

Нефтехимия, 2020, T. 60, № 4, стр. 488-500
Обнаружение множественности стационарных состояний в процессе превращения диметилового эфира, катализируемого композицией ZnO/γ-Al2O3. Влияние кокса и пероксида водорода
А. Л. Максимов 1, В. Ф. Третьяков 1, *, Р. М. Талышинский 1
1 Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Россия
* E-mail: tretjakov@ips.ac.ru
Поступила в редакцию 18.10.2019
После доработки 29.11.2019
Принята к публикации 12.03.2020
Аннотация
Изучено гетерогенно-каталитическое превращение диметилового эфира (ДМЭ) на катализаторе ZnO/γ-Al2O3 в 1,3-бутадиен и бутилены в широком интервале температур, объемных скоростей сырьевого потока и концентраций реагентов. Установлено, что области температур 380–420 и 440–480°С, условно обозначенные, как низкотемпературная и высокотемпературная, отличаются закономерностями протекания процесса, связанными со спецификой зауглероживания поверхности катализатора и перераспределением поверхностной концентрации кислотных центров бренстедовского и льюисовского типа. Выдвинута гипотеза о смене механизма каталитического процесса при вхождении реакции в высокотемпературную область, подтвержденная обнаруженным гистерезисом в рассмотренной области температур. Показано влияние пероксида водорода на параметры гистерезиса и устойчивость стационарных состояний в процессе превращения ДМЭ.
ВВЕДЕНИЕ
Образование на поверхности гетерогенных катализаторов при эксплуатации продуктов уплотнения (кокса), блокирующих их активные центры, является причиной проявляющейся потери активности, избирательности и нестабильности, что представляет собой одну из острых проблем каталитической химии.
Специфика процессов зауглероживания, протекающих на неоднородной поверхности твердых контактов, зачастую приводит к возникновению множества стационарных состояний реакционных систем и сложностью управления технологическими параметрами реакций в оптимальном режиме.
Ключевыми задачами устранения этих проблем с целью восстановления главных эксплуатационных характеристик гетерогенных катализаторов являются стабилизация, либо восстановление их химического и фазового состава, а также структурных и текстурных характеристик.
Следует, однако, констатировать, что разработанные к настоящему времени методы регенерации поверхности отработанных катализаторов в полной мере не удовлетворяют всем требованиям, предъявляемым современными технологиями каталитического производства химической продукции.
Так, например, широко применяемый способ восстановления отработанных катализаторов путем периодической термоокислительной обработки, т.е. выжигания кокса в токе воздуха (в присутствии, или в отсутствие водяного пара) зачастую приводит к спеканию каталитически активного компонента. Следствие этого – уменьшение удельной поверхности и изменение пористой структуры катализатора, что, в свою очередь, требует экономически не целесообразного частого обновления каталитической шихты [1–3].
В указанном аспекте весьма актуальным представляется поиск принципиально новых путей продления времени безрегенерационной эксплуатации дезактивирующихся катализаторов, не приводящих к нежелательным и необратимым изменениям их характеристик. К таким путям относится, к примеру, метод саморегенерации каталитической системы за счет введения в состав реакционной среды соединений (например, пероксида водорода), ингибирующих образование продуктов уплотнения и кокса на гетерогенной поверхности и стабилизирующих стационарное состояние в ходе протекания процесса [4].
В настоящее время в силу сложившегося дефицита в производстве мономеров синтетического каучука существенно расширяется использование альтернативного нефти углеродсодержащего сырья, в частности таких оксигенатов, как этанол, метанол и диметиловый эфир [5–7].
Ранее в работе [8–10] нами впервые была продемонстрирована принципиальная возможность получения одного из важных мономеров основного органического синтеза – 1,3-бутадиена из ДМЭ в присутствии модифицированного ZnO/γ-Al2O3-катализатора путем промежуточного его превращения в этанол и дальнейшего протекания реакции в бутадиен по известному механизму, установленному С.В. Лебедевым [11]. В работе [10] предложена кинетическая модель, удовлетворительно описывающая поученные закономерности в рассмотренном диапазоне температур.
Следует, однако, отметить, что в отличие от упомянутой конверсии этилового спирта в бутадиен, превращения ДМЭ сопровождаются относительно высокой степенью зауглероживания поверхности катализатора, что приводит к быстрой потере первоначальной активности, образованию катализата переменного состава и значительному сокращению времени безрегенерационного цикла.
Цель данной работы – исследование взаимосвязи характеристик предложенного нами ZnO/ γ-Al2O3-катализатора конверсии ДМЭ в дивинил с процессом зауглероживания его поверхности, а также изучение влияния добавок пероксида водорода к сырьевому потоку на регенерацию активных центров катализатора от углистых отложений с установлением соответствующего стационарного состояния при протекании реакции.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Изучение влияния режимных параметров в процессе превращения ДМЭ проводили в кварцевом реакторе при загрузке 10 мл катализатора на установке, приведенной на рис. 1, в температурном диапазоне 380–480°С и объемной скорости GHSV по ДМЭ в интервале 100–190 ч–1 при мольном разбавлении сырья азотом от 2.0 : 1 до 11.5 : 1. Градиент температуры по слою катализатора при загрузке 5–10 мл составлял ±5°С. Качественный и количественный состав полученных катализатов анализировали хроматографически по методике, приведенной в [8].
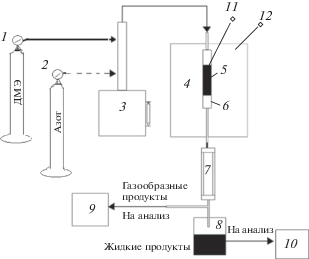
Рис. 1.
Схема проточной установки для испытания активности катализатора: 1, 2 – баллоны ДМЭ и азота; 3 – насос-дозатор для подачи жидких реагентов; 4 – печь; 5 – слой катализатора; 6 – реактор; 7 – холодильник; 8 – конденсатор; 9, 10 – хроматографы “Кристалл Люкс-4000М” для анализа газовой и жидкой фаз; 11, 12 – термопары – контрольная в слое катализатора и регулирующая в печи.
