

Нефтехимия, 2020, T. 60, № 2, стр. 199-205
Синергетический эффект Bi–Mo-катализаторов, нанесенных на TiO2, в мягком и экологически безопасном синтезе пиридилальдегидов окислением пиколинов
Jie Yu 1, 3, *, Viachaslau Zhylko 2, Liyan Dai 3, **
1 College of Biology and Environmental Engineering, Zhejiang Shuren University
310015 Hangzhou, China
2 Faculty of Environmental Monitoring of International Sakharov Environment Institute of Belarusian State University
220070 Minsk, Republic of Belarus
3 College of Chemical and Biological Engineering, Zhejiang University
310027 Hangzhou, China
* E-mail: yj313513@sina.com
** E-mail: dailiyan@zju.edu.cn
Поступила в редакцию 05.04.2019
После доработки 23.05.2019
Принята к публикации 14.10.2019
Аннотация
В данной работе было изучено окисление пиколинов до пиридилальдегидов в присутствии висмут-молибдатных катализаторов, нанесенных на TiO2. Показано, что α-Bi2Mo3O12 превосходит β-Bi2Mo2O9 и γ-Bi2MoO6 с точки зрения реакционной способности. Дальнейшее допирование α-Bi2Mo3O12/TiO2 триоксидом молибдена приводит к повышению каталитической активности, что обусловлено синергетическим эффектом между α-Bi2Mo3O12 и MoO3. Данный эффект, с одной стороны, проявляется в стабилизации кристаллической структуры катализатора за счет тесного взаимодействия между α-Bi2Mo3O12 и MoO3, а также в сохранении удельной поверхности катализатора, о чем свидетельствует низкотемпературная адсорбция азота (БЭТ) и рентгенофазовый анализ (РФА). Кроме того, результаты температурно-программируемой десорбции (ТПД) аммиака демонстрируют влияние модификации на поверхностную кислотность катализатора, и, таким образом, на ускорение адсорбции субстрата, поскольку пиколины являются веществами с основными свойствами. Помимо этого, синергизм между α-Bi2Mo3O12 и MoO3, как показано в результатах температурно-программируемого восстановления (ТПВ) и ТПД O2, приводит к изменению электронных свойств катализатора, и, соответственно, к его способности десорбировать кислород.
Пиридилальдегиды имеют первостепенное значение в химии в качестве промежуточных продуктов синтеза ввиду их широкого применения в области фармацевтики, агрохимии, хирального катализа, функциональных полимеров, и других областях [1–5]. По этой причине интенсивное изучение методов их получения становится актуальным. В окислительных процессах в качестве “зеленого” окислителя предпочтительно использование молекулярного кислорода [6], поскольку он безопасен для окружающей среды, особенно в рамках крупнотоннажной промышленной химии. Вместе с тем, вода, как нетоксичная, экономичная и экологически безопасная среда, представляет большой интерес для органического синтеза [7]. Исходя из этого, крайне желательно создать метод “зеленого” синтеза пиридилальдегидов из пиколинов посредством гетерогенного катализа, используя кислород в качестве конечного окислителя и воду в качестве реакционной среды. Наиболее часто используются катализаторы на основе оксида ванадия [8–10], однако выход пиридилальдегида низок и неудовлетворителен. Таким образом, разработка эффективного и высокопроизводительного катализатора остается по-прежнему сложной задачей.
Обычно синергетический эффект между двумя металлическими активными компонентами может приводить к удивительной каталитической активности в окислительных реакциях, катализируемых системами Cu–Ce [11], Fe–V [12] и Bi–Mo [13]. C 1960-х гг. в качестве катализатора окисления широко используется каталитическая система Bi–Mo благодаря высокой производительности в реакциях окисления углеводородов [14–16], особенно в реакции окисления пропилена в акролеин [17], которая была успешно реализована в промышленности. На данный момент продолжаются исследования данной системы в области новых свойств и применения [18–20]. Хотя до настоящего момента редко сообщалось об исследованиях, касающихся окисления пиколинов в пиральдегиды на данной системе, мы предположили, что Bi–Mo может быть эффективным кандидатом на роль катализатора окисления пиколинов по причине того, что они похожи на пропилен ненасыщенной структурой и наличием подвижных атомов водорода. В нашем предыдущем исследовании [21], в котором как модельное сырье использовался 2-пиколин, показано, что α-Bi2Mo3O12 и MoO3, нанесенные на диоксид титана, обладали большей производительностью, чем V2O5 и MoO3, также нанесенные на диоксид титана [22]. При этом причины, объясняющие сокатализ в системе α-Bi2Mo3O12 и MoO3, до сих пор остаются до конца непонятными.
