

Нефтехимия, 2020, T. 60, № 3, стр. 251-269
Лигнин – возобновляемый ресурс углеводородных продуктов и энергоносителей (обзор)
О. В. Арапова 1, *, А. В. Чистяков 1, М. В. Цодиков 1, И. И. Моисеев 1
1 Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Россия
* E-mail: arapova@ips.ac.ru
Поступила в редакцию 15.11.2019
После доработки 18.11.2019
Принята к публикации 18.11.2019
Аннотация
Представлен обзор современных промышленных и развиваемых экспериментальных подходов к выделению и переработке лигнина, как возобновляемого и накапливаемого сырья растительного происхождения в важные органические продукты. На основании литературного анализа показана перспективность использования каталитических систем для повышения селективности и глубины превращения лигнина. В работе рассматриваются основные принципы использования микроволнового стимулирования в процессах деструкции устойчивой структуры лигнина, в том числе, новый подход к проведению плазменно-каталитического скоростного превращения лигнина в синтез-газ.
Интерес к химии важнейших компонентов парниковых газов (СО2 и метану) в конце 60-х гг. стимулировался не столько проблемами защиты окружающей среды, сколько желанием сократить потребление нефти. Появилось направление, получившее название “химия С1” [1]. Это направление в наши дни дополнило развитие методов “зеленой химии” [2, 3], в которой в качестве основного источника возобновляемого углерода рассматривают биомассу. Задача защиты окружающей среды стимулировала интерес промышленности к замене ископаемого сырья возобновляемым. В результате 21 в. ознаменован бурным развитием исследований, направленных на разработку эффективных подходов к превращению возобновляемого сырья в компоненты топлив, энергоносители и важные мономеры химии. К возобновляемым источникам углеводородов относятся продукты ферментативного брожения биомассы, селекционированные микроводоросли с повышенным содержанием липидов, и лигнин (от лат. lignum – дерево, древесина), который представляет собой cложный трехмерный полимер, содержащийся в клетках сосудистых растений и некоторых водорослях.
Лигнин – побочный продукт деревообрабатывающей промышленности [4]. Полимерно-сшитая устойчивая молекулярная структура лигнина затрудняет его переработку. Специфика лигнина как сырья – постоянное накопление его запасов, образование техногенных месторождений, так как он почти не подвергается биохимическому окислению, по последним данным ежегодное количество отходов лигнина составляет 150–200 млн т [5]. Предлагается много методов переработки продуктов брожения биомассы и растительных масел в продукты нефтехимии и энергоносители, в то время как для переработки лигнина в ценные продукты еще не найдены эффективные подходы.
Лигнин – второй после целлюлозы наиболее распространенный углеродсодержащий ресурс на Земле – является перспективным источником природных аренов [4]. В составе лигнина содержится достаточно большое количество водорода и углерода, что позволяет рассматривать его в качестве перспективного сырьевого источника для получения не только аренов, но также синтез-газа и водорода, являющихся ценными продуктами нефтехимии [6]. Лигнин представляет собой трехмерный полимер с молекулярной массой 1000–15 000 Да, проявляющий высокую устойчивость к химическому и физическому воздействию [7]. Температура самовоспламенения лигнина: аэрогеля – 300°C, аэровзвеси – 450°C; нижний концентрационный предел распространения пламени – 40 г/м3; максимальное давление взрыва – 710 кПа; максимальная скорость нарастания давления – 35 МПа/с; минимальная энергиия зажигания – 20 мДж; минимальное взрывоопасное содержание кислорода – 17 об. % [4]. Однако пожары на техногенных месторождениях лигнина представляют угрозу для окружающей среды, поскольку в отвалах хранится лигнин, содержащий целлюлозу. Например, возгорание на лигнинохранилище Зиминского гидролизного завода (25 га, 10 м высотой, около 262 тыс. т) в 2005 г. переросло в техногенную катастрофу, предотвращение которой потребовало больших усилий [8].
Исследования в области разработки новых технологий, обеспечивающих получение важных продуктов и энергоносителей из лигнина, актуальны не только для устойчивого развития общества, но и для оздоровления окружающей среды и снижения зависимости от невозобновляемых углеродсодержащих депозитов.
Цель настоящего обзора – анализ современного состояния процессов переработки растительного сырья с выделением лигнина и рассмотрение существующих и перспективных подходов к его переработке.
ОСНОВНЫЕ СВОЙСТВА И ОСОБЕННОСТИ СТРОЕНИЯ ЛИГНИНА
Лигнин – один из трех основных компонентов сосудистых растений, другими двумя являются целлюлоза и гемицеллюлоза [9]. Лигнин составляет 30% от всего неископаемого углерода на Земле. Это рентгеноаморфный трехмерный полимер, состоящий из метоксилированных фенилпропановых структур [9]. В клеточной ткани растений лигнин заполняет пространство между целлюлозой и гемицеллюлозой, и выполняет, как смола, функцию связывания и упрочнения лигноцеллюлозной матрицы [10]. Сшиваясь с углеводным полимером, лигнин, формирует структуру и добавляет прочность клеточным стенкам, контролирует потоки жидкости и защищает от биохимических стрессов, путем подавления ферментативной деградации структурных полисахаридов [11].
Точная структура протолигнина, (необработанного лигнина), найденного в растениях (также обозначаемого, как “природный лигнин”), еще до конца неизвестна. Лигнин – сетчатый сополимер (рис. 1), биосинтез которого протекает через радикальную полимеризацию трех основных мономеров: п-кумарилового, кониферилового и синапилового спиртов [12, 13].
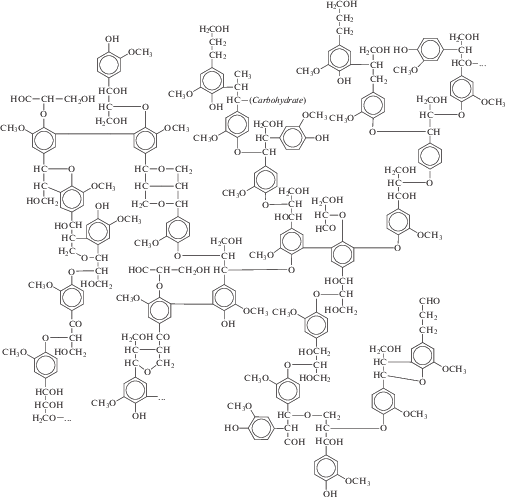
Когда они включены в полимер лигнина, единицы, которые образованы из монолигнолов, называются п-гидроксифениловая (Н), гвайациловая (G) и сирингиловая (S), соответственно (рис. 2) [12–17]. Полимеризация идет за счет радикальной поликонденсации нескольких монолигнолов – паракумарового, конеферилового, синапилового спиртов под действием ферментов, таких как пероксидазы и лакказы, с образованием в растениях сосудистого лигнина [18, 19]. В растениях лигнин находится в виде комплексов с целлюлозой. Состав, молекулярная масса и количество лигнина зависят от вида растений: в целом, количество лигнина в растениях уменьшается в следующем порядке: хвойные (мягкая древесина) > лиственные (твердая древесина) > трава. Древесина лиственных пород содержит 18–25% лигнина, хвойных – 23–50%, в соломе злаков 12– 20% от массы [20].
Рис. 2.
Основные предшественники лигнина (монолигнолы) и соответствующие им структуры в полимере лигнина.
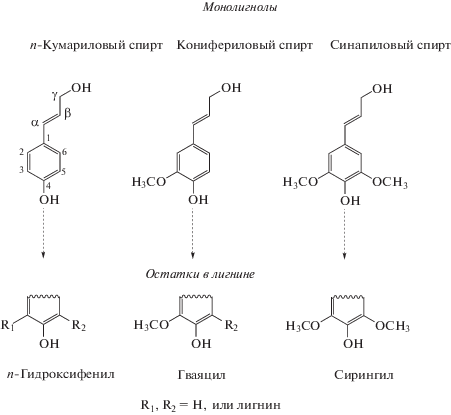
В ходе естественного процесса лигнификации растений, а также промышленных процессов, применяющихся для извлечения лигнина из растительного материала, образуются различные типы связей между строительными блоками монолигнолов: β-O-4', углерод–углеродные 5–5, β-5, 4-O-5, β-1, дибензодиоксицин и β-β-связи [21–27].
Монолигнолы связаны в основном эфирными связями β-O-4', число которых составляют более 50% структуры лигнина [9, 28]. Фрагментация этих связей, как правило, приводит к образованию водорастворимых аренов, содержащих свободные фенольные группы [9]. Углерод–углеродные (5–5) связи в лигнине проявляют высокую устойчивость к разрыву, и большинство этих связей, как правило, сохраняются в ходе предварительной механической обработки [9]. Помимо существующей в природном лигнине связи углерод–углерод во время обработки лигнина NaOH и Na2SO4 (или Na2S) при 165–170°С и давлении 0.6–0.8 МПа (крафт-процесс) могут возникать дополнительные С–С-связи.
Состав лигнина хвойных, лиственных и травянистых пород отличается также по относительному содержанию п-кумариловых, конифериловых и синапиловых монолигнолов. Конифериловый спирт составляет приблизительно 90% лигнина хвойных пород, в то время как относительно равные пропорции кониферилового и синапилового спиртов содержатся в лигнине лиственных пород, хотя существует много исключений [29]. Лигнин травянистых пород имеет относительно неоднородный состав со средним отношением кониферилового, синапилового, п-кумарилового спиртов 1 : 0.8 : 0.6 [30, 31]. Дополнительное количество метокси-групп на синапиловых ароматических кольцах предотвращает образование 5–5 или дибензодиоксициновых связей; таким образом, из лигнинового полимера лиственных пород образуется более линейная структура по сравнению с хвойными породами.
Как и другие биореактивные полимеры, лигнин проявляет антиоксидантную, противогрибковую и противомикробную активности. По сравнению с целлюлозой/гемицеллюлозой лигнин более устойчив к химическим и биологическим воздействиям и обладает более высокой прочностью, которая обеспечивает структурную целостность клеточной стенки, защищая другие компоненты от ферментативной деградации [32–35]. Лигнин также способен поглощать УФ-излучение и имеет низкую теплопроводность [36, 37]. Состав и физико-химические свойства лигнина делают его перспективным сырьем для возобновляемой энергетики. Наличие реакционно-способных функциональных групп позволяет модифицировать молекулу с использованием целевых фрагментов, таких как пептиды или антитела для получения привитых сополимеров. Другие полезные свойства лигнина – гидрофильность или гидрофобность, в зависимости от источника лигнина; хорошие реологические характеристики, такие как вязкость и упругость; пленкообразующая способность; и совместимость с широким спектром промышленных химикатов [38].