Расчет скорости превращения диметилового эфира осуществлялся по формуле
где $\alpha $ – конверсия ДМЭ, %, $Wo$ – скорость подачи ДМЭ, $\left( {\frac{{{\text{моль}}}}{{{\text{л}}\,\,{\text{ч}}}}} \right){\text{.}}$Опыты повторяли на воспроизводимость, а полученные данные усреднялись. Среднеквадратичные отклонения от средних значений скоростей по компонентам системы не превышали ∆ = ±7%. Скорость реакции нормировалась к 1 л катализатора в час и представлена в размерности моль/л ч.
Катализатор готовили методом диффузионной пропитки γ-оксида алюминия растворами нитратов цинка и алюминия при 200–500°С последующей сушкой и прокаливанием прекурсоров в условиях, приведенных в работах [9, 10].
Нитрат алюминия из расчета 10 мас. % в конечной рецептуре вводили в систему для создания прочной связи оксидов цинка и алюминия между собой.
Перед использованием γ-оксид алюминия прокаливали 6 ч при 600°С и помещали в эксикатор, где он остывал до комнатной температуры. Пропитку растворами активных компонентов осуществляли при 80°С в течение 3 ч. Количество раствора активных компонентов рассчитывали по влагоемкости с добавкой требуемого количества дистиллированной воды.
Пропитанный влажный носитель эмульгировали при 20°С 12 ч, после чего образец разогревали до 120°С и сушили при перемешивании для предотвращения слипания частиц прекурсора 2 ч. Затем, после добавления по влагоемкости водного раствора винной кислоты при рН 2.5, в сушильном шкафу производили досушивание катализатора в течение 10 ч.
Синтезированный образец помещали в муфельную печь, где прокаливали по 2 ч при 200, 300°С, остужая его в эксикаторе до комнатной температуры. По влагоемкости добавляли аммиачную воду. После отдушки оксидов азота в муфельной печи при 350°С, прекурсор катализатора в требуемом количестве пересыпали в реактор. В реакторе осуществляли прокаливание образца с одновременной активацией при 400 и 550°С в чередующихся потоках (2 л/ч на 5 мл образца) азота, воздуха и водорода в течение 3 ч. Рецептуру катализатора рассчитывали, исходя из содержания оксидов, мас. %: ZnO – 22.5, γ-Al2O3 –77.5.
Специальными экспериментами [8], по изменению линейной скорости сырьевого потока в интервале 5–10 см/с и фракционного состава гранул катализатора в пределах (0.2–0.3)–(0.8–1.0) мм, было продемонстрировано отсутствие влияния внешней и внутренней диффузии при превращении ДМЭ в изученном диапазоне варьирования технологических параметров реакции.
Текстурные характеристики образцов катализаторов исследовали на унифицированной установке “Сорби-MS” методом низкотемпературной адсорбции азота и рассчитывали по методу Брунауэра–Эммета–Тейлера (БЭТ). Общий сорбционный объем пор VƩ (суммарный объем микро- и мезопор) определяли по количеству азота, адсорбированного при относительном давлении, близком к единице, исходя из предположения, что в результате капиллярной конденсации все поры заполнены азотом в конденсированном состоянии. В этом случае объем адсорбированного азота (VƩ) пересчитывался в объем жидкого азота (Vж) с помощью уравнения:
(2)
${{V}_{{\text{ж}}}} = \frac{{{{P}_{{\text{A}}}}{{{\text{V}}}_{\Sigma }}{{V}_{{\text{M}}}}}}{{RT}},$Кислотные свойства поверхности катализатора определяли методом температурно-программированной десорбции (ТПД) аммиака и ИК-спектрального анализа адсорбированных молекул аммиака и монооксида углерода, в качестве молекул-зондов.
Общую поверхностную кислотность образцов оценивали в предположении об одноцентровой адсорбции аммиака по количеству хемосорбированных молекул, десорбция которых практически завершалась при подъеме температуры в колонке с катализатором до 600°С.
Суммарное количество кислотных центров: NΣ (единиц/м2), рассчитывали из значений площади под десорбционными кривыми по формуле:
(3)
${{N}_{\Sigma }} = \frac{{6.02 \times {{{10}}^{{23}}}\sum {{{T}_{{{{{\max }}_{i}}}}}} }}{{22{\kern 1pt} {\kern 1pt} 400 \times {{S}_{{{\text{уд}}}}}G}},$Дифференциально-термический анализ образцов носителей и катализаторов (свежих и зауглероженных), а также их предшественников осуществляли на дериватографе
Q 1500 D (F. Paulik, J. Paulik, L. Erdey; МОМ, Венгрия); скорость линейного нагрева 7.5–10.0°С/мин.
Термогравиметрический анализ (ТГА) проводили в воздушной атмосфере или в атмосфере азота. Навески образцов составляли 120–250 мг (погрешность взвешивания ±0.4 мг). Температуру измеряли термопарой платина–платинородий (ПП-1) с погрешностью ±2°C в интервале температур от 22 до 900°С.
Рентгеновские дифрактограммы получали на автоматизированном рентгеновском дифрактометре ДРОН-3 с графитовым монохроматором. Измерения проводили с использованием CuKα излучения в режиме пошагового сканирования с шагом 2θ = 0.1°. Время экспозиции на одну точку составляло 3 с. Обработку дифрактограмм проводили с использованием программ PHAN и PHAN (%) для качественного и количественного рентгенофазового анализов.