В данной работе были раскрыты причины синергизма между α-Bi2Mo3O12 и MoO3. Кроме того, принимая во внимание целостность процесса, также в деталях были изучены каталитические свойства трех фаз молибдата висмута и характеристики нанесенных на диоксид титана Bi2Mo3O12 и MoO3 в реакции окисления пиколинов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализатора. Катализатор был приготовлен методом соосаждения. Были использованы коммерчески доступные реагенты без предварительной очистки. (NH4)6Mo7O24 · 4H2O растворяли в деионизированной воде, затем нагревали до 70°C при перемешивании, затем по каплям добавляли раствор 1 M Bi(NO3)3 · 5H2O в 2M HNO3. После этого добавляли необходимое количество TiO2 (анатаз), полученную суспензию перемешивали в течение 3 ч, после чего на роторном испарителе удаляли воду, остаток экструдировали в гранулы с диаметром 2 мм и длинной 2.5 мм, сушили при 80°C в течение 2 ч, а затем прокаливали при 550°C в течение 10 ч.
Исследование катализатора. Значение удельной поверхности (БЭТ) было определено методом адсорбции азота при 77 K с помощью прибора Micromeritics TriStar II 3020.
Дифрактограммы образцов по РФА были получены с помощью дифрактометра Rigaku D/max-2550pc с CuKα излучением (40 кВт, 250 мА) и углами съемки (2θ) от 20° до 90°.
ТПД аммиака проводили с помощью адсорбционного газоанализатора PX200. Образец (100 мг) предварительно нагревали при 500°C в течение 1 ч в атмосфере гелия, затем охлаждали до 40°C, наполняли газовой смесью 4% NH3/He в течение получаса, а затем газ переключали на чистый гелий (расход газа 30 мл/мин) на 1 ч для удаления слабо адсорбированного азота, и, наконец, нагревали образец до 500°C со скорость 10°C/мин в атмосфере чистого He (расход газа 30 мл/мин).
Температурно-программируемое восстановление (ТПВ) проводили с помощью газоанализатора Micromeritics AutoChem II 2920: образец катализатора (100 г) измельчали до фракции 60 меш и предварительно нагревали в атмосфере аргона до 400°C в течение 1 ч, затем охлаждали до 80°C и восстанавливали в токе газовой смеси 10% H2/Ar (расход газа 30 мл/мин) в температурном интервале 80–800°C со скоростью нагрева 15°C/мин.
Температурно-программируемая десорбция кислорода (ТПД O2) проводилась с использованием прибора GC1690: образец катализатора (100 мг) обрабатывали газовой смесью 5% O2/Ar (расход газа 30 мл/мин) при 300°C в течение 1 ч, затем охлаждали до 50°C, и нагревали до 900°C со скоростью нагрева 20°C/мин в атмосфере чистого гелия (расход газа 40 мл/мин).
Испытание активности. Испытание каталитической активности проводили в трубчатом реакторе (внутренний диаметр – 10 мм, длина – 600 мм) с неподвижным слоем катализатора. Температуру в зоне катализатора поддерживали постоянной и измеряли с помощью термопары; когда температура достигала необходимого значения и стабилизировалась, кислород, подачу которого регулировали массовым расходомером, подавали в течение часа. Затем в систему с помощью насоса с постоянным расходом в течение 1 ч вводили деионизированную воду, после чего вместо чистой воды был использован водный раствор пиколина. Неочищенные продукты собирали и анализировали методом газовой хроматографии на приборе Agilent 6820. Конверсию пиколинов и селективность образования пиридилальдегидов рассчитывали на основе площадей их пиков на хроматограмме (с использованием метода внешнего стандарта).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Наши первоначальные усилия были сфокусированы на скрининге трех молибдатов висмута, а именно Bi2Mo3O12 (α), Bi2Mo2O9 (β) и Bi2MoO6 (γ), используя 2-пиколин в качестве модельного сырья. В табл. 1 представлены результаты испытания их каталитической активности. Известно, что все три фазы могут быть легко получены при 600°C (строки 1–3 и 6–8), в то время как при 550°C (строки 5 и 10) β-фаза была нестабильна, и, таким образом, разлагалась на α- и γ-фазы [23] в независимости от того, была ли она нанесена на диоксид титана или нет. Очевидно, что катализаторы с чистой α- и β-фазами обладают схожей активностью, которая выше, чем у γ-фазы (например, строки 6, 7 и 8); присутствие γ-фазы в катализаторе вызывает снижение конверсии 2-пиколина и селективности образования 2-формилпиридина (строки 5 и 10). Кроме того, для α- и β-фаз значение конверсии почти удваивается при нанесении на диоксид титана, при этом селективность изменяется лишь незначительно, не более чем на 7%, что иллюстрируют примеры MB32/550 (α, конверсия – 15.5%, селективность – 87.3%) и MB32T550 (α, конверсия – 29.5%, селективность – 81.3%).