Природный лигнин почти бесцветен, но после обработки кислотой или щелочью он приобретает коричневый или темно-коричневый цвет [39, 40]. На содержание фенольных, гидроксильных, карбоксильных и сульфонатных функциональных групп влияет способ экстракции лигнина [12]. Однако разработка универсальных методов получения промышленно важных продуктов на базе лигнина затруднено из-за его непостоянной молекулярной структуры [41].
МЕТОДЫ ВЫДЕЛЕНИЯ ЛИГНИНА
Существует несколько способов, с помощью которых лигнин можно выделить из биомассы. Эти способы можно разделить на две группы: 1) процессы, в которых лигнин разрушается до растворимых фрагментов и удаляется путем отделения твердого остатка из отработанного щелока; 2) процессы, в которых в процессе превращения целлюлозы избирательно гидролизуются полисахариды, а лигнин остается вместе с твердыми продуктами. В первую группу входят процессы варки целлюлозы, такие как крафт, лигносульфонатный, щелочной и органосольвентный (рис. 3), а во вторую – кислотный гидролиз лигноцеллюлозы с получением мономеров сахарозы, фурфурола и левулиновой кислоты. Во время отделения от других компонентов структура лигнина обычно претерпевает значительные изменения и, следовательно, поведение изолированного лигнина может существенно отличаться от поведения природного, содержащегося, например, в древесине. В дополнение к структурным изменениям, вызванным жесткими условиями экстракции, в структуру лигнина вводится сера в процессах крафт- и лигносульфонатной варки целлюлозы, которые в настоящее время используются наиболее часто.

Крафт-процесс. Крафт (сульфатная) варка – основной метод обработки лигноцеллюлозы, на него приходится около 85% произведенного лигнина [42, 43]. При переработке целлюлозы лигнин, как правило, вступает в два типа реакций: 1) реакции разложения, приводящие к образованию меньших фрагментов лигнина и повышению растворимости в водно-щелочном растворе; 2) реакции конденсации, в которых образуются связи, устойчивые к разложению в щелочах, что уменьшает эффективность переработки.
В этом процессе выделяют целлюлозные волокна, а лигнин и гемицеллюлоза древесной щепы переходят в раствор гидроксида натрия и сульфида натрия (белый щелок) [22, 44]. В начале процесса рН, равный 13–14, постепенно снижается в результате высвобождения органических кислот из гемицеллюлозы и лигнина. После предварительной выдержки варочного раствора при 170°C растворенные органические и неорганические соединения выгружают из реактора в виде основного водного слоя, содержащего до 15% твердого вещества (слабый черный щелок).
Из-за последовательности реакций разрушения структуры и конденсации природный лигнин подвергается серьезным химическим и структурным изменениям [25]. Например, процент прочных C–C-связей между пропилфенольными мономерами становится значительно выше, чем в лигнине, первоначально содержащемся в древесине [9], что затрудняет дальнейшую деполимеризацию крафт-лигнина. Хотя крафт-лигнин с высоким выходом можно выделить с помощью подкисления черного щелока, эта технология используется всего лишь на ~2% деревоперерабатывающих предприятий [44], производящих коммерческий крафт-лигнин для продажи, а большинство предприятий сжигает органическую составляющую черного щелока в котлах-утилизаторах для производства электроэнергии и пара. Крафт-лигнин в своей структуре обычно содержит небольшое количество серы (1.5–3.0 мас. % [45]), что значительно ниже, чем лигнин, полученный из лигносульфонатной целлюлозы (4–8% [46]). Тем не менее, поскольку крафт-лигнин растворим только при рН >10, с целью повышения растворимости в воде большую часть коммерчески доступного крафт-лигнина обрабатывают сульфитом натрия при температурах не превышающих 200°C [25, 46]. Из-за отсутствия достоверной методики определение молекулярной массы лигнина является дискуссионным вопросом; считается, что среднее значение молекулярной массы Мw крафт-лигнина обычно колеблется от 1000 до 3000 Да, но также она может достигать 15 000 Да [44, 47]. Несмотря на упомянутые недостатки, большое количество лигнина, произведенного с использованием этого процесса, востребовано для получения из него ряда важных продуктов [48].
Лигносульфонатный процесс. Этот процесс представляет собой метод экстракции, обычно используемый в целлюлозно-бумажной промышленности [22]. Включает в себя реакцию между лигнином, сульфитом металла и диоксидом серы; при этом, зачастую используемые ионы кальция, магния или натрия действуют как противоионы с целью обеспечения нейтрального рН среды [25, 38]. В зависимости от состава химических соединений и их концентрации pH системы может варьироваться в пределах от 2 до 12. Процесс проводят при 120–180°C в течение 1–5 ч [22, 46]. В этих условиях лигносульфонатная делигнификация приводит к расщеплению α-эфирной (α-O-4') и β-эфирной (β-O-4') связей лигнина. Содержание серы в лигносульфонатах, полученных этим способом, может составлять от 4 до 8%, а из-за низкого значения pKa (менее 2) сульфонатных групп, изолированный лигнин в большинстве случаев водорастворим [25, 46], в то время как целлюлоза остается в твердом состоянии [44, 49]. Лигносульфонаты имеют более высокую Mw по сравнению с лигнином, экстрагированным крафт-процессом, из-за включения сульфонатных групп в полимер лигнина. В зависимости от источника лигнина Mw варьируется от 1000 до 50 000 Да при выделении лигнина из лиственных или хвойных пород, соответственно [22, 44, 46]. Однако лигносульфонатный процесс имеет ряд недостатков, таких как образование новых С–С-связей, повышенную зольность, высокое содержание серы и других загрязняющих веществ [44].
Щелочной процесс, или щелочная варка, используется для обработки недревесных материалов, таких как трава, солома и сахарный тростник, что составляет 5% от общего объема производства целлюлозы [25, 44]. Способ перевода лигнина в раствор аналогичен методу солюбилизации в крафт-процессе. Биомассу перерабатывают в присутствии 13–16 мас. % водного раствора гидроксида натрия при 140–170°C. Каталитические количества антрохинона заметно снижают деградацию углеводов и растворение лигнина; а скорость удаления лигнина еще ниже по сравнению с крафт-процессом [25, 38, 44]. Полученный щелочной лигнин трудно выделить путем фильтрации или центрифугирования из-за высокого содержания в нем карбоксильных групп, возникающих в результате окисления алифатических гидроксильных групп, что придает конечному продукту свойства диспергента [38]. Среднее значение Mw щелочного лигнина составляет 2400 Да и варьирует от 1000 до 3000 Да в зависимости от типа исходного сырья [25, 44, 50].
Однолетние растения легче делигнифицировать. Делигнификация биоресурсов в среде щелочи или в смеси щелочь/антрахинон кажется предпочтительней описанных ранее способов, поскольку отработанный раствор и, следовательно, осажденный лигнин, не содержат серы, Следует отметить, что присутствие серы в структуре крафт- и лигносульфонатных лигнинов всегда было препятствием для более эффективного использования лигнина в последующих каталитических процессах. Отсутствие серы делает щелочной лигнин потенциально перспективным источником для производства высокоценных продуктов [38].
Органосольвентный процесс. Для получения органосольвентного лигнина, главным образом, используют волокнистые остатки древесины или жмых сельскохозяйственных культур, оставшийся после извлечения сока [51]. Существует ряд процессов переработки целлюлозы с использованием низкокипящих органических растворителей, в том числе, органосольвентный, в большинстве которых для делигнификации используют химические соединения, не содержащие серы. Наиболее часто используемые растворители: водные растворы этанола, метанола, уксусной и муравьиной кислот, которыми обработку лигнина проводят при температурах 170–190°C [44, 52, 53]. Применение высококипящих растворителей для органосольвентной варки позволяет проводить делигнификацию при атмосферном давлении, однако в этих условиях извлечение целлюлозы из отработанного щелока затруднено [54]. Во время органосольвентного процесса разрушаются главным образом связи α-O-4' лигнин−углевод, а связи β-O-4' в меньшей степени подвергаются расщеплению [44, 55]. При обработке сохраняется структура природного лигнина, что позволяет считать этот способ наиболее эффективным для дальнейшего увеличения стоимости лигнина [25, 44]. Основные преимущества органосольвентного процесса заключается в возможности организации отдельных потоков целлюлозы, гемицеллюлозы и лигнина, что позволяет увеличить добавленную стоимость всех компонентов биомассы.
Более того, этот процесс считается более экологически чистым, поскольку он не требует внесения серосодержащих соединений или жестких условий проведения, которые применяются в крафт- или лигносульфонатном процессах. Выделенный таким способом лигнин обладает низкой Mw (500–5000 Да), не содержит серы и характеризуется более высокой степенью чистоты из-за низкого содержания золы и углеводов [44, 52, 55, 56].
Однако, несмотря на эти преимущества, данный процесс делигнификации не получил широкого развития из-за обширной коррозии оборудования, более низкого качества производимой целлюлозы по сравнению с методами щелочной и крафт-варки и высокой стоимостью извлечения растворителей [38]. Кроме того, при повышении кислотности могут возникать случайные реакции конденсации вместо реакций деполимеризации. Например, в кислых условиях может происходить образование бензильных катионов, что приводит к образованию новых C–C-связей [44].
На сегодняшний день не существует экономически обоснованного метода делигнификации растений, позволяющего использовать все три их основных компонента – лигнин, целлюлозу и гемицеллюлозу. Наиболее востребован крафт-процесс, позволяющий производить самую качественную целлюлозу и получать остаточное содержание серы в лигнине не более 3 мас. %.
ДЕПОЛИМЕРИЗАЦИЯ – ОСНОВНОЙ СПОСОБ ПЕРЕРАБОТКИ ЛИГНИНА
В последнее время значение лигнина в качестве источника полезных химических соединений возрастает, поскольку ископаемое сырье становится все более дорогостоящим [57]. Деполимеризация (или фрагментация) лигнина обусловлена двумя целями: 1) выяснение состава и структуры лигнина; 2) получение полезных продуктов, таких как ванилин, простые и гидроксилированные ароматические соединения, хинины, альдегиды, алифатические кислоты и многие другие [58, 59].
Почти 100 лет химики исследовали разложение лигнина на фенольные производные, в результате чего было предложено большое количество методов термохимической деполимеризации [60]. В течение последних десятилетий большое внимание уделяется развитию таких процессов переработки лигнина, как гидрогенолиз, пиролиз, гидролиз, газификация и окисление (рис. 4) [22, 61–65]. Наряду с описанными выше химическими методами переработки лигнина большое внимание уделяется биологическим методам. Процессам деполимеризации подвергаются различные типы лигнинов (крафт, щелочной, лигносульфонатный, органосольвентный) как из хвойных, так и из лиственных пород.