Запись ИК-спектров осуществляли при температуре адсорбции на Фурье-спектрометре Shimadzu 8300 с разрешением 4 см–1 и числом накоплений спектров 50. Свежеприготовленные образцы предварительно прессовали в виде таблеток плотностью 7– 17 мг/см2, прокаливали в ИК-кювете при 450°C в течение 1 ч в вакууме (0.013–0.001 МПа) и охлаждали до 110°C жидким азотом. При определении кислотных центров в качестве молекул-зондов использовали аммиак и монооксид углерода.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В табл. 1 приведены характеристики удельной поверхности и пористости синтезированных для испытания в процессе превращения ДМЭ образцов ZnO/γ-Al2O3-катализаторов. Видно, что их итоговые текстурные характеристики определяются параметрами использованного носителя. Тем не менее, введение в матрицу носителя оксида цинка приводит к некоторому уменьшению удельной поверхности и суммарного объема пор. С увеличением содержания в образцах катализаторов оксида цинка наблюдается, также перераспределение объема пор по размерам, а именно, снижение количества микро- и мезопор и рост числа макропор.
Таблица 1.
Зависимость текстурных характеристик γ-Al2O3-носителя и ZnO/γ-Al2O3-катализаторов от условий термической обработки
| Образец ZnO/Al2O3-катализатор |
Условия термообработки Т, °С/время, мин | Sуд, м2/г | Vпор, см3/г | Распределение объема пор по размерам, нм | |||
|---|---|---|---|---|---|---|---|
| термолиз | прокаливание | 5–10 | 10–102 | 102–103 | |||
| γ-Al2O3* ZnO (5.0%) ZnO (10%) ZnO (15%) ZnO (20%) |
– 320/120 320/150 320/180 320/150 |
– 430/240 450/240 450/300 450/300 |
188 178 168 165 153 |
0.58 0.55 0.52 0.50 0.47 |
0.23 0.21 0.18 0.16 0.13 |
0.27 0.24 0.22 0.21 0.15 |
0.08 0.10 0.12 0.13 0.19 |
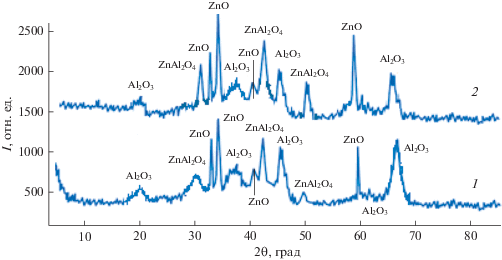
Наблюдаемое изменение текстурных характеристик ZnO/γ-Al2O3-катализаторов с увеличением содержания оксида цинка, вероятно, связано, как вытекает из дифрактограмм приведенных на рис. 2, с образованием и ростом относительного содержания фазы алюмината цинка, влияющей на степень развитости активной поверхности образцов.
Рис. 2.
Дифрактограммы образцов ZnO/Al2O3-катализаторов. Содержание Zn в образцах (мас. %): 1 – 10%, 2 – 20%, в пересчете на оксиды.
В качестве исходных данных для расчета значений скорости суммарного и частных превращений ДМЭ использовалась выборка результатов хроматографического анализа компонентного состава продуктов реакции при варьировании температуры реакции в диапазоне 380–480°С, объемной скорости подачи ДМЭ 100–190 ч–1, мольном отношении азот : сырье = = (1–10) : 1 (табл. 2).
Таблица 2.
Зависимость скорости суммарного превращения ДМЭ $\left( {W_{{{\text{ДМЭ}}}}^{\Sigma }} \right)$ и выхода продуктов реакции (α $ \times {\text{\;}}$ Si) от объемной скорости подачи ДМЭ при варьировании температуры реакции в области 420–480°С, мольном разбавлении ДМЭ : N2 = 1 : 10 в присутствии ZnO/γ-Al2O3-катализатора.
| GHSV, ч–1 | Скорость суммарного превращения $W_{{{\text{ДМЭ}}}}^{\Sigma },$$\left( {\frac{{{\text{моль}}}}{{{\text{л}}\,\,{\text{ч}}}}} \right)$ |
Конверсия, ДМЭ α,% |
Выход продуктов реакции в контактном газе, (α × Si), % | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| дивинил | н-бутилены | этанол | ацетальдегид | этилен | пропилен | диэтиловый эфир | формальдегид | СО + СН4 | |||
| Температура в контактной зоне, 380°С | |||||||||||
| 100 | 1.96 | 44.0 | 19.3 | 0 | 4.5 | 0 | 10.0 | 0 | 0 | 0.34 | 4.3 |
| 150 | 2.28 | 34.0 | 20.0 | 3.0 | 5.9 | 0.44 | 33.5 | 2.9 | 4.6 | 0.29 | 6.1 |
| 190 | 2.54 | 30.0 | 19.2 | 3.3 | 8.3 | 0.23 | 38.8 | 2.3 | 4.0 | 0.27 | 7.0 |
| Температура в контактной зоне, 400°С | |||||||||||
| 100 | 2.14 | 48.0 | 20.0 | 0 | 27.1 | 0 | 19.6 | 0 | 0 | 0.52 | 19.4 |
| 150 | 2.55 | 38.0 | 20.2 | 3.8 | 4.3 | 0.5 | 28.3 | 3.4 | 3.9 | 0.40 | 19.3 |
| 190 | 2.97 | 35.0 | 18.3 | 12.0 | 2.86 | 0.29 | 27.8 | 2.86 | 2.2 | 0.34 | 21.0 |
| Температура в контактной зоне, 420°С | |||||||||||
| 100 | 2.39 | 53.5 | 19.4 | 3.7 | 15.9 | 0 | 18.9 | 0 | 0.2 | 0.37 | 17.2 |
| 150 | 2.88 | 43.0 | 19.5 | 9.3 | 6.42 | 0.42 | 24.0 | 3.2 | 3.5 | 0.19 | 29.6 |
| 190 | 3.22 | 38.0 | 18.9 | 11.8 | 5.18 | 0.23 | 21.7 | 3.15 | 1.3 | 0.16 | 31.3 |
| Температура в контактной зоне, 440°С | |||||||||||
| 100 | 2.55 | 57.2 | 15.3 | 0 | 12.3 | 0 | 18.0 | 0.1 | 0.3 | 0.2 | 23.5 |
| 150 | 3.25 | 48.5 | 15.6 | 6.7 | 0.3 | 0.35 | 22.5 | 3.2 | 3.4 | 0.1 | 30.4 |
