.58 | 0.81 | 0.88 | 0.78 | 0.82 |
| толуол | 10.27 | 9.12 | 8.10 | 8.08 | 6.99 |
| ксилолы | 26.28 | 25.32 | 25.24 | 24.45 | 23.39 |
| триметилбензол | 39.90 | 40.85 | 41.23 | 41.96 | 42.47 |
| диметилэтилбензол | 20.11 | 21.08 | 21.70 | 22.33 | 23.19 |
| алкилнафталин | 2.85 | 2.82 | 2.85 | 2.41 | 3.14 |
Как видно из данных табл. 4, этанол в водных растворах в присутствии цеолитсодержащего катализатора превращается в широкий набор УВ, состоящих из фракций алканов и олефинов С3–С8 и ароматических УВ С6–C12. Важно отметить, что увеличение воды до 50 об. % приводит к снижению образования газообразных продуктов и, главным образом, метана. Этот результат указывает, что присутствие паров воды снижает вклад процесса термолиза органических субстратов. Другим важным результатом, является снижение выхода этан-этиленовой фракции газов, образующихся при дегидратации этанола и являющихся нецелевыми продуктами. Превращение концентрированного этанола приводит к образованию, главным образом, ароматических УВ [52, 55]. В этой связи, важным результатом, полученным при использовании обводненного этанола, является то, что в составе органических продуктов количество алифатических УВ превышает ароматические, как известно, образующиеся на металлсодержащих цеолитных катализаторах типа ZSM [56, 66].
Ранее было показано, что превращение этанола протекает по двум маршрутам, с образованием алкилароматических и алифатических непредельных УВ [48, 50, 52, 53, 66–68]. Образование ароматики протекает по, так называемому, механизму pool hydrocarbon mechanism [66]. Образование алифатических УВ, наиболее вероятно, протекает по маршруту олигомеризации этилена, образуемому при дегидратации этанола [54, 65, 67].
Важно отметить, что алифатические УВ, образуемые из обводненного этанола, содержат до 80% алканов с разветвленной структурой, являющиеся ценными компонентами моторного топлива. Ароматические УВ состоят, главным образом, из алкилзамещенных бензолов. Присутствует также небольшое количество метилнафталина (табл. 4).
Неожиданный результат был получен в экспериментах по изучению стабильности работы в присутствии 70%-ного раствора этанола в воде. Установлено, что в ходе конверсии 95%-го этанола катализатор стабильно работает в течение 20 ч, после чего его активность резко снижается, что выражается в уменьшении выхода алифатических и ароматических УВ С3–С10 с 90 до 8 мас. % и резком увеличении выхода этилена с 15 до 71 мас. % (рис. 4). При превращении обводненного этанола активность каталитической системы снижается не более чем на 10–15% после проведения эксперимента, по крайней мере, в течение 100 ч (рис. 4).
Рис. 4.
Стабильность работы Pd–Zn/MFI/Al2O3-катализатора (350°С, 0.6 ч–1) в ходе конверсии 95% (синяя) и 70% этанола (красная).
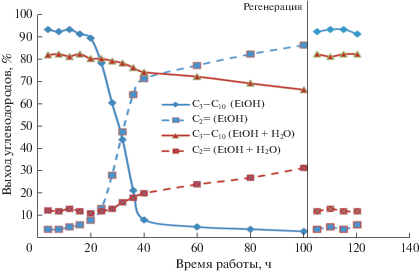
Обработка катализатора водяным паром при 450°С в течении 1–2 ч с последующим восстановлением в атмосфере водорода при давлении 50 атм позволяет полностью восстановить активность Pd–Zn/MFI/Al2O3-катализатора (рис. 4).
Основные трудности и возможности нового процесса
Трудности в получении целлюлозного этанола заключаются в высокой стоимости ферментов-целлюлаз для энзиматического гидролиза целлюлозы и относительно низкой концентрации результирующих сахаров, что выражается затем в невысоких концентрациях образуемого микробами биоэтанола, делающими последующие процессы простой дистилляции экономически необоснованными. Поэтому для выделения и концентрирования лигноцеллюлозного этанола должен быть применен новый, более эффективный с точки зрения затрат энергии, способ разделения водно-этанольных смесей и их концентрировании, который заключается в применении мембранных парофазных технологий.
Присутствие воды снижает активность катализатора в реакции термолиза углеводородов, образующихся из этанола, и существенным образом повышает стабильность работы цеолитсодержащего катализатора в процессе превращения этанола в алифатические и ароматические УВ фракции С3–С12.