Таблица 1.
Скрининг Bi–Mo-катализаторов окисления 2-пиколинаa
| № | Катализаторб | Активная фаза | Конверсия 2-пиколина, % | Селективность образования 2-формилпиридина, % |
|---|---|---|---|---|
| 1 | MB32/600 | α | 8.6 | 94.1 |
| 2 | MB11/600 | β | 9.9 | 93.2 |
| 3 | MB12/600в | γ | N.a. | N.a. |
| 4 | MB32/550 | α | 15.5 | 87.3 |
| 5 | MB11/550 | α и γ | 12.2 | 78.5 |
| 6 | MB32T600 | α | 17.7 | 87.2 |
| 7 | MB11T600 | β | 19.1 | 86.6 |
| 8 | MB12T600 | γ | 13.1 | 63.5 |
| 9 | MB32T550 | α | 29.5 | 81.3 |
| 10 | MB11T550 | α, β и γ | 19.7 | 68.1 |
| 11 | MB12T550 | γ | 16.0 | 52.1 |
a Условия реакции: 4 г катализатора, 270°C, объемный расход 10% водного раствора 2-пиколина 0.5 мл/мин, объемный расход кислорода 0.1 л/мин. б Катализаторы MBxy/z представляют собой массивные катализаторы, а MBxyTz – нанесенные на TiO2, M = Mo, B = Bi, xy = молярное соотношение Mo/Bi, а z = температура прокаливания. в Катализатор был непригоден для испытания активности ввиду низкой механической прочности.
Исходя из изложенного выше, впоследствии для дальнейших исследований была использована α-фаза, нанесенная на диоксид титана (MB32T550), из-за ее стабильности, а также наибольшей, по сравнению с другими образцами, производительности. С точки зрения реакционной способности, одним из важнейших параметров является температура реакции. При проведении реакции при 290°C наблюдалось увеличение конверсии до 45.2%, сопровождаемое снижением селективности до 70.3%, дальнейшее увеличение температуры до 310°C обеспечивает увеличение конверсии до 57.2%, но также приводит к снижению селективности до 61.0%. С точки зрения экономии атомов, а также выхода продукта, температура 290°C была принята в качестве обоснованного и целесообразного значения для последующих экспериментов.
В предыдущих исследованиях мы обнаружили, что введение дополнительного количества MoO3 в состав катализатора α-Bi2Mo3O12/TiO2 значительно увеличивает его активность. Поэтому было приготовлено пять образцов катализатора (табл. 2) и, соответственно, была испытана их каталитическая активность (табл. 3) [21]. Было показано, что конверсия в присутствии MB32T и MT, содержащих только один активный компонент, была намного ниже, чем конверсия в присутствии трех других катализаторов с двумя активными компонентами. Похожая зависимость наблюдалась в отношении селективности. Более того, катализатор MB21T с молярным соотношением α-Bi2Mo3O12/MoO3, равным 1, обеспечивал наибольшую производительность. При этом можно полагать, что причиной высокой производительности катализатора Bi2Mo3O12/TiO2, допированного MoO3, является синергетический эффект между α-Bi2Mo3O12 и MoO3.
Таблица 2.