| 190 | 3.60 | 42.4 | 14.8 | 7.0 | 0.2 | 0.15 | 19.3 | 2.9 | 1.0 | 0 | 33.6 |
| Температура в контактной зоне, 460°С | |||||||||||
| 100 | 2.70 | 60.5 | 10.2 | 0 | 0 | 0 | 17.6 | 0 | 0 | 0.1 | 28.3 |
| 150 | 3.44 | 51.3 | 10.5 | 6.2 | 0.1 | 0.21 | 21.0 | 3.0 | 3.2 | 0 | 36.7 |
| 190 | 3.82 | 45.0 | 9.6 | 5.9 | 0 | 0.1 | 16.6 | 2.8 | 0.5 | 0 | 40.2 |
| Температура в контактной зоне, 480°С | |||||||||||
| 100 | 2.79 | 62.4 | 7.8 | 1.8 | 5.2 | 2.6 | 7.3 | 1.6 | 0.9 | 0 | 31.2 |
| 150 | 3.66 | 54.6 | 8.0 | 5.6 | 5.8 | 2.9 | 11.0 | 2.5 | 2.3 | 0 | 39.6 |
| 190 | 4.07 | 48.0 | 7.4 | 5.1 | 3.3 | 2.7 | 8.0 | 2.0 | 0.1 | 0 | 42.4 |
Из приведенных данных, а также результатов исследования кинетических закономерностей реакции, приведенных в работе [10], наиболее вероятной представляется следующая последовательность превращения ДМЭ в целевой продукт реакции 1,3-бутадиен, реализуемая в высокотемпературной области 420–480°С:

В низкотемпературной области протекания реакции, по-видимому, реализуется последовательность образования дивинила по Лебедеву [11]:
(5)
$\begin{gathered} 2{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OC}}{{{\text{H}}}_{3}} \to 2{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{5}}}}{\text{OH}} \to {\text{2C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}} \to \\ \to {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHOH}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}} \to \\ \to {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CH = CHCHO}} \to {\text{C}}{{{\text{H}}}_{2}}{\text{ = CH}}{\kern 1pt} - {\kern 1pt} {\text{CH = C}}{{{\text{H}}}_{2}}. \\ \end{gathered} $При сопоставлении активности свежеприготовленных образцов катализатора и образцов, “проработавших” в течение 7 ч и более (табл. 3), был выявлен факт изменения скорости и направлений превращения ДМЭ, не наблюдавшийся в раннем периоде проявления активности. Установлено, что наибольшие различия указанных закономерностей проявляются при смене температурных областей протекания процесса (380–420°С) и (440–480°С), условно обозначенных, как низкотемпературная и высокотемпературная.
Таблица 3.
Зависимость скорости суммарного превращения ДМЭ $\left( {W_{{{\text{ДМЭ}}}}^{\Sigma }} \right)$ и выхода продуктов реакции (α × Si) от объемной скорости подачи ДМЭ при варьировании температуры реакции в области 420–480°С, мольном разбавлении ДМЭ: N2 = 1 : 10 в присутствии ZnO/γ-Al2O3-катализаторов после 7 часовой непрерывной эксплуатации свежеприготовленных образцов
| GHSV, ч–1 | Скорость суммарного превращения $W_{{{\text{ДМЭ}}}}^{\Sigma },$$\left( {\frac{{{\text{моль}}}}{{{\text{л}}\,\,{\text{ч}}}}} \right)$ |
Конверсия, ДМЭ α, % |
Выход продуктов реакции в контактном газе, (α ×Si), % | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| дивинил | н-бутилены | этанол | ацетальдегид | этилен | пропилен | дэтиловый эфир | формальдегид | СО + СО2 + СН4 | |||
| Температура в контактной зоне, 420°С | |||||||||||
| 100 | 2.05 | 37.0 | 13.5 | 2.2 | 13.7 | 1.2 | 14.2 | 2.0 | 0.4 | 0.22 | 18.3 |
| 150 | 2.23 | 32.0 | 14.3 | 3.5 | 6.2 | 0.24 | 20.5 | 2.8 | 2.7 | 0.15 | 30.3 |
| 190 | 2.58 | 27.0 | 13.2 | 6.3 | 4.1 | 0.15 | 17.7 | 3.0 | 1.2 | 0.11 | 28.3 |
| Температура в контактной зоне, 480°С | |||||||||||
| 100 | 2.23 | 44.0 | 5.5 | 0 | 27.1 | 0 | 19.6 | 0 | 0 | 0.52 | 19.4 |
| 150 | 2.56 | 33.0 | 4.9 | 3.8 | 4.3 | 0.5 | 28.3 | 3.4 | 3.9 | 0.40 | 19.3 |
| 190 | 2.97 | 28.0 | 4.3 | 12.0 | 2.86 | 0.29 | 27.8 | 2.86 | 2.2 | 0.34 | 21.0 |
| Температура в контактной зоне, 480°С* | |||||||||||
| 100 | 2.57 | 58.0 | 7.4 | 1.6 | 5.0 | 2.3 | 6.8 | 1.5 | 0.7 | 0.2 | 38.4 |
| 150 | 3.08 | 48.0 | 7.6 | 5.3 | 5.4 | 2.2 | 9.6 | 2.4 | 2.0 | 0 | 43.2 |
| 190 | 3.72 | 45.0 | 7.2 | 4.7 | 2.9 | 2.5 | 7.8 | 1.9 | 0 | 0 | 48.6 |
Причем, при переходе от граничной точки низкотемпературной области (420°С) в высокотемпературную область (480°С) относительное снижение скорости суммарного превращения ДМЭ и выхода продуктов реакции наблюдается более рельефно.
Примечательно, что при максимальной в изученном диапазоне температуре реакции (480°С) введение в сырьевой поток пероксида водорода в количестве 1.5 мас. % по отношению к ДМЭ способствует восстановлению активности катализатора, – и значения скорости суммарного и частных превращений ДМЭ практически приближаются к таковым, приведенным в табл. 2, а в газообразной части катализата идентифицируется незначительное количество диоксида углерода.