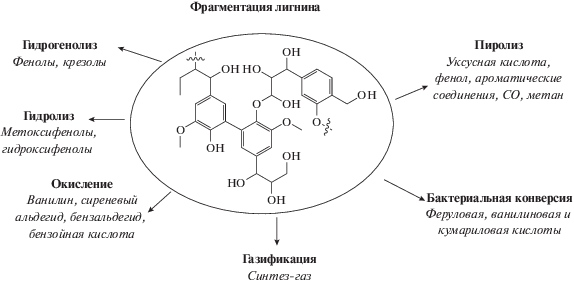
Пиролиз. Процесс пиролиза лигнина представляет собой разложение путем нагревания биомассы до 500–1000°С в отсутствие кислорода с катализатором или без него [62, 66–68]. Лигнин начинает разлагаться уже с 280°С за счет расщепления эфирных и мостиковых углерод-углеродных связей. В зависимости от условий [59, 63, 69] в различных пропорциях образуются жидкость (пиролизное масло), твердый остаток и газ (СО, СО2, СН4 и Н2). Жидкий продукт, полученный в результате пиролиза лигнина, состоит на 20% из органических соединений (метанол, уксусная кислота, ацетон), 15% смолы (конденсация горячих летучих веществ, дающая в основном фенольные соединения) и воды [70, 71]. Газовая фракция составляет около 10% от массы исходного материала и содержит, главным образом, оксиды углерода, метан и водород, а также небольшое количество легких алифатических углеводородов [72]. Твердый продукт представляет собой обогащенный углеродом остаток (содержание углерода ≥90%). Содержание минеральных примесей в углеродном остатке меньше, чем в ископаемых УВ, что делает его экологически более чистым топливом [73, 74]. Кроме того, твердый остаток может быть использован в качестве предшественников в производстве активированного углеродного адсорбента [75].
Существуют две основные разновидности пиролиза: традиционный (также называемый медленным пиролизом) и быстрый [33, 76]. При традиционном пиролизе лигнин медленно (от 5 до 30 мин) нагревают до 500°С. Быстрый пиролиз проводят при температурах от 600 до 1000°С с более высокой скоростью нагрева и коротким временем реакции (0.5–5 с), чем при традиционном пиролизе [26, 33, 76–80]. Быстрый пиролиз используют преимущественно для получения смеси жидких органических продуктов (био-масло) с суммарным выходом углеводородных соединений до 80%. Обычно продукты быстрого пиролиза состоят из 60–75 мас. % био-масла, 15–25 мас. % твердого остатка и 10–20 мас. % неконденсируемых газов [81]. Благодаря этому методу практически не образуются отходы, био-масло и углеродный остаток могут быть использованы по отдельности как топливо, а газ может быть возвращен обратно в процесс. Изменяя параметры пиролиза, можно варьировать состав продуктов. Так, с целью получения в качестве топлива угля, следует использовать низкие температуры и низкие скорости нагрева. Высокие температура, скорость нагрева и короткое время реакции позволяют достигнуть максимального выхода жидких продуктов. Чтобы получить максимальный выход газа требуется высокая температура при длительном времени реакции и низкой скорости нагрева [76, 77].
Для переработки лигнина используют также быстрый пиролиз с использованием различных катализаторов, таких как цеолиты (ZSM-5), оксиды переходных металлов (Co3O4, MoO3, NiO, Fe2O3, MnO3 и CuO) и переходные металлы (Mo, Cu, Mn, Fe и Ni), нанесенные на различные носители [82, 83].
В течение последних 20 лет были разработаны технологии быстрого пиролиза для преобразования биомассы всего растения в био-масло с использованием непрерывных или периодических реакторов с псевдоожиженным слоем (от лабораторного до стендового масштаба) [84, 85].
Высокое содержание кислородсодержащих соединений в составе пиролизного био-масла и значительное количество твердого остатка (≤20%) в продуктах пиролиза, препятствует реализации этого процесса в промышленности. Для увеличения выхода пиролизного масла, в основном состоящего из фенольных соединений, и снижения в нем содержания кислорода рассматривается возможность совместного пиролиза лигнина с полиолефинами, а также совместного пиролиза щелочного лигнина с синтетическими полимерами, имеющими фенольные структуры [86].
Окисление. Процесс окисления лигнина используют для получения альдегидов, таких как ванилин и сиреневый альдегид, и соответствующих им ванилиновой и сиреневой кислот. Наличие в структуре лигнина гидроксильных (фенольных) групп делает его перспективным сырьем для получения перечисленных продуктов. Скорость окисления можно контролировать количеством фенольных групп, значением рН (щелочное или кислотное окисление) и концентрацией окислителя [60, 87]. Применяют окислители, позволяющие сохранять ароматические фрагменты в структуре лигнина, и катализаторы (соли Cu2+ и Co2+) [86–94]. Наиболее часто для деполимеризации лигнина используют нитробензол, оксиды металлов, кислород и перекись водорода [60].
Наиболее крупнотоннажный продукт окисления лигнина – ванилин (4-гидрокси-3-метоксибензальдегид), – применяют в основном в качестве ароматизатора и как сырье в фармацевтической промышленности [94]. Первые предположения о возможности получения ванилина из лигнинсодержащих отходов были опубликованы в 1875 г. – отработанный кислотно-сульфитный раствор при выделении лигнина источал запах ванилина [95]. Эта идея получила развитие в реализации промышленного процесса, и в течение длительного времени ванилин получали исключительно окислением лигносульфонатов. В последнее время была разработана технология по эффективному разделению ванилина и сиреневого альдегида, как структурно схожих соединений, используя одностадийную кристаллизацию с высоким выходом (90%) и чистотой более 98% [96].
Исследован процесс окисления лигнина пеpоксидом водорода в щелочных и кислых средах [97]. При высоком значении рН окислительную деструкцию проводили при низких температурах, от 80 до 90°С. В кислых средах для достижения той же степени разложения лигнина требуется нагрев до 130–160°С. Основные продукты окислительной деструкции лигнина – моно- и дикарбоновые кислоты, а промежуточные – альдегиды [97]. В ряде исследований было показано, что добавление ионов переходных металлов, таких как Fe2+, Cu+, Mn3+ и Co2+ может ускорять разложение пероксида водорода, участвуя в переносе атомарного кислорода [98, 99].
Однако, получение альдегидов окислением лигнина пероксидом водорода не является оптимальным методом [60]. В отличие от него, альдегиды (ванилин, сиреневый альдегид и п-гидроксибензальдегид) наряду с соответствующими им кислотами (ванилиновую кислоту, сиреневую кислоту и п-гидроксибензойную кислоту) можно получать действием нитробензола на лигнин в щелочной среде при 170–190°C и времени реакции 1–4 ч, хотя условия могут меняться в зависимости от источника лигнина [100]. Несмотря на то, что нитробензол является эффективным окислителем, его канцерогенные свойства существенно ограничивают возможность использования этого метода [101]. Для замены нитробензола при окислении лигнина до альдегидов было выбрано окисление кислородом и воздухом в присутствии каталитических систем на основе переходных металлов и их оксидов, что позволило увеличить производство фенольных соединений [60]. Так, ионы переходных металлов, такие как Cu2+, Fe3+, Mn2+/3+, Co2+ и Zr 4+, используют в качестве катализаторов для производства бензальдегида и бензойной кислоты при окислении лигнина кислородом [99, 102]. Установлено, что значение рН среды влияет на селективность процесса. Например, щелочная среда предпочтительна для образования бензальдегида, тогда как кислая среда благоприятна для получения бензойной кислоты [99]. CuO и другие оксиды металлов, такие как TiO2 и ZnO могут быть использованы в качестве катализаторов фотохимического разложения лигнина в смесь альдегидов (ванилин, сиреневый альдегид и 3,4,5-триметоксибензальдегид), кислот (ванилиновая кислота и 4-гидроксибензойная кислота) и спиртов (2-гидроксибензиловый спирт и катехол) [99, 103–105].
В природных условиях деполимеризация лигнина происходит под действием окислительных ферментов, включая лакказы и пероксидазы (пероксидаза лигнин и пероксидаза марганца), которые секретируются некоторыми грибами и бактериями. Методы окисления, использующие природные ферменты, рассматриваются в качестве перспективных промышленных процессов с использованием биокатализаторов. В этом направлении заслуживают внимания гибридные процессы, в которых окислительно-восстановительный потенциал ферментов используют совместно с другими катализаторами, что позволяет достигнуть синергетического эффекта в деполимеризации лигнина [106–108].
Гидрогенолиз и гидрирование. Эти процессы направлены на получение органических продуктов, образующихся в результате насыщения водородом C–O–C-связей в лигнине. По сравнению с пиролизом, гидрогенизационные процессы обычно проводят при более низких температурах от 300 до 600°С. Выход монофенолов в этих условиях выше, в то время как твердого углеродного остатка образуется меньше [60, 66]. Реакцию проводят в среде таких растворителей, как тетралин, формиат натрия и муравьиная кислота, служащих жидкофазными донорами водорода [60, 109–113]. В этом направлении наибольшее внимание уделяется каталитическим процессам, позволяющим проводить превращение лигнина в более мягких условиях.
Каталитический гидрогенолиз лигнина с использованием водорода в присутствии катализатора NiMo на алюмосиликате позволяет достигать выход био-масла до 65 мас. % [114]. При гидрогенизации органосольвентного лигнина повышение давления водорода с 5 до 14 МПа позволило увеличить выход био-масла с 20 до 57 мас. %, а фенольной фракции с 7 до 12.3 мас. % [115].
Эффективный метод гидрогенизации лигнина в среде изопропанола с использованием катализатора Ni–Mo2C/C (Т = 250°С, время реакции 2 ч и начальное давление Н2 2.0 МПа), позволяет получать фенолы, гваяколы и триметоксибензолы с суммарной селективностью до 60% [111]. Лигнин гидрируют до мономерных фенолов (например, гваяколов и сиринголов) с использованием других типов катализаторов, включая благородные металлы, такие как Pt, Ru, Pd и Rh, оксиды металлов, такие как CoMo/Al2O3 и активированный уголь [113, 117–120].
В присутствии катализатора Ru/C сравнили состав продуктов каталитического гидрогенолиза органосольвентных лигнинов, полученных из китайской пихты и клена в смеси закритического этанола и 1-бутанола при 300°С, давлении 40 МПа и времени реакции 40 мин [121]. Оказалось, что лигнин китайской пихты дает меньше био-масла (32 мас. %), чем лигнин, полученный из клена (41 мас. %). Это объясняют различным строением лигнина, выделенного из различных сортов древесины. Биметаллические катализаторы AuxPd1 – x использовали для гидрогенолиза лигнина и модельных соединений, выделяемых из лигнина, в присутствии муравьиной кислоты при низкой температуре. В этих работах был достигнут 44.1% выхода множества ценных монофенолов [122]. Превращение щелочного лигнина в суперкритическом этаноле в присутствии катализатора CuMgAlOх приводит к высокому выходу ароматических углеводородов (23 мас. %) без образования твердого остатка [123].