Свойства нанесенных на TiO2Bi−Mo-катализаторовa
| Катализатор | Состав катализатора | Удельная поверхность, м2/г | Расход водорода по ТПВ, ммоль/г кат | |
|---|---|---|---|---|
| α-фаза/MoO3, моль/моль | cодержание MoO3,% | |||
| MT | 0 : 1 | 15 | 4.1 | 1.56 |
| MB32T | ∞ | 0 | 3.1 | 1.77 |
| MB21T | 1 : 1 | 2.1 | 4.0 | 1.96 |
| MB74T | 2 : 1 | 1.1 | 4.0 | 1.98 |
| MB85T | 5 : 1 | 0.5 | 3.9 | 1.96 |
Таблица 3.
Активность различных Bi−Mo-катализаторов в реакции окисления 2-пиколинаa
| № | Катализатор | Конверсия 2-пиколина, % | Селективность образования 2-формилпиридина, % |
|---|---|---|---|
| 1 | MT | 51.8 | 78.1 |
| 2 | MB32T | 45.2 | 70.3 |
| 3 | MB21T | 70.9 | 83.1 |
| 4 | MB74T | 63.6 | 80.3 |
| 5 | MB85T | 61.0 | 81.7 |
Для объяснения причин возникновения синергетического эффекта, в первую очередь были проведены анализы БЭТ (табл. 2) и РФА (рис. 1). Низкая активность MB32T может быть частично объяснена сниженной удельной поверхностью (3.0 м2/г) примерно на 25% в сравнении с другими образцами (около 4.0 м2/г), что, в результате, способствует образованию меньшего количества активных центров. Из рис. 2 видно, что катализатор MT состоит из MoO3 и анатаза (TiO2), а остальные 4 – из α-Bi2Mo3O12 и анатаза (TiO2). Согласно [23], катализаторы со значением мольного соотношения Bi/Mo меньшим, чем 2/3, состоят как из α-Bi2Mo3O12, так и из MoO3, причем, из-за крайне малого количества, MoO3 невозможно обнаружить в висмутсодержащих катализаторах методом РФА, но можно обнаружить с помощью СЭМ [21]. Кроме того, на дифрактограммах видно, что пик рутила (TiO2), который отсутствовал в остальных трех висмутсодержащих катализаторах, имел сопоставимую с анатазом (TiO2) интенсивность сигнала в образце MB32T. Поскольку Bond G.C. сообщил, что в чистых образцах анатаза (TiO2) не наблюдается фазового перехода в рутил при температурах ниже 600°C [24], одновременное присутствие анатаза и рутила в MB32T, прокаленного при 550°C, означает, что наличие α-Bi2Mo3O12 упрощает фазовый переход анатаза в рутил, что приводит к снижению удельной поверхности катализатора (на 25% по сравнению с другими образцами, как показано в табл. 2). Присутствие MoO3, видимо, предотвращает этот фазовый переход, поскольку в трех других образцах с избытком MoO3 образование рутила не наблюдалось. Можно сделать вывод, что наличие Mo ослабляет взаимодействие между α-Bi2Mo3O12 и TiO2, тем самым сдерживая изменение кристаллической структуры TiO2. Иначе говоря, этот синергетический эффект, с одной стороны, объясняется стабилизацией кристаллической структуры катализатора и, следовательно, сохранением удельной поверхности.
Рис. 1.
Дифрактограммы катализаторов MT, MB32T, MB21T, MB74T и MB85T, где (a) – анатаз TiO2, (a*) – рутил TiO2, (b) MoO3 проб и (c) α-Bi2Mo3O12.