Причем, при переходе от граничной точки низкотемпературной области (420°С) в высокотемпературную область (480°С) относительное снижение скорости суммарного превращения ДМЭ и выхода продуктов реакции наблюдается более рельефно.
Примечательно, что при максимальной в изученном диапазоне температуре реакции (480°С) введение в сырьевой поток пероксида водорода в количестве 1.5 мас. % по отношению к ДМЭ способствует восстановлению активности катализатора, – и значения скорости суммарного и частных превращений ДМЭ практически приближаются к таковым, приведенным в табл. 2, а в газообразной части катализата идентифицируется незначительное количество диоксида углерода.
Так как при вхождении в высокотемпературный режим реакции переход превращений ДМЭ в диффузионную область, либо наличие гетерогенно-гомогенных закаталитических объемных превращений после постановки специальных экспериментов были исключены, то наиболее вероятная причина изменения проявляемой каталитической активности связана со спецификой зауглероживания поверхности катализатора.
Для подтверждения упомянутой причины изменения закономерности превращений ДМЭ при переходе в высокотемпературную область, нами были проведены сопоставительные испытания активности катализаторов, по прошествии некоторого времени контактирования с реакционной средой.
В результате обработки экспериментальных данных, в координатах WДМЭ/ТРЕАК нами был обнаружен гистерезис (рис. 3), очевидно, связанный со сменой механизма поверхностных превращений ДМЭ определяемого изменением характера активной поверхности катализатора.
Рис. 3.
Температурная зависимость скорости суммарного превращения ДМЭ: 1 – исходный образец ZnO/γ-Al2O3-катализатора; 2 – образец катализатора после завершения реакционного цикла; 3 – превращение ДМЭ после завершения реакционного цикла при введении 1.5 мас. % пероксида водорода. Условия реакции: GHSV, ч–1 = 190; временной интервал превращения ДМЭ до перехода к следующему измерению скорости превращения ДМЭ – 1 ч.
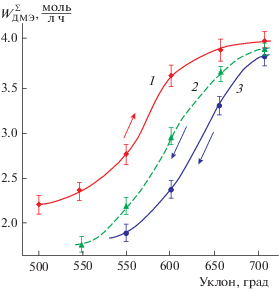
При реализации экспериментов изначально определялась скорость суммарного превращения ДМЭ в присутствии свежеприготовленного катализатора при 380°С (первая точка на кривой 1). Далее, по истечении 1 ч, переходили к следующему измерению скорости (вторая точка на кривой 1). При достижении температуры реакции 440°С, при четвертой точке на кривой 1 (вхождение реакции в высокотемпературную область), наблюдается резкое отклонение от близкой к экспоненциальной зависимости скорости превращения ДМЭ от температуры и дальнейший прирост скорости снижается. Очевидно, что за время окончания реакционного цикла (шестая точка на кривой 1) успевает накопиться определенное количество кокса, блокирующего активные центры поверхности катализатора, что и является причиной снижения его активности. Характерно, что при ступенчатом снижении температуры реакции (кривая 2) и возвращении к точкам, зафиксированным на температурной кривой 1, наблюдается значительное расхождение скорости превращения ДМЭ, т.е. имеет место S-образный температурный гистерезис.
Установлено, что введение в состав реакционной смеси Н2О2 в количестве 1.5 мас. % и последующее пошаговое снижение температуры приводит к частичному восстановлению активности катализаторов (кривая 3). Разница между активностью исходных и зауглероженных образцов при этом становится не столь значительной, т.е. в данном случае проявляется регенерирующая функция пероксида водорода, как окислителя углеродистых отложений.
Непосредственное доказательство образования углистых отложений при конверсии ДМЭ в присутствии ZnO/Al2O3-катализаторов было получено с помощью дифференциально-термического анализа образцов катализаторов, после 10-ти часового непрерывного пробега установки (рис. 4).
Рис. 4.
Кривые ТГ и ДТА ZnO/γ-Al2О3-катализаторов после 10 часовых пробегов установки в отсутствие добавок Н2О2 (1) и при введении в зону реакции пероксида водорода в количестве 1.5 мас. % (2).
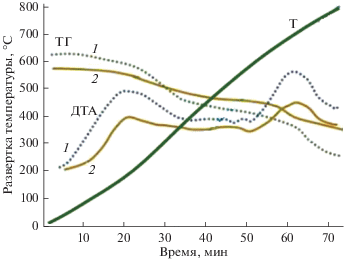
Из полученных результатов видно, что в процессе нагрева образцов наблюдается поэтапное уменьшения их веса, которое сопровождается несколькими экзотермическими эффектами, связанными с выгоранием поверхностных углистых отложений. Проявление двух максимумов на кривых ДТА в диапазоне температуры 450–500°С и 500–550°С (образец 1), а также 350–400°С и 400–450°С (образец 2) свидетельствует о наличии двух типов отложений, отличающихся прочностью связи с поверхностью образцов катализатора.
В результате ДТА образцов катализатора после 10-ти часового пробега установки, осуществляемого с добавлением в сырьевой поток пероксида водорода, установлено снижение экзотермических пиков, соответствующих двум типам углеродистых отложений, что подтверждает наличие регенерирующей коксообразование функции Н2О2 при конверсии ДМЭ.
Приведенные результаты дают основание полагать, что образование углистых отложений на поверхности катализаторов происходит при участии по меньшей мере двух типов активных центров, предположительно кислотной природы, локализованных в матрице γ-Al2O3-носителя.
В ТПД-спектрах аммиака (рис. 5 и табл. 4), адсорбированного на поверхности γ-Al2O3-носителя активной массы потенциальных ZnO/γ-Al2O3-катализаторов проявляются три десорбционных максимума, изменяющие положение на температурной координате в зависимости от условий эксперимента, °С: ${{T}_{{{{{\max }}_{1}}}}}$ (145–235); ${{T}_{{{{{\max }}_{2}}}}}$ (240–290); ${{T}_{{{{{\max }}_{3}}}}}$ (500–550).