Основная проблема производства монофенильных продуктов из лигнина – это нежелательная реполимеризация фрагментированных структур лигнина. Другая проблема – контроль степени реакции гидрогенизации, так как полная гидрогенизация ароматических колец из практических соображений нежелательна [124].
Гидролиз. Лигнин гидролизуют при 280–400°C в докритической или сверхкритической воде. Время реакции варьируют от нескольких минут до нескольких часов при 20–25 МПа общего давления [26, 60]. Вблизи критической точки (22 МПа и 374°C) вода обладает необычными свойствами, такими как низкая вязкость и высокая способность к растворнению органических веществ, что делает околокритическую воду благоприятной средой для эффективных и быстрых реакций. Высокая концентрация H+ и OH– в околокритической воде может ускорять реакции гидролиза, катализируемые кислотой или основанием [26, 125–127]. Оказалось, однако, что более высокая конверсия в метоксифенолы или гидроксифенолы достигается в докритических условиях, при которых связи лигнина расщепляются при умеренных температурах (280–370°C) и сохраняется ароматический характер молекул монолигнолов.
Кроме того, различные катехины и гваяколы образуются путем гидролиза эфирных связей. Однако с увеличением времени гидротермальной обработки растворимые продукты превращаются в нерастворимые из-за реакций реконденсации. Чтобы преодолеть эту проблему и повысить выход жидких продуктов, скорость гидролиза увеличивают путем введения таких катализаторов, как K2CO3, Ca(OH)2 и NaOH [26, 128–131]. Катализируемая основанием деполимеризация лигнина с использованием щелочей может эффективно превращать лигнин в различные ароматические соединения [132].
Повышение концентрации K2CO3 приводит к уменьшению образования углеродного остатка и росту выхода жидкого продукта, количество катехола в водорастворимых продуктах превышает количество гваякола [133]. ZrO2 и K2CO3 в качестве катализаторов используют совместно для превращения крафт-лигнина в ароматические УВ в смеси воды и фенола в околокритических условиях (350°C, 25 МПа). Образующиеся при этом однокольцевые ароматические соединения представляют собой в основном анизолы, алкилфенолы, гваяколы и катехины, их общий выход при повышенном содержании K2CO3 увеличивается с 17 до 27% [134].
Смеси воды с другими растворителями использовали для улучшения растворимости лигнина и предотвращения повторной полимеризации продуктов [26, 135]. Смеси, которые содержали 50/50 (об./об.) вода/сорастворитель (фенол, этанол, ацетон или муравьиная кислота), позволяли более эффективно превращать лигнин в целевые продукты [26, 136–139]. Использование кислотного цеолитного катализатора НВКЦ-30 в среде этанола при 350°С позволяет достичь максимального значения конверсии лигнина (71 мас. %) с выходом легкокипящей (<180°С) фракции жидких продуктов до 44 мас. % [140].
Добавление фенола в реакционную систему уменьшает образование углеродного остатка [136, 141, 142]. Влияние фенола особенно подробно изучено в [142], где в сверхкритической воде разлагали лигнин при температуре 400–600°C. Согласно данным этой работы, фенол способствует улучшению гомогенности среды и предотвращает образование твердого остатка. Поэтому большая часть продуктов деполимеризации имеет дифениловые димерные структуры, в которых один фенольный фрагмент происходит от лигнина, а второй − от сорастворителя. Полученные продукты можно использовать в качестве высококипящих растворителей.
Газификация – процесс, в котором лигнин превращается, главным образом, в смесь CO2, CO, H2 и CH4 с различными соотношениями компонентов в зависимости от температуры и давления, наличия пара и кислорода, скорости нагрева и элементного состава исходного реагента [66, 143]. Могут также быть получены небольшие количества C2H4 и C2H6. Поскольку технические лигнины часто содержат неорганические примеси и серу, включенную в структуру, продукты газификации лигнина также содержат небольшое количество золы и H2S. Полученные газообразные продукты имеют несколько применений, включая использование в топливных элементах, газовых турбинах и в качестве синтез-газа [144]. Для полной газификации лигнина необходимо последовательно провести гидролиз, деполимеризацию, гидрирование и конверсию водяного газа, эффективность которых зависят от температуры, времени реакции, давления и типа катализатора. Образование газов из лигнина может быть достигнуто путем различных процессов: обычная газификация кислородом и/или водяным паром при высоких температурах и атмосферном давлении. В некоторых исследованиях сообщается, что температура, необходимая для полной газификации лигнина, составляет по меньшей мере 700°С [144, 145]. Каталитическая газификация в сверхкритической воде протекает при умеренных температурах и высоких давлениях.
Газификация лигнина может происходить в котле-утилизаторе как неотъемлемая часть крафт-варки. Черный щелок также можно газифицировать [126]. Конечные продукты пиролиза – СО, Н2, СН4. Иногда газы сжигают с внесением в систему кислорода или воздуха, выделившееся тепло потребляет реакция эндотермического пиролиза. Основная цель газификации лигнина – получение монооксида углерода и водорода (синтез-газа). Синтез-газ может быть непосредственно применен в газовых двигателях и газовых турбинах, преобразован в жидкое топливо в процессе Фишера–Тропша [66], а также направлен на производство диметилового эфира, который можно использовать в дизельных двигателях [146].
При газификации биомассы количество выделившегося водорода зависит от содержания лигнина. Установлено, что из лигнина образуется в четыре раза больше водорода, чем из целлюлозы и практически в три раза больше, чем из гемицеллюлозы. Повышенное содержание водорода, в дополнение к более высокому количеству метана, также образуемому при газификации, можно объяснить высоким содержанием ароматических колец и метокси-групп в лигнине [147].
Каталитическая газификация лигнина в водных растворах (также известная как гидротермальная газификация) перспективна для производства водорода и метана из органического сырья с высоким содержанием воды, поскольку нет необходимости в сушке сырья [148–150]. Эффективность газификации лигнина в гидротермальных условиях возрастает при повышенном давлении (15–27.5 МПа), приводя к более высоким выходам H2 и CH4 [148–150]. Использование катализаторов в гидротермальной газификации лигнина, в основном щелочей и солей щелочных металлов (NaOH, KOH, Na2CO3, CaO, NaHCO3 и т.д.), переходных металлов (Ni, Pt, Ru и Rh) и оксидов металлов (например, Ni/MgAl2O3), позволяет повысить выход водорода и ускорить конверсию водяного газа, метанирование и гидрирование [144, 145, 151–156].
Каталитическая газификация водой в сверхкритических условиях происходит при более низких температурах и более высоком давлении (например, от 350 до 500°С и 25 МПа) по сравнению с обычной газификацией (800°C и 0.1 МПа), что, в свою очередь, благоприятствует высокому метанообразованию и низким выходам водорода при сравнимых условиях. Однако можно повысить селективность по водороду путем предотвращения реакций метанирования с помощью катализаторов. Считается, что катализаторы для получения водорода из лигнина должны быть гидротермально стабильны, облегчать раскрытие фенольного кольца и последующее расщепление связей С–С, активно катализировать конверсию водяного газа и не участвовать в расщеплении связи С–О [151–156]. Никелевые и рутениевые каталитические системы проявляют активность в реакции гидрирования двуокиси углерода и, следовательно, необходимо подавить их активность в расщеплении связи С–О при помощи промоторов [157, 158] или изменяя подложки и размер кристаллитов [159]. Добавление щелочных солей, таких как NaOH, может несколько увеличить выход газа [153]. Сверхкритическая газификация природного лигнина в лигнинсодержащем сырье, таком как древесина и кора, оказалась проще по сравнению с изолированным лигнином из крафт-процесса, который содержит более высокий процент устойчивых межцепных C–C-связей [22].
СТИМУЛИРОВАНИЕ КАТАЛИТИЧЕСКИХ РЕАКЦИЙ МИКРОВОЛНОВЫМ ИЗЛУЧЕНИЕМ
Особенности воздействия микроволнового излучения на материалы. В последнее время микроволновое излучение все чаще используют в практике химического синтеза различных веществ и стимулирования каталитических реакций. Микроволновое излучение, сверхвысокочастотное излучение (СВЧ-излучение) — электромагнитное излучение, включающее в себя дециметровый, сантиметровый и миллиметровый диапазоны радиоволн, частоты микроволнового излучения изменяются от 300 МГц до 300 ГГц (длина волны от 1 м до 1 мм). К микроволнам относятся УВЧ-диапазон (дециметровые волны) и КВЧ-диапазон (миллиметровые волны), в радиолокации микроволновым диапазоном принято обозначать волны с частотами от 1 до 100 ГГц (с длинами волн от 300 до 3 мм). В обоих определениях микроволновое излучение включает в себя СВЧ-диапазон. В рамках этого диапазона частот распределяются рабочие частоты сотовых телефонов, радаров и спутниковой связи. Во избежание помех в телекоммуникационных устройствах для промышленной микроволновой (МВ) обработки различных материалов выделены частоты 0.915, 2.45, 5.8 и 24.124 ГГц, в то время как для бытовых целей эта частота составляет 2.45 ГГц (λ = ~ 12 см) [160], выбранная, в частности, для кухонных печей, как оптимальная по скорости нагрева воды, основной составляющей продуктов питания. Техника микроволнового излучения была введена после Второй мировой войны и, соответственно, после разработки магнетронов в качестве источника излучения. По характеру восприимчивости к микроволновому излучению (МВИ) материалы можно разделить на три основных типа: прозрачные и не взаимодействующие с излучением; материалы, отражающие излучение и материалы, обладающие способностью к поглощению МВИ [161]. Микроволновое облучение диэлектрических материалов приводит к поляризации молекулярных единиц материала. В печи происходит диэлектрический нагрев веществ, содержащих полярные молекулы. Электрическая компонента электромагнитных волн ускоряет движение молекул, обладающих дипольным моментом, а межмолекулярное трение приводит к поглощению электромагнитного излучения и повышению температуры [162]. Этот метод нагрева имеет много преимуществ по сравнению с традиционными методами термического нагрева.
При традиционном нагреве с использованием внешнего источник нагрева тепло передается через поверхность; в результате нагрев регулируется температурой этой поверхности, а также ограничен физическими свойствами нагреваемых материалов, такими как плотность, теплоемкость и теплопроводность материала. Большинство твердых продуктов биомассы обладают низкой теплопроводностью [74]. Твердые частицы вблизи нагретой поверхности быстрее достигают условий пиролиза, чем частицы в объеме материала, что приводит к медленному и дифференциальному разогреву биомассы. Поглощение микроволнового излучения твердым материалом соответствует приближенно половине длины волны излучения. В пределах этой величины микроволновый нагрев действует на весь объем нагреваемого материала [163]. Такой способ нагрева позволяет удалять влагу из диспергированных частиц биомассы и генерировать пар, который повышает температуру твердых частиц биомассы [164].