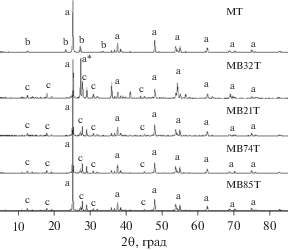
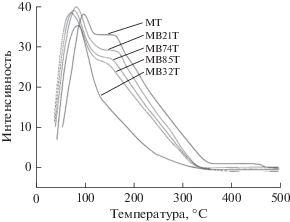
Реакции окисления углеводородов на гетерогенных катализаторах – процесс, который следует по механизму Марса–ван-Кревелена [25, 26], включающему в себя много стадий: 1 – хемосорбция углеводорода на поверхности катализатора и отщепление α-протона с образованием интермедиата; 2 – внедрение решеточного кислорода в интермедиат с образованием продукта; 3 – наполнение решетки кислородом, удаленным с поверхности в ходе второй стадии, для восстановления активного центра. На образовании аллильного интермедиата за счет отщепления α-протона основан общепринятый подход к окислению пропилена [27]. Bi рассматривали как центр отщепления α-протона для генерирования аллильных частиц, в то время как Mo – центр хемосорбции и введения кислорода, поэтому относительная активность α-Bi2Mo3O12 была выше, чем у MoO3, так как MoO3 не имеет центров для отщепления α-протона [28]. На основании вышенаписанного было предложено образование пиколинового интермедиата в результате отщепления α-протона в ходе окисления 2-пиколина в 2-формилпиридин. Однако, в случае окисления 2-пиколина в 2-формилпиридин факт того, что активность MT была выше, чем активность MB32T, доказывает, что MoO3 все-таки обладает активным центром для отщепления α-протона. Правдоподобным объяснением этого является то, что пиколин – вещество с основными свойствами, так что кислотность катализатора благоприятна для его адсорбции, и, соответственно, взаимодействия с катализатором, а затем, возможно, отщепления α-протона. Для определения кислотности катализатора было проведено ТПД аммиака, что показано на рис. 2. Очевидно, что слабые кислотные центры присутствуют во всех образцах. Значения площади под пиками десорбции аммиака показывают, что плотность поверхностных кислотных центров уменьшается в ряду MB21T > MT > > MB74T > MB85T > MB32T, свидетельствуя о том, что поверхностная кислотность катализаторов, содержащих α-Bi2Mo3O12, возрастает с увеличением количества MoO3. Кроме того, немного большая плотность кислотных центров в MB21T по сравнению с MT показывает синергизм между α-Bi2Mo3O12иMoO3 в изменении поверхностной кислотности катализатора.
Наряду с этим, отщепление α-протона в пиколине легко осуществить, потому что пиколиновый интермедиат должен образовываться гораздо легче и быть более стабильным, чем аллильный интермедиат, поскольку электрон пиколинового радикала после отщепления α-протона делокализован сопряженным гетероциклом, в то время как аллильный радикал стабилизирован только за счет кратной связи. Кроме того, атом азота, на котором происходит сближение электронов, из-за его большой электроотрицательности, может дополнительно стабилизировать пиколиновый радикал.
В литературе показано, что подвижность решеточного кислорода играет ключевую роль в определении активности висмут-молибдатных катализаторов [29]; добавление небольшого количества оксида проводящего металла может способствовать переносу решеточного кислорода [30], и, следовательно, увеличению активность катализаторов. В этом случае, MoO3 является оксидом проводящего металла [31], хотя связь между проводимостью и каталитической активностью не была предметом настоящего исследования.
В анализе ТПВ (табл. 2) факт того, что количество потребляемого водорода для MB21T, MB74T и MB85T было почти одинаковым и большим, чем для MTи MB32T, свидетельствует о том, что α-Bi2Mo3O12 и MoO3 в синергии могут повысить подвижность кислорода в решетке катализатора, и таким образом улучшить его способность к восстановлению, о чем в дальнейшем свидетельствуют результаты ТПД O2, показанные на рис. 3.
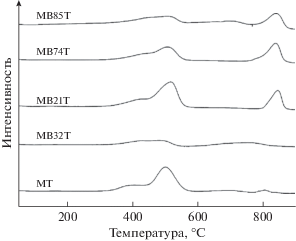
В спектре образца MT наблюдался один десорбционный пик в области около 500°C с плечом и слабый десорбционный пик около 800°C, у MB32T – два слабых широких пика около 500 и 750°C, а у образцов MB21T, MB74T и MB85T – один пик около 500°C с плечом и один около 850°C. Также видно, что общая интенсивность пиков десорбции в спектрах MB21T, MB74T и MB85T была выше, чем для MB32T и MT, а для MB21T она была наибольшей. Это является показателем того, что α-Bi2Mo3O12 и MoO3 обладали синергетическим взаимовлиянием на увеличение подвижности кислорода в решетке катализатора.