Рис. 5.
Термодесорбционные спектры аммиака с поверхности образца носителя γ-Al2О3 (1) и ZnO/Al2О3-катализатора с содержанием ZnO в матрице носителя 10 – 2 и 20 мас. % – 3.
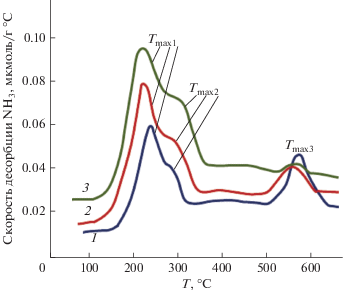
Таблица 4.
Сопоставление характеристик кислотности поверхности γ-Al2O3-носителя и ZnO/γ-Al2O3-катализаторов вычисленных по термодесорбционным спектрам аммиака. Начальная температура адсорбции NH3 – в пределах 50–65°C, скорость программированного нагрева β = 10–20°С/мин, дозировка NH3 – в пределах 0.1–0.5 ммоль
| Образцы носителя и катализатора ZnO/Al2O3* | Температура ${{T}_{{{{{\max }}_{i}}}}}$ | Кислотность, соответствующая ${{T}_{{{{{\max }}_{i}}}}}$ | $\sum {{{N}_{i}}} $ × 1019 ед./м2 | ||||
|---|---|---|---|---|---|---|---|
| ${{T}_{{{{{\max }}_{1}}}}}$ | ${{T}_{{{{{\max }}_{2}}}}}$ | ${{T}_{{{{{\max }}_{3}}}}}$ | N1 | N2 | N3 | ||
| γ-Al2O3 | 145–235 | 240–290 | 500–550 | 1.20 × 1019 | 6.72 × 1018 | 9.58 × 1018 | 2.83 |
| ZnO, 10% | 140–225 | 233–285 | 490–545 | 1.26 × 1019 | 7.08 × 1018 | 3.52 × 1018 | 2.32 |
| ZnO, 20% | 133–210 | 230–280 | 485–540 | 1.25 × 1019 | 6.25 × 1018 | 2.75 × 1018 | 2.15 |
| ZnO, 10%** | 135–220 | 225–275 | 470–520 | 1.12 × 1019 | 5.75 × 1018 | 1.25 × 1018 | 1.82 |
| ZnO, 10%*** | 138–220 | 235–280 | 492–547 | 1.28 × 1019 | 6.65 × 1018 | 3.25 × 1018 | 2.27 |
Наблюдаемые максимумы соответствуют десорбции аммиака с поверхностных кислотных центров, условно ранжированных как “слабые”, “умеренные” и “сильнокислотные”.
Из приведенных данных следует, что введение в матрицу алюмооксидного носителя оксида цинка приводит к количественному перераспределению кислотных центров. Так, с увеличением содержания активной массы (ZnO) наблюдается блокировка и уменьшение числа кислотных центров всех трех типов, что проявляется также в сдвиге максимумов десорбционных пиков аммиака (${{T}_{{{{{\max }}_{i}}}}}$) в низкотемпературную область (см. рис. 5). При этом в наибольшей степени происходит снижение числа “сильных” кислотных центров.
Аналогичный факт перераспределения кислотных центров был установлен при термодесорбции аммиака с зауглероженной поверхности ZnO/Al2O3 катализаторов, находившихся в контакте с реакционной средой после 10-ти часового пробега установки.
Для определения бренстедовской кислотности исходных образцов носителя γ-Al2О3 и ZnO/γ-Al2О3-катализаторов анализировали спектры поглощения ОН-групп, проявляющихся в диапазоне 3400–3800 см–1 (мостиковые и терминальные ОH-группы) (рис. 6).
Рис. 6.
Фрагменты ИК-спектров образцов носителя γ-Al2О3 (1) и ZnO/γ-Al2О3-катализаторов в области волновых чисел 3900–3400 см–1 (поглощение ОН-группами). Содержание ZnO в матрице γ-Al2О3 10 – 2 и 20 мас. % – 3; после напуска в кювету аммиака – 4.
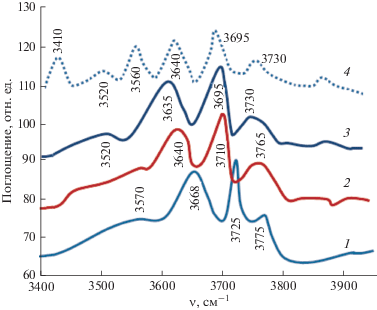
Как видно из параметров спектра носителя, до нанесения активной массы оксида цинка (1), наиболее высокочастотные ОН-группы (νОН = = 3775 см–1), вероятно связанные с тетраэдрическими ионами алюминия, а также ОН-группы (νОН = = 3725 см–1), связанные с октаэдрическими ионами алюминия, можно отнести к терминальным.
Остальные ОН-группы, полосы которых наблюдаются в низкочастотном участке спектра (νОН = 3670–3570 см–1), являются мостиковыми, расположенными между различно координированными ионами алюминия.
С повышением количества наносимой активной массы ZnO (спектры 2 и 3) наблюдается смещение полос поглощения ОН-групп в низкочастотную область при одновременном росте их относительной интенсивности, что обусловлено частичной нейтрализацией более сильных бренстедовских кислотных центров на поверхности образцов.
После напуска в измерительную кювету дозированного количества аммиака, в ИК-спектрах образца с содержанием ZnO в матрице носителя 20 мас. % наблюдается снижение интенсивности полос поглощения свободных гидроксильных групп при 3695 и 3730 см–1, при одновременном появлении полос 3560 и 3410 см–1, что характерно для образования водородной связи между молекулами NH3 и протоно-донорными бренстедовскими кислотными центрами на поверхности катализаторов.