Ранее, методом программированной термодесорбции аммиака было показано, что после предварительного восстановления металлсодержащих катализаторов на основе цеолита и оксида алюминия обработка поверхности парами воды приводит к существенному возрастанию силы кислотности активных центров [67, 69, 70]. На основании полученных результатов можно предположить, по крайней мере два фактора, обусловливающих влияние паров воды на активность катализатора. Воздействие паров воды в процессе превращения этанола, вероятно, приводит к формированию сильных кислотных центров в полости пор цеолита на алюминий-содержащих центрах, обеспечивающих возрастание селективности в реакции олигомеризации этилена, образуемого из этанола, в алифатические УВ и изомеризации их структуры. Этим, по всей вероятности, можно объяснить, что в присутствии воды содержание алифатических УВ превышает содержание ароматических. Другим фактором, повышающим стабильность работы катализатора, является так называемая отпарка поверхности, препятствующая развитию реакции конденсации ароматических УВ в порах цеолитного катализатора и формированию коксовых отложений.
Возможность эффективной каталитической переработки разбавленных водных растворов этанола существенным образом упрощает технологию получения топливных компонентов и мономеров на базе возобновляемого сырья по технологической схеме нового поколения: микробиологическая ферментация–мембранное концентрирование этанола–каталитическое превращение в энергоносители и ценные продукты.
ЗАКЛЮЧЕНИЕ
Вследствие объективных причин технологии брожения, конечная концентрация биоэтанола, получаемого из лигноцеллюлозы, не превышает 3%, что заставляет искать новые пути для его концентрирования, и эти пути связаны с применением мембранных методов, в частности, парофазной технологии, аналогов которой в мире не существует. Полученные результаты убедительно демонстрируют преимущества нового способа водной отпарки для продления времени активной стадии работы и регенерации катализатора для получения компонентов моторных топлив на основе лигноцеллюлозного биоэтанола.
Проведенные исследования показали существенную научную новизну, прокладывая новый путь к дальнейшим разработкам в области получения возобновляемой энергии, основанной на использовании растительной биомассы и органических сельскохозяйственных отходов (солома), что приведет к созданию высокоэффективных и экономически выгодных производств моторных топлив будущего, со значительным снижением эффекта глобального потепления.
Исследования были финансово поддержаны грантом РНФ № 16-14-00098. Авторы благодарят АО “РМ Нанотех” за предоставление образцов мембран для исследований.
Список литературы
Kang Q., Appels L., Tan T., Dewil R. // Sci. World. J. 2014. Article ID 298153, http://dx.doi.org/1–1310.1155/2014/298153.
Souza G.M., Victoria R.L., Verdade L.M., Joly P.A., Netto P.E.A., Cantarella H., Chum H.L., Diaz-Chavez R., Fernandes E., Fincher G. // Bioenergy numbers. In: Bioenergy & Sustainability: Bridging the Gaps. Ed. Souza G.M., Paris: SCOPE, 2015. P. 28.
Fulton L.M., Lynd L.R., Körner A., Greene N., Tonachel L.R. // Biofuels Bioprod. Biorefin. 2015. V. 9. P. 476.
Jordan N., Boody G., Broussard W., Glover J.D., Keeney D., McCown B.H., McIsaac G., Muller M., Murray H., Neal J. // Science. 2007. V. 316. P. 1570.
Werling B.P., Dickson T.L., Isaacs R., Gaines H., Gratton P., Gross K.L., Liere H., Malmstrom P.M., Meehan T.D., Ruan L. // Proc. Natl. Acad. Sci USA. 2014. V. 111. P. 1652.
Lynd L.R., Sow M., Chimphango A.F., Cortez L.A., Brito Cruz P.H., Elmissiry M., Laser M., Mayaki I.A., Moraes M.A., Nogueira L.A. // Biofuels Biotechnol. 2015. V. 8. P. 18.
Naqvi M., Yan J. / First-Generation Biofuels. Handbook of Clean Energy Systems. John Wiley & Sons, 2015. 312 p.
Gupta A., Verma J.P. // Renew Sustainable Energy Rev. 2015. V. 41. P. 550.
Searchinger T., Edwards R., Mulligan D., Heimlich R., Plevin R. // Science. 2015. V. 347 (6229). P. 1420.
Enquist-Newman M., Faust A.M.E., Bravo D.D., Santos P.N.S., Raisner R.M., Hanel A., Sarvabhowman P., Le P., Regitsky D.D., Cooper S.R. // Nature. 2014. V. 505 (7482). P. 239.
Lynd L.R., Liang X., Biddy M.J., Allee A., Cai H., Foust T., Himmel M.E., Laser M.S., Wang M., Wyman P.E. // Curr. Opin. Biotechnol. 2017. V. 45. P. 202.
Bondesson P.-M., Galbe M. // Biotechnol. Biofuels. 2016. V. 9. P. 222.