Синергетический эффект может быть связан либо с образованием новой фазы, либо с “механизмом дистанционного управления”, в котором нет образования новых фаз [32]. Поскольку РФА анализ не дал никаких признаков образования новой фазы, увеличение собственной активности катализаторов α-Bi2Mo3O12/TiO2, допированных MoO3, можно объяснить с точки зрения “механизма дистанционного управления”, при котором активируется “вторичный кислород” посредством миграции “донорной фазы” в “акцепторную”, где и создаются новые каталитически активные центры [33]. Предыдущие исследования показали, что Bi способствует диссоциации газообразного кислорода в решеточный и его дальнейшей диффузии к активным центрам [34]. Подвижность решеточного кислорода в MoO3 очень высока [35], поэтому решеточный кислород, дополнительно активированный посредством α‑Bi2Mo3O12, должен легко диффундировать в MoO3, где создаются новые активные каталитические центры, что и приводит к увеличению активности. Можно сделать вывод о том, что тесное взаимодействие между α-Bi2Mo3O12 и MoO3 приводит к изменению электронных свойств катализатора, вслед за этим – к модификации способности десорбировать кислород и восстановительных свойств катализаторов, что приводит к увеличению их производительности.
Также, для исследования каталитической активности в реакции окисления двух других изомеров пиколина был использован MB21T, и, как показано в табл. 4, его применение также было эффективно в окислении 4-пиколина. Низкая конверсия 3-пиколина главным образом была связанна с тем, что метильный заместитель в ме-та-положении к атому азота имеет большую электронную плотность, чем у заместителей в о-рто- и пара-положениях, что неблагоприятно для его депротонирования.
Таблица 4.
Окисление пиколинов в присутствии катализатора MB21Ta
| № | Субстрат | Конверсия пиколина, % | Селективность образования пиридилальдегида, % |
|---|---|---|---|
| 1 | 2-Пиколин | 70.9 | 83.1 |
| 2 | 3-Пиколинб | 18.9 | 93.7 |
| 3 | 4-Пиколин | 73.1 | 80.3 |
В данной работе изучено окисление пиколинов в пиридилальдегиды в присутствии висмут-молибденовых катализаторов, нанесенных на TiO2. Было показано, что α-Bi2Mo3O12 превосходит β-Bi2Mo2O9 и γ-Bi2MoO6 с точки зрения реакционной способности. Допирование α-Bi2Mo3O12/ TiO2 с помощью MoO3 приводит к увеличению каталитической активности, что обусловлено синергетическим эффектом между α-Bi2Mo3O12 и MoO3. Катализатор с мольным соотношением α-Bi2Mo3O12 к MoO3 равным 1 : 1 обладал наибольшей активностью. 2-Пиколин, и 4-пиколин могут быть легко окислены в 2-формилпиридин и 4-формилпиридин соответственно. Гораздо меньшая конверсия 3-пиколина главным образом была связанна с тем, что метильный заместитель в ме-та-положении к атому азота имеет большую электронную плотность, чем у заместителей в ор-то- и пара-положениях, что неблагоприятно для его депротонирования.
Список литературы
Liu S., Wei W., Li Y., Liu X., Cao X., Lei K., Zhou M. // Eur. J. Med. Chem. 2015. V. 95. P. 473.
Aggarwal P.S., Tiwari A.K., Kumar V., Pratap R., Singh G., Mishra A.K. // RSC Adv. 2015. V. 5. P. 23 471.
Zhang M., Chen G.Y., Li T., Liu B., Deng J.Y., Zhang L., Yang L.Q., Xu X.H. // J. Heterocycl. Chem. 2015. V. 52. P. 1887.
Zi G., Zhang F., Liu X., Ai L., Song H. // J. Organomet. Chem. 2010. V. 695. P. 730.
Zhang L., Murphy C.S., Kuang G.C., Hazelwood K.L., Constantino M.H., Davidson M.W., Zhu L. // Chem. Comm. 2009. V. 47. P. 7408.
Trach Yu.B., Makota O.I. // Petrol. Chemistry. 2009. V. 49. P. 413.
Mel’nik L.V., Meshechkina A.E., Rybina G.V., Srednev S.S., Moskvichev Yu.A., Kozlova O.S. // Petrol. Chemistry. 2012. V. 52. P. 313.
Tewary P.K., Lal G., Ganesan K. // React. Kinet. Catal. Lett. 1990. V. 41. P. 283.
Chen Y.Q., Dai L.Y., Yang N.W. // J. Chem. Eng. Chin. Univ. 2002. V. 16. P. 436.
Takehira K., Shishido T., Song Z., Matsushita T., Kawabata T., Takaki K. // Catal. Today. 2004. V. 91–92. P. 7.
Jia A.P., Hu G.S., Meng L., Xie Y.L., Lu J.Q., Luo M.F. // J. Catal. 2012. V. 289. P. 199.
Zhang G.Q., Zhang X., Lin T., Gong T., Qi M. // Chin. Chem. Lett. 2012. V. 23. P. 145.