Степень поглощения МВИ характеризуется диэлектрическими потерями [165, 167]. Скорость поглощения электромагнитного излучения микрочастицами, в том числе металлов, существенно отличается от таковой в массивных сплошных образцах [167]. В частности, помимо диэлектрической проницаемости вещества ε(ω), на скорость поглощения влияет также соотношение между размером частиц, длиной волны излучения λ и глубиной проникновения электромагнитного поля в частицу δ.
Материалы нагреваются в микроволновом поле благодаря дипольной поляризации, ионной проводимости и поверхностной поляризации [168, 169].
По механизму дипольной поляризации в основном нагреваются вещества, содержащие полярные группы. При воздействии МВ-поля электроны вокруг ядер (электронная поляризация) или сами атомные ядра (поляризация атомов) смещаются из положения равновесия, образуя индуцированные диполи. В некоторых материалах (например, в воде) существуют постоянные диполи из-за асимметричного распределения заряда в молекуле. Индуцированные или постоянные диполи имеют тенденцию переориентироваться под влиянием переменного электрического поля. Химические связи индуцированных или постоянно поляризованных молекул перестраиваются под действием флуктуаций поля. Эта перестройка происходит триллионы раз в секунду [170].
Поверхностная поляризация или поляризация Максвелла–Вагнера возникает вследствие накопления заряда в области контакта или поверхности раздела между различными компонентами в гетерогенных системах. Поляризация создается из-за разницы в проводимостях и диэлектрических постоянных веществ на границах раздела фаз. Накопление пространственного заряда приводит к искажениям поля и диэлектрическим потерям, которые способствуют нагреванию.
По механизму ионной проводимости в электропроводящем материале электромагнитное облучение приводит к возникновению зон проводимости, когда заряженные частицы или носители (электроны, ионы и т.д.) переносятся через материал под воздействием внешнего электромагнитного поля. Поскольку электрические токи возникают внутри структуры материалов, которые в большинстве случаев имеют относительно высокое удельное электрическое сопротивление, материал нагревается генерируемым возникающим потоком заряженных частиц [171].
Удивительно высокая эффективность преобразования электрической энергии в тепловую с КПД до 80–85% микроволнового излучения позволяет достичь высоких температур и скорости нагрева [172, 173].
Диэлектрические свойства вещества, определяющие степень нагрева материала при воздействии электромагнитного излучения, зависят от двух ключевых параметров: действительной величины диэлектрической постоянной (ε') и мнимой (ε"). Диэлектрическая постоянная отражает способность материала поляризоваться в электрическом поле, определяя, сколько электромагнитной энергии отражается и сколько поглощается, тогда как мнимая величина диэлектрических потерь определяет эффективность, с которой электромагнитная энергия преобразуется в тепло. Отношение мнимой величины к действительной обозначает тангенс угла диэлектрических потерь или коэффициент диссипации материала:
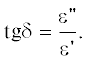
Тангенс угла диэлектрических потерь материала определяет способность материала поглощать и преобразовывать электромагнитную энергию в тепловую при заданной частоте. В процессе микроволнового нагрева этот параметр выступает как фактор, определяющий скорость нагрева и конечную температуру, которая может быть достигнута [174]. Таким образом, материал с небольшим значением ε' и высоким значением ε" (и поэтому высоким значением tg δ) рассматривается как обладающий большими диэлектрическими потерями и высокой способностью к преобразованию электромагнитной энергии в тепловую. Углеродные материалы и ряд неорганических оксидов характеризуются большими диэлектрическими потерями с высоким значением tg δ [175]. Силикатная керамика и некоторые пластмассы, не обладающие способностью к поглощению МВИ, считаются “прозрачными” для микроволн и характеризуются низким коэффициентом диэлектрических потерь (ε") [170].
Предельный случай накопления энергии при МВИ характеризуется накоплением зарядов и возникновением каналов проводимости, приводящих к пробою и генерированию плазмы [176]. При обработке микроволновым излучением ν = 34 МГц и 2.45 ГГц прозрачной силикатной керамики, на поверхность которой были нанесены частицы сажи, за несколько секунд возникают разряды и плазма, приводящая к быстрому и локальному выжиганию частиц сажи с образованием в этих местах малых отверстий [176]. При этом температура поверхности керамики, не содержащей частицы сажи, оставалась комнатной. Разработан способ формирования катализаторов, содержащих наноразмерные железосодержащие компоненты, путем микроволнового облучения пористого носителя, обладающего высокими диэлектрическими потерями, на который предварительно был нанесен ацетилацетонат железа в качестве предшественника [177]. Полученный таким способом катализатор показал существенно более высокую каталитическую активность в реакции окислительного разложения сероводорода в серу по сравнению с каталитической системой, полученной традиционным способом нанесения активных компонентов, с последующей их термической обработкой.
Зависимость скорости нагрева высокодисперсных частиц углерода от частоты электромагнитного поля анализировали с помощью закона Кирхгофа и комплексной диэлектрической проницаемости сажи при различных частотах ω, и рассматривая частицу как абсолютное черное тело [177]. Было найдено, что при микроволновом облучении с частотой 2.45 ГГц время нагрева до 800–850°С составляет 25–30 с. Этот результат хорошо коррелирует с данными, полученными при облучении ряда углеродных пористых материалов на установке с мощностью магнетрона 0.5 кВт. При этом нагрев материалов в среде Ar сопровождался возникновением свечения, что указывало на генерирование плазмы
Микроволновый нагрев углеродных материалов и его применение в процессах пиролиза. Микроволновый нагрев часто применяют для обработки углеродных материалов. Пористые углеродные адсорбенты эффективно поглощают микроволновое излучение. Тангенс угла диэлектрических потерь (tg δ) ряда углеродных материалов, таких как древесный уголь, сажа и активированный уголь ≥8, что практически на два порядка выше tg δ дистиллированной воды (~ 0.1), которая широко известна, как поглотитель микроволнового излучения [170]. Таким образом, углеродные материалы можно использовать в качестве эффективного поглотителя МВИ для нагрева веществ, прозрачных для МВИ. Нагрев происходит, когда достаточная энергия МВИ поглощается углеродными частицами и преобразуется в тепловую энергию путем дипольной переориентации и ионной проводимости [178]. Недавно было показано, что микроволновый нагрев углеродных материалов также происходит через комбинацию поляризации Максвелла−Вагнера и ионной проводимости [170]. Многократная переориентация под влиянием переменного электрического поля приводит к накоплению заряда. В ряде работ утверждают, что в некоторых случаях кинетическая энергия π-электронов в углеродном материале может увеличиться до уровня пробоя и генерирования каналов проводимости [170]. Это явление воспринимается как искры или электрическая дуга на макроскопическом уровне, а на самом деле представляет собой горячие точки или плазму.
Микроволновый нагрев с совместным использованием углеродных материалов применяется в пиролизе биомассы [179–182], сланцев [183], глицерина [184] и различных органических отходов [185]. В химических процессах для достижения более высоких температур, необходимых для протекания глубокого пиролиза предпочитают использовать углеродные материалы [179, 180, 183, 184, 186], а не дорогостоящие оксиды металлов 170, 181]. “Микроволновый пиролиз” обладает рядом преимуществ по сравнению с традиционными методами пиролиза.
Для пористых углеродных сорбентов характерны высокие значения тангенса диэлектрических потерь порядка 8–12 [187, 188]. Поглощение ими МВИ в токе аргона и диоксида углерода генерирует пробойные явления и плазму на поверхности углеродных сорбентов с последующим переходом в объем. Было установлено, что увеличение отношения объема макропор к объему микропор приводит к усилению эффектов пробоя и более быстрому нагреву материала. Авторы предполагают, что макропоры несут функцию псевдоконденсаторов, поскольку их размеры достаточны для генерирования объемных носителей заряда и формирования каналов проводимости (рис. 5). В то же время, микропоры скорее всего могут быть рассмотрены как неровности поверхности, которые согласно закону Фаулера–Нортгейма существенно усиливают эффект поглощения.

Поверхность таких углеродных сорбентов можно эффективно использовать для разложения адсорбированных органических или металлсодержащих молекул при действии МВИ [187, 188]. Таким образом, были разработаны способы получения катализаторов, содержащих на поверхности наноразмерные частицы активных компонентов, для разложения ряда токсичных соединений и нефтяных выбросов, предварительно адсорбированных в порах углеродных адсорбентов, обладающих большим значением tg δ.
ПРИМЕНЕНИЕ МИКРОВОЛНОВОГО ИЗЛУЧЕНИЯ В ПЕРЕРАБОТКЕ ЛИГНИНА
Микроволновый пиролиз крафт-лигнина при различных температурах, мощности МВИ и содержании поглотителей электромагнитного излучения был проведен в [189]. Изучали влияние количества внесенного поглотителя МВИ (20–40 мас. %) при мощности микроволнового облучения (1.5–2.7 кВт). Продукты пиролиза присутствовали в нескольких фазах: водная фаза, масляная фаза, неконденсируемый газ и твердый остаток; их содержание составило 17–21, 15–20, 21–27 и 32–40% соответственно. Один из основных выводов этой работы заключается в обнаружении того, что количество добавленного поглотителя МВИ оказывает значительно большее влияние на скорость нагрева реакционной массы, чем увеличение мощности излучения. Увеличение скорости нагрева реакционной массы путем ввода поглотителя МВИ приводит к увеличению выхода жидкой фракции, так при оптимальных условиях в зависимости от типа сырья концентрация ароматических соединений в составе масляной фракции составила 264–353 мг/г, а в водной фазе 15–38 мг/г.
Выход фенола и его замещенных гомологов в каталитическом микроволновом пиролизе лигнина с использованием активированного углерода в качестве катализатора и поглотителя достигало максимума при 550°С и обьемной скорости 2.18 ч–1 [190]. Основными продуктами превращения лигнина были фенолы, гваяколы, гидрокарбонаты и сложные эфиры. Доля фенола и фенольных соединений в био-масле достигала 38.9 и 66.9%, соответственно.
Пиролиз крафт-лигнина с использованием угля в качестве поглотителя МВИ и катализатора приводит к образованию неконденсируемого газа, тяжелой масляной фракции, легкой масляной фракции и твердого уголя с выходами 21.4, 19.4, 16.7 и 42.5% соответственно [191].