Chen M., Zhao J., Xia L. // Biomass Bioenergy. 2009. V. 33. P. 1381.
Alvira P., Tomas-Pejo E., Ballesteros M., Negro M.J. // Bioresour. Technol. 2010. V. 101. P. 4851.
Galbe M., Zacchi G. // Biomass Bioenergy 2012. V. 46. P. 70.
Meng X., Ragauskas A.J. // Curr. Opin. Biotechnol. 2014. V. 27. P. 150.
Gerbrandt K., Chu P.L., Simmonds A., Mullins K.A., MacLean H.L., Griffin W.M., Saville B.A. // Curr. Opin. Biotech. 2016. V. 38. P. 63.
Guilliams A., Pattathil S., Willies D., Richards M., Pu Y., Kandemkavil S., Wiswall E. // Biotechnol. Biofuels. 2016. V. 9. P. 30.
Choi S., Song P.W., Shin J.H., Lee S.Y. // Metab. Eng. 2015. V. 28. P. 223.
Joelsson E., Erdei B., Galbe M., Wallberg O. // Biotechnol Biofuels. 2016. V. 9. P. 1.
Mukherjee V., Steensels J., Lievens B., Van de Voorde I., Verplaetse A., Aerts G., Willems K.A., Thevelein J.M., Verstrepen K.J., Ruyters S. // Appl. Microbiol. Biotechnol. 2014. V. 98. P. 9483.
Nielsen J., Keasling J.D. // Cell. 2016. V. 164. P. 1185.
Ledesma-Amaro R., Nicaud J.M. // Trends Biotechnol. 2016. V. 34. P. 798.
Vane L.M. // Biofuels. Bioprod. Bioref. 2008. V. 2. P. 553.
Ezeji T., Milne P., Price N.D. // Appl. Microbiol. Biotechnol. 2010. V. 85. P. 1697.
Fadeev A.G., Selinskaya Ya.A., Kelley S.S., Meagher M.M., Litvinova E.G., Khotimsky V.S., Volkov V.V. // J. Membr. Sci. 2001. V. 186. P. 205.
Teplyakov V.V., Khotimskii V.S., Yakovlev A.V., Shalygin M.G., Gasanova L.G., Zen’kevich V.B., Netrusov A.I. // Catalysis in Industry. 2011. V. 3. P. 62.
Vane L.M., Alvarez F.R. // J. Chem. Technol. Biotechnol. 2013. V. 88. P. 1436.
Vane L.M., Alvarez F.R., Rosenblum L., Govindaswamy Sh. // J. Chem. Technol. Biotechnol. 2013. V. 88. P. 1448.
Pierrot P., Fick M., Engasser J.M. // Biotechnol. Lett. 1986. V. 8. P. 253.
Vane L.M. // J. Chem. Technol. Biotechnol. 2005. V. 80. P. 603.
Freeman B., Yampolskii Y., Pinnau I. / Materials Science of Membranes for Gas and Vapor Separation. Chichester, West Sussex, England: John Wiley & Sons Ltd. The Atrium, Southern Gate. 2006. 440 p.
Baker R.W. / Membrane Technology and Applications. Chichester: John Wiley & Sons Ltd., 2012. 484 p.
Sommer S., Melin Th. // Chem. Eng. Sci. 2005. V. 60. P. 4509.
Sommer S., Melin Th. // Chem. Eng. Sci. 2005. V. 60. P. 4525.
Zhou H., Su Y., Chen X., Wan Y. // Sep. Pur. Tech. 2011. V. 79. P. 3375.
Sirkar K.K. // Ind. En. Chem. Res. 2008. V. 47. P. 5250.
Kujawski W. // Polish J. Env. Studies. 2000. V. 9. P. 13.
Neubauer K., Dragomirova R., Stöhr M., Lubenau U., Mothes R., Paschek D., Wohlrab S. // J. Mem. Sci. 2014. V. 453. P. 100.
Yakovlev A.V., Shalygin M.G., Matson S.M., Khotimskiy V.S., Teplyakov V.V. // J. Mem. Sci. 2013. V. 434. P. 99.
Teplyakov V.V., Shalygin M.G. Pervaporation, Vapor Permeation and Membrane Distillation. Principles and applications. Chapter 7: Integrated systems involving membrane vapor permeation and applications: A volume in Woodhead Publishing Series in Energy, 2015. P. 177.
Borisevich O.B., Syrtsova D.A., Teplyakov V.V., Khotimsky V.S., Roziard D. // Desalination 2004. V. 163. P. 267.
Gonsalez-Marcos J.A., Lopez-Dehesa P., Gonsalez-Velasco J.R. // J. App. Polymer Sci. 2004. V. 94. P. 1395.