Nuankaeoa P., Phuruangratb A., Kuntaluec B., Dumrongrojthanathd P., Thongtemd T., Thongteme S. // Russ. J. Appl. Chem. 2016. V. 89. P. 830.
McCain C.C., Gough G., Godin G.W. // Nature. 1963. V. 198. P. 989.
Kremenic G., Nieto J.M.L., Weller S.W., Tascon J.M.D., Tejuca L.G. // Ind. Eng. Chem. Res. 1987. V. 26. P. 1419.
Bettahar M.M.,Costentin G., Savary L., Lavalley J.C. // Appl. Catal. A: Gen. 1996. V. 145. P. 1.
Moro-Oka Y., Ueda W. // Adv. Catal. 1994. V. 40. P. 233.
Zhai Z., Getsoian A.B., Bell A.T. // J. Catal. 2013. V. 308. P. 25.
Zhai Z., Wang X., Licht R., Bell A.T. // J. Catal. 2015. V. 325. P. 87.
Aiswarya P.M., Ganesan R., Rajamadhavan R., Gnanasekaran T. // J. Alloy. Compd. 2018. V. 745. P. 744.
Zhang L.L., Yu J., Wang X.Z., Zhang S.J., Dai L.Y. // J. Chem. Eng. Chin. Univ. 2016. V. 30. P. 1082.
Yu J., Dai L. Y., Wang X.Z., Chen Y.Q., Zhang L.L. // J. Chem. Eng. Chin. Univ. 2013. V. 27. P. 604.
Egashira M., Matsuo K., Kagawa S., Seiyama T. // J. Catal. 1979. V. 58. P. 409.
Bond G.C., Tahir S.F. // Appl. Catal. 1991. V. 71. P. 1.
Wachs I.E., Weckhuysen B.M. // Appl. Catal. A: Gen. 1997. V. 157. P. 67.
Usachev N.Ya., Gerzeliev I.M., Kharlamov V.V., Kalinin V.P., Belanova E.P., Kanaev S.A., Kazakov A.V., Starostina T.S. // Pet. Chem. 2016. V. 56. P. 841.
Burrington J.D., Kartisek C.T., Grasselli R.K. // J. Org. Chem. 1981. V. 46. P. 1877.
Burrington J.D., Kartisek C.T., Grasselli R.K. // J. Catal. 1980. V. 63. P. 235.
Jung J.C., Lee H., Kim H., Chung Y.M., Kim T.J., Lee S.J., Oh S.H., Kim Y.S., Song I.K. // Catal. Lett. 2008. V. 124. P. 262.
Le M.T., Do V.H., Truong D.D., Bruneel E., Driessche I.V., Riisager A., Fehrmann R., Trinh Q.T. // Ind. Eng. Chem. Res. 2016. V. 55. P. 4846.
Bespalova Z.I., Khramenkova A.V. // Russ. J. Appl. Chem. 2013. V. 86. P. 539.
Mazumder B., Védrine J.C. // Appl. Catal. A: Gen. 2003. V. 245. P. 87.
Védrine J.C. // Appl. Catal. A: Gen. 2014. V. 474. P. 40.
Jiang Z.C., An L.D., Chen Z.S., Zhang B., Gao L., Yin Y.G. // Sci. China B Chem. 1992. V. 35. P. 28.
Kühn S., Weber D., Lerch M., Ressler T. // ChemCatChem. 2016. V. 8. P. 758.
Дополнительные материалы
- скачать JieYu_1.1.tiff
- Рис. S1. РФА образцов (а) MB11/600, (б) MB32/600 и MB32/550, (в) MB12/600
- скачать JieYu_1.2.tiff
- Рис. S1. РФА образцов (г) MB11/550, (д) MB32T600, (е) MB11T600
- скачать JieYu_1.3.tiff
- Рис. S1. РФА образцов (ж) MB12T600, (з) MB32T550, (и) MB11T550
- скачать JieYu_1.4.tiff
- Рис. S1. РФА образцов (к) MB12T550
- скачать JieYu_2.tiff
- Рис. S2. Масс-спектры 2-формилпиридина
- скачать JieYu_3.tiff
- Рис. S3. Масс-спектры 3-формилпиридина
- скачать JieYu_4.tiff
- Рис. S4. Масс-спектры 4-формилпиридина