При совместном пиролизе лигнина и полиэтилена низкой плотности (ПЭНП) с катализаторами HZSM-5 и MgO, стимулируемом МВИ, выход ароматических соединений возрастал с увеличением содержания полиэтилена. Кроме того, с добавлением ПЭНП (лигнин/ПЭНП = 1/2), было обнаружено, что доля ароматических соединений увеличивается, а доля алкилированных фенолов уменьшается с ростом отношения HZSM-5 к MgO. Выход продуктов пиролиза биомассы растет с увеличением содержания катализатора, при этом также возрастает доля алкилированных фенолов [192].
Ожижение лигноцеллюлозных материалов, стимулируемое МВИ, проводили в метаноле при 180°С в течение 15 мин, конверсия сырья составляла ~75% [193]. Осажденные из водной фазы продукты в основном состояли из фенольных производных, таких как 2-метокси-4-пропилфенол и 4-гидрокси-3-метоксибензойная кислота метилового эфира.
Конверсия лигноцеллюлозной биомассы в фураны в ионной жидкости в микроволновом реакторе привела к образованию 5-гидроксиметилфурфурола и фурфурола с выходами 45–52 и 23–31% соответственно [194].
Деполимеризация в микроволновой печи органосольвентного лигнина в присутствии металлсодержащих наночастиц на мезопористом Al-SBA-15, включая никель (2, 5 и 10 мас. %), палладий (2 мас. %), платину (2 мас. %) и рутений (2 мас. %) за 30 мин, при средней температуре 140°С приводит к образованию био-масла [195]. Наибольший выход продукта, 30 мас. %, наблюдали при использовании 10 мас. % Ni на Al, тогда как катализатор на основе палладия показал самый низкий выход био-масла 5 мас. %.
При окислении органосольвентного лигнина с использованием H2O2 в качестве донора атомов кислорода при микроволновом облучении в присутствии соединений La выход 9.56% ванилина и выход 15.66% сиреневого альдегида достигался после 10 мин реакции при мощности 200 Вт микроволновой энергии [194].
Выход био-масла в каталитическом сольволизе лигнина с использованием МВИ (мощность: 600 Вт, температура реакции: 130°С и время реакции: 30 мин) увеличивался с добавлением катализаторов HUSY, модифицированных щавелевой кислотой в разных концентрациях. Максимальные значения составляли 15.36% фракции ароматических мономеров и 67.52% фракции олигомеров, когда в качестве катализатора использовали HUSY, модифицированный 0.2 моль/л щавелевой кислоты [197].
При микроволновом сольволизе трех типов лигнина в присутствии твердых кислотных катализаторов максимальные выходы био-масла составили от 57.4 до 82.9%, все продукты были получены в метаноле [198].
Предложен подход к скоростному каталитическому углекислотному риформингу лигнина при микроволновом стимулировании [199–201]. В качестве катализаторов были использованы наноразмерные моно никель-, железо- и биметаллические никель-железосодержащие каталитические системы, сформированные непосредственно на поверхности лигнина древесного происхождения. Металлсодержащие системы обладали двумя важными функциями: способностью к поглощению МВИ и каталитической активностью. При микроволновом облучении углекислотный риформинг протекал в плазменно-каталитическом режиме при видимой температуре в реакционной зоне 750–800°С. Время превращения лигнина составляло 10–15 мин; за это время конверсия лигнина составила 63–65% при 92–94%-ной селективности в образовании синтез-газа состава Н2/СО ~ 1 [202–204].
ЗАКЛЮЧЕНИЕ
В составе лигнина содержится достаточно высокое содержание углерода (до 80%) и водорода (до 6%), что делает перспективным использование его в качестве сырьевого источника для получения ароматических углеводородов, синтез-газа и водорода, которые на сегодняшний день являются важными компонентами топлив и реагентами в процессах нефтехимии и основного органического синтеза.
На основании анализа научной и научно-технической литературы можно отметить, что переработке лигнина посвящены многие исследования, но до настоящего времени еще не найдено эффективного подхода, обеспечивающего его высокую конверсию в требуемые продукты. Из литературных данных четко прослеживается перспективность использования каталитических подходов и микроволнового стимулирования с целью интенсификации процессов переработки органической массы лигнина в важные продукты.
Список литературы
Кайм В. Катализ в С1-химии. Л.: Химия, 1987. С. 296.
Моисеев И.И. // Кинетика и катализ. 2011. Т. 52. № 3. С. 347.
Моисеев И.И. // Кинетика и катализ. 2016. Т. 57. № 4. С. 411.
Hill C.A.S. Wood Modification: Chemical, Thermal and Other Processes. John Wiley & Sons, 2007. V. 5. P. 260.
Liu W.-J., Jiang H., Yu H.-Q. // Green Chem. 2015. V. 17. P. 4888.
Edwards P.P., Kuznetsov V.L., David W.I.F., Brandon N.P. // Energy Policy. 2008. V. 36. № 12. P. 4356.
Strassberger Z., Tanase S., Rothenberg G. // RSC Adv. 2014. V. 4. №. 48. P. 25310.
Кузнецов Г. // Восточно-Сибирская правда. От 01.12. 2005. № 25180. С. 28.
Chakar F.S., Ragauskas A.J. // Ind. Crops and Products. 2004. V. 20. №. 2. P. 131.
Ritter S.K. // Chemical & engineering news. 2008. V. 86. № 49. P. 10.
Boerjan W., Ralph J., Baucher M. // Annual review of plant biology. 2003. V. 54. № 1. P. 519.
Norgren M., Edlund H. // Current Opinion in Colloid & Interface Science. 2014. V. 19. № 5. P. 409.
Vanholme R., Demedts B., Morreel K., Ralph J., Boerjan W. // Plant Physiology. 2010. V. 153. № 3. P. 895.
Constant S. et al. // Green Chem. 2016. V. 18. № 9. P. 2651.
Vanholme R. et al. // Plant Cell. 2012. V. 24. P. 3506.
Van Acker R. et al. // Biotechnol. Biofuels. 2013. V. 6. P. 46.
Li L. et al. // Proc. Natl. Acad. Sci. USA. 2003. V. 100. P. 4939.
Liu C.J. // Molecular Plant. 2012. V. 5. № 2. P. 304.
O’Brien J. A., Daudi A., Butt V.S., Bolwell G.P. // Planta. 2012. V. 236. № 3. P. 765.
Хвиюзов С.С., Боголицын К.Г., Гусакова М.А., Зубов И.Н. // Фундаментальные исследования. 2015. № 9. С. 87.
Upton B.M., Kasko A.M. // Chem. Rev. 2015. V. 116. №. 4. P. 2275.
Zakzeski J., Bruijnincx P.C., Jongerius A.L., Weckhuysen B.M. // Chem. Rev. 2010. V. 110. № 6. P. 3552.
Xu C., Arancon R.A.D., Labidi J. // Chem. Society Rev. 2014. V. 43. № 22. P. 7485.
Li C., Zhao X., Wang A., Huber G.W., Zhang T. // Chem. Rev. 2015. V. 115. P. 11559.
Azadi P., Inderwildi O.R., Farnood R., King D.A. // Renewable and Sustainable Energy Reviews. 2013. V. 21. P. 506.
Joffres B., Laurenti D., Charon N., Daudin A., Quignard A., Geantet C. // Oil & Gas Science and Technology – Revue d’IFP Energies nouvelles. 2013. V. 68. № 4. P. 753.
Grabber J.H. // Crop Sci. 2005. V. 45. P. 820.
Hu T.Q. // Blackwell. 2008. P. 148.
Kamm B., Gruber P.R., Kamm M. // Wiley-VCH Verlag. 2006. V. 2. P. 151.
Kocheva L.S., Karmanov A.P., Kuz’min D.V., Dalimova G.N. // Chemistry of Natural Compounds. 2011. V. 47. № 5. P. 792.
Wen J.L., Xue B.-L., Xu F., Sun R.-C., Pinkert A. // Industrial Crops and Products. 2013. V. 42. P. 332.
Thakur V.K., Thakur M.K. // Intern. J. Biological Macromolecules. 2015. V. 72. P. 834.
Laurichesse S., Avérous L. // Progress in Polymer Science. 2014. V. 39. № 7. P. 1266.
Pan X., Kadla J.F., Ehara K., Gilkes N., Saddler J.N. // J. Agric Food Chem. 2006. V. 54. P. 5806.
Cruz J.M., Domínguez J.M., Domínguez H., Parajó J.C. // J. Agric Food Chem. 2001. V. 49. P. 2459.
Toh K., Nakano S., Yokoyama H., Ebe K., Gotoh K., Noda H. // Polym J. (Tokyo). 2005. V. 37. № 8. P. 633.
Réti C., Casetta M., Duquesne S., Bourbigot S., Delobel R. // Polym. Adv. Technol. 2008. V. 19. № 6. P. 628.
Doherty W.O.S., Mousavioun P., Fellows C.M. // Ind Crops Prod. 2011. V. 33. № 2. P. 259.
Agrawal A., Kaushik N., Biswas S. // The SciTech J. 2014. V. 1. № 7. P. 30.
Wang J., Deng Y., Qian Y., Qiu X., Ren Y., Yang D. // Green Chem. 2016. V. 18. № 3. P. 695.
Bruijnincx P.C.A., Weckhuysen B.M. // Nat. Chem. 2014. V. 6. № 12. P. 1035.
Tejado A., Pena C., Labidi J., Echeverria J.M., Mondragon I. // Bioresource Technology. 2007. V. 98. № 8. P. 1655.
Xiao B., Sun X.F., Sun R.C. // Polym. Degrad. Stab. 2001. V. 74. P. 307.
Galkin M.V., Samec J.S.M. // ChemSusChem. 2016. V. 9. № 13. P. 1544.
Svensson S. // Stcokholm: STFI-Packforsk, 2008.
Saake B., Lehnen R. // Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2012. P. 21.
Glasser W.G., Dave V., Frazier C.E. // J. Wood Chemistry and Technology. 1993. V. 13. № 4. P. 545.
Duval A., Lawoko M. // React. Funct. Polym. 2014. V. 85. P. 78.
Marques A.P., Evtuguin D.V., Magina S., Amado F.M.L., Prates A. // J. Wood Chem. Technol. 2009. V. 29. P. 337.
Prinsen P., Rencoret J., Gutiérrez A., Liitiä T. et al. // Ind. Eng. Chem. Res. 2013. V. 52. P. 15702.
Holladay J.E., Holladay J.E., Johnson D., White J.F. // Pacific Northwest National Lab. (PNNL), Richland, WA (United States). 2007. № PNNL-16983.
de la Torre M.J., Moral A., Hernández M.D., Cabeza E., Tijero A. // Ind. Eng. Chem. Res. 2013. V. 45. P. 58.
El Hage R., Brosse N., Sannigrahi P., Ragauskas A. // Polym. Degrad. Stab. 2010. V. 95. № 6. P. 997.