Teplyakov V.V., Khotimsky V.S., Matson M., Litvinova E.G., Shalygin M.G. Пaтeнт PФ № 2248341. 2016.
Yampol’skii Yu.P., Volkov V.V. // J. Mem. Sci. 1991. V. 64. P. 191.
Aguayo A.T., Gayubo A.G., Atutxa A., Olazar M., Bilbao J. // Ind. Eng. Chem. Res. 2002. V. 41. P. 4216.
Tsodikov M.V., Yandieva F.A., Kugel V.Ya., Chistyakov A.V., Gekhman A.E., Moiseev I.I. // Catal. Lett. 2008. V. 121. P. 199.
Johansson R., Hruby S.L., Rass-Hansen J., Christensen P.H. // Catal. Lett. 2009. V. 127. P. 1.
Inoue K., Inaba M., Takahara I., Murata K. // Catal. Lett. 2010. V. 136. P. 14.
Tsodikov M.V., Chistyakov A.V., Netrusov A.I. / Catalytic Valorization of Biomass into Fuel Components and Chemicals. Saarbrucken: Lambert Academic Publishing, 2017. 161 p.
Nikolaev S.A., Chistyakov A.V., Chudakova M.V., Kriventsov V.V., Yakimchuk E.P., Tsodikov M.V. // J. Catal. 2013. V. 297. P. 296.
Chistyakov A.V., Murzin V.Yu., Gubanov M.A., Tsodikov M.V. // Chem. Eng. Transaction 2013. V. 32. P. 619.
Yandieva F.A., Tsodikov M.V., Moiseev I.I., Gekhman A.E. // Russian Chemical Bulletin. 2012. V. 61. P. 1669.
Zharova P., Chistyakov A., Tsodikov M., Corbetta M., Manenti F. // Chem. Eng. Transact. 2015. V. 43. P. 415.
Chistyakov A., Gubanov M., Zharova P., Tsodikov M. // Chem. Eng. Transact. 2014. V. 37. P. 547.
Derouane E.G., Nagy J.B., Dejaifve P., van Hoff J.H., Spekrnan B., Vedrine P., Naccache P. // J. Catal. 1978. V. 53. P. 40.
Ingram P.W., Lancashire R.J. // Catal. Lett. 1995. V. 31. P. 395.
Ni Y., Sun Z. // Appl. Microbiol. Biotechnol. 2009. V. 83. P. 415.
Green E. // Curr. Opin. Biotech. 2011. V. 22. P. 337.
Oudejans J.P., Van Den Oosterkamp P.F., Van Bekkum H. // Applied Catalysis. 1982. V. 3. P. 109.
Schulz J., Bandermann F. // Chem. Eng. Technol. 1994. V. 17. P. 179.
Talukdar A.K., Bhattacharyya K.G., Sivasanker S. // Applied Catalysis. A: General. 1997. V. 148. P. 357.
Хаджиев С.Н., Колесниченко Н.В., Маркова Н.А., Букина З.М., Ионин Д.А., Графова Г.М., Лин Г.И. Патент РФ № 2442767. 2010.
Kolesnichenko N.V., Bukina Z.M., Kitaev L.E., Kurumov S.A., Peresypkina E.G., Khadzhiev S.N. // Petrol. Chemistry. V. 56. № 9. P. 2016.
Zhang K., Курумов С.А., Su X., Снатенкова Ю.М., Букина З.М., Колесниченко Н.В., Wu W., Хаджиев С.Н. // Нефтехимия. 2017. Т. 57. № 6. C. 656.
Seiler M., Wang W., Buchholz A., Hunger M. // Catal. Lett. 2003. V. 88. P. 187.
Yandieva F.A., Tsodikov M.V., Chistyakov A.V., Kugel V.Ya., Moiseev I.I., Zubavichus Ya.V., Veligzhanin A.A., Kitaev L.E., Yushchenko V.V., Gekhman A.E. // Kinetics and Catalysis. 2010. V. 51. P. 548.
Chistyakov A.V., Tsodikov M.V., Chudakova M.V., Gubanov M.A., Zharova P.A., Bukina Z.Ya., Kolesnichenko N.V., Gekhman A.E., Khadzhiev S.N. // Petroleum Chemistry. 2018. V. 58. P. 32.
Yushchenko V.V., Romanovsky B.V. // J. Phys. Chem. 1997. V. 71. P. 2048.
Kubasov A.A., Kitaev L.E., Yuschenko V.V., Tikhiy Ya.V. // Moscow University Chemistry Bulletin. 2005. V. 46. P. 236.
Дополнительные материалы отсутствуют.