Спектры поглощения монооксида углерода, адсорбированного на поверхности γ-Al2О3-носителя и ZnO/Al2О3-катализаторов, предварительно прокаленных при 500–550°C, свидетельствуют о наличие двух типов различающихся по силе льюисовских кислотных центров, первые из которых характеризуются полосами поглощения CO в диапазоне 2150–2170 см–1, а вторые в интервале 2180–2210 см–1 (рис. 7).
Рис. 7.
ИК-спектры: монооксида углерода, адсорбированного на поверхности γ-Al2О3-носителя (1) и ZnO/Al2О3-катализатора, с содержанием ZnO в образцах: 2 – 10; 3 – 20 мас. %.
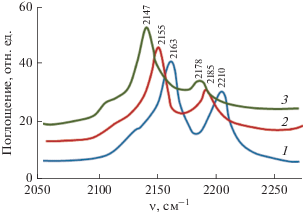
При увеличении концентрации катионов цинка видно, что полосы поглощения соответствующие как сильным, так и слабым льюисовским кислотным центрам сдвигаются в низкокочастотную область. При этом интенсивность полос поглощения, характерных для “сильнокислотных” центров исходного γ-Al2O3 уменьшается, происходит перераспределение поверхностной концентрации кислотных центров, и появляются серии новых полос поглощения, соответствующих более слабым акцепторным центрам (2155 и 2147 см–1).
Таким образом, на основании результатов по конверсии ДМЭ в присутствии свежеприготовленных и зауглероженных в ходе процесса ZnO/γ-Al2О3-катализаторов, ИК-спектрального исследования адсорбированных на их поверхности молекул – зондов (NH3 и CO), а также, принимая во внимание данные дифференциального термического анализа образцов, можно прийти к выводу о наличие двух температурных областей протекания реакции (380–420°С) и (440–480°С). В каждой из этих областей доминирует определенная последовательность образования целевого продукта реакции – 1,3-бутадиена, согласно приведенным схемам (3) и (4) , т.е. в зависимости от условий проведения реакции, проявляется наличие множественности стационарных состояний (МСС), в рассматриваемом случае двух.
Наличие двух стационарных состояний реакции при превращении ДМЭ позволяет считать, что и ее механизм в рассмотренных температурных областях имеет многоканальный характер, и не исключен факт смены медленных стадий при варьировании температуры. При этом в низкотемпературной области реакции, постулируется доминирующее участие активных кислотных центров бренстедовского типа, в высокотемпературной области – льюисовского.
Подобный механизм, скорее всего, имеет место, несмотря на обнаруженное с помощью РФА постоянство фазового состава образцов при разной степени зауглероженности поверхности катализатора после реакции во всем изученном температурном интервале.
ЗАКЛЮЧЕНИЕ
Наряду с изучением кинетического механизма для реализации процессов необходимо тщательно изучать их динамику, связанную как с макрофакторами, так и с критическими явлениями в катализе, вызванными множественностью стационарных состояний, коксованием поверхности, автоколебательными режимами и т.п. В настоящей работе показано, что макрокинетические факторы не оказывают существенного влияния на процесс превращения ДМЭ. Обнаруженный гистерезис в рассмотренном интервале температур свидетельствует о наличии множественности стационарных состояний, вызванный наличием двух типов центров (бренстедовских и льюисовских).
Полученные результаты позволяют ускорить прогнозирование дизайна катализатора и эффективно реализовать в промышленном масштабе процесс. Отметим, что в большинстве работ в области гетерогенного катализа отсутствует изучение нелинейных явлений и МСС, что тормозит развитие науки и производства в химической промышленности.
Эволюция гетерогенно-каталитических превращений в пористых средах связана с многофазными процессами [12–15], проявляющимися в динамике реакций, связанных, по-видимому, с взаимным влиянием катализатора и инициатора, формирующем каталитическую систему в условиях коксования поверхности катализатора. Углубление представлений о механизме каталитического процесса позволяет прогнозировать генезис катализатора, определяемый его химическим составом и алгоритмом синтеза.
Изучению связи генезиса катализатора с кинетическими параметрами реакций посвящены работы [17–21], в которых показана возможность путем изменения параметров синтеза катализаторов влиять на энергию активации и факторы взаимодействия реагентов с активными центрами поверхности.
Отметим, что в работах [5–7] алюминий используется как связующее в бинарной системе ZnO + Al2O3. Имеются общие приемы в синтезе, находящиеся в соответствии с результатами синтезированных нами образцов катализатора. Так, например, в случае пропитки используется прием диффузионной пропитки смешанного раствора активных компонентов, содержащих цинк и алюминий, которые наносятся на оксид алюминия, а также вяление 12 ч при комнатной температуре. Целью этого приема является упрочнение связи между оксидами цинка и алюминия. Желаемым итоговым результатом упрочнения связи между металлами являлось бы образование шпинели ZnAl2O4. Шпинели обычно образуются при достаточно высоких температурах, выше 1000°С. Однако в [6] упоминается о ее наличии, несмотря на наивысшую точку прокаливания, не превышающую 500°C. Тем не менее, независимо от наличия в системе шпинели, цинк, судя по работам [5–7, 22] растворяется в смеси цинка с алюминием, усиливая связь между оксидами металлов.
Таким образом, наряду с решением практической задачи по синтезу катализатора, углубленное представление о механизме, включающем кинетику и динамику процесса, позволяет расширить и развить современные фундаментальные представления о гетерогенном катализе [21].
Отметим, что изучение химии каталитического превращения ДМЭ в аспекте влияния на механизм множественности стационарных состояний в представленных нами ранее работах [8–10] не рассматривалась, в связи с чем, обнаруженные закономерности требовали углубленного изучения.
Полученные в настоящей работе результаты свидетельствуют о необходимости детального изучения динамики процессов, катализируемых композициями на основе оксидов алюминия и цинка, используемых в различных реакциях, в которых участвуют оксигенаты [7, 22–24]. Обычно объектами для изучения подобных явлений являются многомаршрутные реакции, протекающие по нелинейным механизмам, осложненным значительным выходом побочных продуктов и коксованием поверхности катализаторов [25–33], что необходимо учитывать при реализации промышленных нефтехимических процессов.