Sarkanen K. // Pulping Conference. 1990.
Lora J.H., Glasser W.G. // J. Polym. Environ. 2002. V. 10. № 1–2. P. 39.
Bajpai P. // NY: John Wiley & Sons, Inc. 2010. P. 264.
Nowakowski D.J., Bridgwater A.V., Elliott D.C., Meier D., de Wild P. // J. Anal. Appl. Pyrolysis. 2010. V. 88. № 1. P. 53.
Erdocia X., Toledano A., Corcuera M.A., Labidi J. // Chem. Eng. Trans. 2012. V. 29. P. 535.
Amen-Chen C., Pakdel H., Roy C. // Bioresource Technology. 2001. V. 79. № 3. P. 277.
Pandey M.P., Kim C.S. // Chemical Engineering & Technology. 2011. V. 34. № 1. P. 29.
Brebu M., Vasile C. // Cellulose Chemistry & Technology. 2010. V. 44. № 9. P. 353.
Swain P.K., Das L.M., Naik S.N. // Renewable and Sustainable Energy Reviews. 2011. V. 15. № 9. P. 4917.
McKendry P. // Bioresour. Technol. 2002. V. 83. P. 47.
Hamaguchi M., Cardoso M., Vakkilainen E. // Energies. 2012. V. 5. № 7. P. 2288.
Zakzeski J., Weckhuysen B. // ChemSusChem. 2011. V. 4. P. 369.
Huber G.W., Iborra S., Corma A. // Chemical Reviews. 2006. V. 106. № 9. P. 4044.
Epshtein Y.V., Akhimina E.I., Raskin M.N. // Khim. Drev. 1977. V. 6. P. 24.
Wienhaus O., Schiene R., Fischer F. // Zellst. Pap. 1980. V. 29. P. 125.
Mohan D., Pittman C.U., Steele P.H. // Energy & Fuels. 2006. 20. № 3. P. 848.
Balat M., Kirtay E., Balat H. // Energy Conv. Manag. 2009. V. 50. P. 3147.
Bridgwater T. Pyrolysis of Biomass. IEA Bioenergy: Task 34. Bioenergy Research Group, Aston University: Birmingham. UK. 2007.
Leonowicz A., Matuszewska A., Luterek J., Ziegenhagen D. // Fungal Genetics and Biology. 1999. V. 27. № 2–3. P. 175.
Di Blasi C. // Progress in energy and combustion science. 2008. V. 34. № 1. P. 47.
Basu P. // Academic Press. 2010. P.65.
Goyal H.B., Seal D., Saxena R.C. // Renew. Sustain. Energy Rev. 2008. V. 12. № 2. P. 504.
Demirbas A. // Energy Conversion and Management. 2009. V. 50. № 11. P. 2782.
Demirbas A. // Energy Sources, Part A: Recovery, Utilization, and Environmental Effects. 2007. V. 30. № 1. P. 38.
Venderbosch R.H., Prins W. // Biofuel. 2010. V. 4. P. 178.
Chiaramonti D., Oasmaa A.l Solantausta Y. // Renew. Sustain. Energy Rev. 2007. V. 11. P. 1056.
Bridgwater A.V. Fast Pyrolysis of Biomass: A Handbook. CRL Press: Newbury. UK, 2005.
Jahirul M.I., Rasul M.G., Chowdhury A.A., Ashwath N. // Energies. 2012. V. 5. P. 4952.
Kalogiannis K.G., Stefanidis S.D., Michailof C.M., Lappas A.A., Sjöholm E. // J Anal. Appl. Pyrol. 2015. V. 115. P. 410.
Ma Z., Custodis V., Hemberger P., Bährle C., Vogel F. et al. // CHIMIA International J. for Chemistry. 2015. V. 69. № 10. P. 597.
Рахманкулов Д.Л., Шавшукова С. Ю., Вихарева И. Н. // Башкирский химический журнал. 2008. Т. 15. № 2. С. 47.
Гафуров Н.М., Хисматуллин Р.Ф. // Инновационная наука. 2016. № 5–2. С. 17.
Brebu M., Spiridon I. // Polym. Degrad. Stab. 2012. V. 97. № 11. P. 2104.
Kalliola A., Kuitunen S., Liitiä T., Rovio S. // Holzforschung. 2011. P. 567.
Araújo J.D.P., Grande C.A., Rodrigues A.E. // Chemical Engineering Research and Design. 2010. V. 88. № 8. P. 1024.
Villar J.C., Caperos A., Garcia-Ochoa F. // Wood Science and Technology. 2001. V. 35. № 3. 245.
Zhou X.F., Qin J.X., Wang S.R. // Drewno. 2011. V. 54. P. 15.
Zoia L., Canevali C., Orlandi M., Tolppa E.-L., Sipila J., Morazzoni F. // Bioresources. 2008. V. 3. P. 21.
Zhou X.F., Liu J. // Hem. Ind. 2012. V. 66. P.685.
Zhou X.F. // J. Appl. Polym. Sci. 2014. V. 131. P.40809.
da Silva E.A.B., Zabkova M., Araújo J.D., Cateto C.A., Barreiro M.F., Belgacem M.N., Rodrigues A.E. // Chemical Engineering Research and Design. 2009. V. 87. № 9. P. 1276.
Hocking M.B. // J. Chemical Education. 1997. V. 74. № 9. P. 1055.
Tarabanko V.E., Chelbina Y.V., Kudryashev A.V., Tarabanko N.V. // Separation Science and Technology. 2013. V. 48. № 1. P. 16.
Xiang Q., Lee Y.Y. // Applied Biochemistry and Biotechnology. 2000. V. 84. № 1–9. P. 153.
Salem I.A., El-Maazawi M., Zaki A.B. // Intern. J. Chemical Kinetics. 2000. V. 32. № 11. P. 643.
Ma R., Xu Y., Zhang X. // ChemSusChem. 2015. V. 8. № 1. P. 24.
Min D., Xiang Z., Liu J., Jameel H., Chiang V., Jin Y. et al. // J. Wood Chemistry and Technology. 2015. V. 35. № 1. P. 52.
Holder J.W. // Toxicology & Industrial Health. 1999. V. 15. № 5. P. 445.
Xiang Q., Lee Y. Y. // Twenty-Second Symposium on Biotechnology for Fuels and Chemicals. Humana Press, Totowa, NJ. 2001. V. 91. P. 71.
Hautala K., Peuravuori J., Pihlaja K. // Chemosphere. 1997. V. 35. № 4. P. 809.
Pan K., Tian M., Jiang Z-H., Kjartanson B., Chen A. // Electrochimica Acta. 2012. V. 60. P. 147.
Ksibi M., Ben Amor S., Cherif S., Elaloui E., Houas A., Elaloui M. // J. Photochemistry and Photobiology A: Chemistry. 2003. V. 154. № 2–3. P. 211.
Ragauskas A.J., Beckham G.T., Biddy M.J., Chandra R., et al. // Science. 2014. V. 344. № 6185. P. 1246843.
Martínez Á.T., Speranza M., Ruiz-Duenas F.J., Ferreira P., Camarero S., Guillén F., Martínez M.J., Gutiérrez, A., Del Rio, J.C. // Int Microbiol. 2005. V. 8. P. 195.
Bugg T.D.H., Ahmad M., Hardiman E.M., Singh R. // Current Opinion in Biotechnology. 2011. V. 22. № 3. P. 394.
Gonçalves A.R., Schuchardt U. // Applied Biochemistry and Biotechnology. 2002. V. 98. № 1–9. P. 1211.
Kudsy M., Kumazawa H., Sada E. // The Canadian J. of Chemical Engineering. 1995. V. 73. № 3. P. 411.
Kleinert M., Barth T. // Chem. Eng. Technol. 2008. V. 31. P. 736.
Zakzeski J., Jongerius A.L., Bruijnincx P.C.A., Weckhuysen B.M. // ChemSusChem. 2012. V. 5. № 8. P. 1602.
Song Q., Wang F., Cai J., Wang Y. et al. // Energy & Environmental Science. 2013. V. 6. № 3. P. 994.
Meier D., Ante R., Faix O. // Bioresource Technology. 1992. V. 40. № 2. C. 171.
Meier D., Berns J., Grünwald C., Faix O. // J. Analytical and Applied Pyrolysis. 1993. V. 25. P. 335.
Wang Y.Y., Ling L.L., Jiang H. // Green Chemistry. 2016. V. 18. № 14. P. 4032.
Yan N., Zhao C., Dyson P.J., Wang C., Liu L.-T., Kou Y. // ChemSusChem: Chemistry & Sustainability Energy & Materials. 2008. V. 1. № 7. P. 626.
Li C., Zheng M., Wang A., Zhang T. // Energy & Environmental Science. 2012. V. 5. № 4. P. 6383.
Dan Y., Jie-Wang Y., Gui-Zhen F., Chun-De J. // 2nd International conference on chemistry and chemical process. (May 5–6, 2012). 2012. V. 3. P. 53.
Jongerius A.L., Jastrzebski R., Bruijnincx P.C.A., Weckhuysen B.M. // J. Catalysis. 2012. V. 285. № 1. P. 315.
Hu J., Shen D., Wu S., Zhang H., Xiao R. // Energy & Fuels. 2014. V. 28. № 7. P. 4260.
Gao X., Zhu S., Li Y. // Molecular Catalysis. 2019. V. 462. P. 69.
Huang X., Koranyi T.I., Boot M.D., Hensen E.J.M. // ChemSusChem. 2014. V. 7. № 8. P. 2276.
Jongerius A.L. Catalytic Conversion of Lignin for The Production of Aromatics: Dis. Utrecht University. 2013.
Toor S.S., Rosendahl L., Rudolf A. // Energy. 2011. V. 36. № 5. P. 2328.
Hunter S.E., Savage P.E. // Chemical Engineering Science. 2004. V. 59. № 22–23. P. 903.
Zakzeski J., Jongerius A.L., Weckhuysen B.M. // Green Chem. 2010. V. 12. P. 1225.
Toledano A., Serrano L., Labidi J. // J. Chemical Technology & Biotechnology. 2012. V. 87. № 11. P. 1593.
Roberts V.M., Stein V., Reiner T., Lemonidou A., Li X., Lercher J. // Chemistry. A European J. 2011. V. 17. № 21. P. 5939.
Vigneault A., Johnson D.K., Chornet E. // The Canadian J. Chemical Engineering. 2007. V. 85. № 6. P. 906.
Heitz M., Wu G., Lapointe J. // J. Wood chemistry and technology. 1995. V. 15. № 4. P. 515.
Косяков Д. С. и др. //Масс-спектрометрия. 2017. Т. 14. № 1. С. 14.
Karagöz S., Bhaskar T., Muto A., Sakata Y. // Bioresource technology. 2006. V. 97. № 1. P. 90.
Nguyen D.H., Maschietti M., Belkheiri T., Amand L.-E. et al. // The J. Supercritical Fluids. 2014. V. 86. P. 67.
Zhang B., Huang H.J., Ramaswamy S. // Appl. Biochem. Biotechnol. 2008. V. 147. P. 119.
Saisu M., Sato T., Watanabe M., Adschiri T., Arai K. // Energy & Fuels. 2003. V. 17. № 4. P. 922.
Yuan Z., Cheng S., Leitch M., Xu C.C. // Bioresource Technology. 2010. V. 101. № 23. P. 9308.
Gosselink R.J.A., Teunissen W., van Dam J.E., de Jong E. // Bioresource Technology. 2012. V. 106. P. 173.
Liguori L., Barth T. // J. Analytical and Applied Pyrolysis. 2011. V. 92. № 2. P. 477.
Шарыпов В.И., Береговцова Н.Г., Барышников С.В., Кузнецов Б.Н. и др. // J. Siberian Federal University. Chemistry 3. 2013. V. 6. P. 241.
Okuda K., Umetsu M., Takami S., Adschiri T. // Fuel processing Technology. 2004. V. 85. № 8–10. P. 803.
Fang Z., Sato T., Smith Jr. R.L., Inomata H. et al. // Bioresource Technology. 2008. V. 99. № 9. P. 3424.
Sricharoenchaikul V. // Bioresource Technology. 2009. V. 100. № 2. P. 638.
Kang S., Li X., Fan J., Chang J. // Renewable and Sustainable Energy Reviews. 2013. V. 27. P. 546.
Guo L.J., Lu Y.J., Zhang X.M., Ji C.M., Guan Y., Pei A.X. // Catalysis Today. 2007. V. 129. № 3–4. P. 275.
Crocker M. // Royal Society of Chemistry. 2010. P. 532.
Хаджиев С.Н., Максимов А.Л., Кротова М.В. // Научный журнал Российского газового общества. 2017. № 1. С. 43.
Ji C., Guo L., Lu Y., Pei A., Guo X. // Taiyangneng Xuebao/Acta Energiae Solaris Sinica. 2007. V. 28. P. 961.
Osada M., Sato T., Watanabe M., Shirai M., Arai K. // Combustion Science and Technology. 2006. V. 178. № 1–3. P. 537.
Osada M., Sato O., Watanabe M., Arai K., Shirai M. // Energy & Fuels. 2006. V. 20. № 3. P. 930.
Yoshida T., Oshima Y., Matsumura Y. // Biomass and Bioenergy. 2004. V. 26. № 1. P. 71.
Guo Y., Wang S.Z., Xu D.H., Gong Y.M., Ma H.H., Tang X.Y. // Renewable and Sustainable Energy Reviews. 2010. V. 14. № 1. P. 334.
Azadi P. Khan S., Strobel F., Azadi F., Farnood R. // Applied Catalysis B: Environmental. 2012. V. 117. P. 330.
Furusawa T., Sato T., Sugito H., Miura Y., Ishiyama Y., Sato M., Itoh N., Suzuki N. // Intern. J. Hydrogen Energy. 2007. V. 32. № 6. P. 699.
Yamaguchi A., Hiyoshi N., Sato O., Bando K., Osada M., Shirai M. // Catalysis Today. 2009. V. 146. № 1–2. P. 192.
Osada M., Sato O., Arai K., Shirai M. // Energy & Fuels. 2006. V. 20. № 6. P. 2337.
Huber G.W., Shabaker J.W., Dumesic J.A. // Science. 2003. V. 300. № 5628. P. 2075.
Davda R.R., Shabaker J., Huber G., Cortright R., Dumesic J. // Applied Catalysis B: Environmental. 2005. V. 56. № 1–2. P. 171.
Azadi P., Afif E., Azadi F., Farnood R. // Green Chemistry. 2012. V. 14. № 6. P. 1766.
Meredith R.J. // The Institution of Electrical Engineers. London. 1998. № 25.
Durka T., Gerven T.V., Stankiewicz A. // Chem. Eng. Technol. 2009. V. 32. № 9. P. 1301.
Robinson J.P., Snape C., Kingman S., Shang H. // J. Analytical and Applied Pyrolysis. 2008. V. 81. № 1. P. 27.
Luque R., Menendez J.A., Arenillas A., Cot J. // Energy & Environmental Science. 2012. V. 5. № 2. P. 5481.
Salema A.A., Ani F.N. // J. Analytical and Applied Pyrolysis. 2012. V. 96. P. 162.
Komarov V.V. Handbook of Dielectric and Thermal Properties of Materials at Microwave Frequencies. Artech House, 2012.
Mijović J., Wijaya J. // Polymer Composites. 1990. V. 11. № 3. P. 184.
Ландау Л.Д., Лифшиц Е.М. Электродинамика сплошных сред. М. :Наука. 1982. С. 327.
Neas E.D., Collins M.J. // American Chemical Soc. Publishing. 1988. P. 7.
Mingos D.M.P. Baghurst D.R. // Chemical Soc. Reviews. 1991. V. 20. P. 1.
Menéndez J.A., Arenillas A., Fidalgo B., Fernández Y. et al. // Fuel Processing Technology. 2010. V. 91. № 1. P. 1.
Liu C.C., Walters A.B., Vannice M.A. // Carbon. 1995. V. 33. № 12. P. 1699.
Ludlow-Palafox C., Chase H.A. // Industrial & Engineering Chemistry Research. 2001. V. 40. № 22. P. 4749.
Osepchuk J.M. // IEEE Transactions on Microwave Theory and Techniques. 2002. V. 50. № 3. P. 975.
Zhang X., Hayward D.O. // Inorganica Chimica Acta. 2006. V. 359. № 11. P. 3421.
Udalov E.I., Bolotov V., Tanashev Y., Chernousov Y., Parmon V. // Theoretical and Experimental Chemistry. 2011. V. 46. № 6. P. 384.
Цодиков М.В., Передерий М.А., Карасева М.С., Гурко А.А., Жеваго Н.К. // Наукоемкие технологии. 2007. № 6–7. С. 55.
Zubko V.G., Menella V., Colangeli L., Bussoletti E. // Mon. Not. R. Astron. Soc. 1996. V. 282. P. 1321.
Menéndez J.A., Menéndez E.M., Garcia A., Parra J.B., Pis J.J. // J. Microwave Power and Electromagnetic Energy. 1999. V. 34. № 3. P. 137.
Dominguez A., Menéndez J.A., Fernández Y., Pis J.J., Nabais J., Carrott P., Carrott M. // J. Analytical and Applied Pyrolysis. 2007. V. 79. № 1–2. P. 128.
Menéndez J.A., Domínguez A., Fernández Y., Pis J.J. // Energy & Fuels. 2007. V. 21. № 1. P. 373.
Monsef-Mirzai P., Ravindran M., McWhinnie W.R., Burchill P. // Fuel. 1995. V. 74. № 1. P. 20.
Monsef-Mirzai P., Ravindran M., McWhinnie W.R., Burchil P. // Fuel. 1992. V. 71. № 6. P. 716.
Mokhlisse A., Chanaa M.B., Outzourhit A. // Fuel. 2000. V. 79. № 7. P. 733.
Fernández Y., Arenillas A., Díez M.A., Pis J.J., Menéndez J.A. // J. Analytical and Applied Pyrolysis. 2009. V. 84. № 2. P. 145.
Holland K.M. // Patent U.S. № 5.387.321. 7 February 1995.
Menendez J.A., Inguanzo M., Pis J.J. // Water Research. 2002. V. 36. № 13. P. 3261.
Цодиков М.В., Передерий М.А., Карасева М.С., Максимов Ю.В., Суздалев И.П., Гурко А.А., Жеваго Н.К. // Российские нанотехнологии. 2006. Т. 1. № 1–2. С. 153.
Цодиков М.В., Передерий М.А., Чистяков А.В., Константинов Г.И., Мартынов Б.И. // Химия твердого толпива. 2012. № 1. С. 39 .
Farag S., Fu D., Jessop P.G., Chaouki J. // J. Analytical and Applied Pyrolysis. 2014. V. 109. P. 249.
Bu Q., Lei H., Wang L., Wei Y., Zhu L., Zhang X. // Bioresource Technology. 2014. V. 162. P. 142.
Fu D., Farag S., Chaouki J., Jessop P.G. // Bioresource Technology. 2014. V. 154. P. 101.
Fan L., Chen P., Zhang Y., Liu S. et al. // Bioresource Technology. 2017. V. 225. P. 199.
Xu J., Jiang J., Hse C., Shupe T.F. // Green Chemistry. 2012. V. 14. № 10. P. 2821.
Zhang Z., Zhao Z. K. // Bioresource Technology. 2010. V. 101. № 3. P. 1111.
Toledano A., Serrano L., Pineda A., Romero A.A. et al. // Applied Catalysis B: Environmental. 2014. V. 145. P. 43.
Gu X., Kanghua C., Ming H., Shi Y. et al. // Maderas. Ciencia y tecnologia. 2012. V. 14. № 1. P. 31.
Shen D., Liu N., Dong C., Xiao R. // Chemical Engineering J. 2015. V. 270. P. 641.
Zhou M., Sharma B.K., Li J., Zhao J., Xu J., Jiang J. // Fuel. 2019. V. 239. P. 239.
Tsodikov M.V., Ellert O.G., Nikolaev S.A., Arapova O.V., Konstantinov G.I., Bukhtenko O.V., Vasil’kov A.Yu.// Chemical Engineering J. 2017. V. 309. P. 628.
Tsodikov M.V., Ellert O.G., Nikolaev S.A., Arapova O.V., Bukhtenko O.V., Maksimov Yu.V., Kirdyankin D.I., Vasil’kov A.Yu. // J. Nanoparticle Research. 2018. V. 20. № 3. P. 86.
Арапова О.В., Бондаренко Г.Н., Чистяков А.В., Цодиков М.В. // Журн. физической химии. 2017. № 9. С. 1520.
Arapova O.V., Tsodikov M.V., Chistyakov A.V., Konstantinov G.I. // Chemical Engineering Transactions. 2017. V. 57. P. 223.
Арапова О.В., Цодиков М.В., Чистяков А.В., Курдюмов С.С., Гехман А.Е. // Доклады Академии наук. 2017. Т. 475. № 4. С. 405.
Tsodikov M.D., Ellert O.G., Arapova O.V., Nikolaev S.A., Chistyakov A.V., Maksimov Yu.V. // Chemical Engineering Transactions. 2018. V. 65. P. 367.
Дополнительные материалы отсутствуют.
Инструменты
Нефтехимия