ВЫВОДЫ
1. Обнаружены две температурные области в процессе превращения ДМЭ, характеризуемые различными тенденциями влияния режимных параметров, что свидетельствует о разных механизмах осуществления реакции и множественности стационарных состояний.
2. О наличии множественности стационарных состояний свидетельствует гистерезисная петля между выделенными температурными областями протекания реакции в пределах температурного диапазона 380–420°С.
3. Обнаруженные стационарные состояния характеризуются устойчивостью, усиливающейся при инициировании процесса пероксидом водорода.
4. Установлена множественность стационарных состояний в процессе превращения ДМЭ, связанная с наличием кокса на поверхности катализатора, содержащего бинарную ZnO/γ-Al2O3-композицию, и регулируемая соотношением льюисовских и бренстедовских центров в зависимости от температуры процесса.
Список литературы
Ламберов А.А., Мухамбетов И.Н., Залялиев Р.Ф. // Катализ в промышленности. 2014. № 1. С. 49.
Хьюз Р. Дезактивация катализаторов: OZON.RU, Химия, ISBN: 5-7245-0154-6, 0-12-360870-8 1989, 280 с.
Островский Н.В. Кинетика дезактивации катализаторов: Математические модели и их применение. М.: Наука, 2001 г. 334 c.
Третьяков В.Ф., Талышинский Р.М. Кинетика и динамика гетерогенных каталитических нефтехимических процессов. М.: МИТХТ, 2012. 190 с.
Di Liu, Yong Men, Jinguo Wang, Xin Liu, Qiuyan Sun // Am. J. of Analytical Chemistry, 2016. № 7. P. 568.
Elam J.W., Routkevitchb A.D, George S.M. // J. of The Electrochemical Soc. 2003. V. 150. № 6. P. 339.
Кипнис М.A., Белостоцкий И.А., Волнина Э.А., Лин Г.И. // Катализ в промышленности. 2018. Т. 18. № 5. С. 12.
Хаджиев С.Н., Максимов А.Л., Третьяков В.Ф., Талышинский Р.М., Илолов А.М. // Нефтехимия. 2018. Т. 58. № 4. С. 405 [Petrol. Chemistry. 2018. V. 58. № 8. P. 613].
Максимов А.Л., Третьяков В.Ф., Талышинский Р.М., Илолов А.М., Хаджиев С.Н. // Нефтегазохимия. 2019. Т. 59. № 2. С. 44–51.
Максимов А.Л., Третьяков В.Ф., Литвишков Ю.Н., Зульфугарова С.М., Талышинский Р.М., Илолов А.М. // Химические проблемы. 2019. № 1 (17). С. 135.
Лебедев С.В. Жизнь и труды. ОНТИ ХИМ. ТЕОРЕТ. Л., 1938. 878 с.
Быков В.И. Моделирование критических явлений в химической кинетике. М.: КомКнига, 2006. 328 с.
Яблонский Г.С., Быков В.И., Горбань А.Н. Кинетические модели каталитических реакций, Новосибирск: Наука, 1983. 255 с.
Гудков Б.С., Субботин А.Н., Якерсон В.И. Температурный гистерезис в гетерогенном катализе. М.: Химия, 2001. 7 с.
Бояринов А.И. Множественность стационарных состояний в системе: смеситель–реактор–узел разделения. М.: Химия, 1982. 288 с.
Хаджиев С.Н., Третьяков В.Ф., Илолов А.М., Талышинский Р.М. // Нанотехнологии. Наука и производство. 2015. Т. 1. № 10. С. 33.
Гюльмалиев А.М., Третьяков В.Ф., Талышинский Р.М., Илолов А.М., Хаджиев С.Н. // Нефтехимия. 2019. Т. 59. № 5. С. 529 [Petrol. Chemistry. 2019. V. 59. № 9. Р. 979].
Литвишков Ю.Н., Мурадова П.А., Эфендиев М.Р., Дадашева Ш.А., Джафарова С.А., Кулиева Л.А. // Химические проблемы. 2009. № 2. С. 290.
Третьяков В.Ф., Талышинский Р.М., Третьяков К.В., Илолов А.М., Литвишков Ю.Н. // Нефтепереработка и нефтехимия. 2011. № 12. С. 44.
Киперман С.Л. Основы химической кинетики в гетерогенном катализе. М.: Химия, 1979. 352 с.
Максимов А.Л., Третьяков В.Ф., Талышинский Р.М. // Нефтегазохимия (Кинетика и катализ). 2018. № 4. С. 34.
Кадиев Х.М., Кадиева М.Х., Зекель Л.А., Хаджиев С.Н. // Коллоидный журнал. 2019. Т. 81. № 2. С. 1.
Cai M., Subramanian V., Sushkevich V.V., Ordomsky V.V., Khodakov A.Y. // https://istina.msu.ru/publications/article/10390799/ Applied Catalysis A: General. 2015. V. 502. P. 370.
Sushkevich Vitaly L., Ivanova Irina I., Taarning Esben // Green Chemistry (Royal Society of Chemistry, United Kingdom). 2015. V. 17. № 4. P. 2552.
Темкин О.Н. Теория механизмов сложных реакций и катализ (конспект лекций). М., 2003. 179 с.
Темкин О.Н., Шестаков Г.К., Трегер Ю.А. Ацетилен. Химия. Механизмы реакций. Технология. М.: Химия, 1991. 416 с.
Брук Л.Г., Темкин О.Н. // Кинетика и катализ. 2016. Т. 57. № 3. С. 275
Дебай П. Избранные труды. Л.: Наука, 1987. 559 с.
Мышлявцев А.В., Мышлявцева М.Д. // Кинетика и катализ. 2007. Т. 48. № 4. С. 576.
Федотов В.Х., Кольцов Н.И., Алексеев Б.В. // ДАН СССР. 1988. Т. 302. № 1. С. 126.
Хейфец Л.И., Неймарк А.В. Многофазные процессы в пористых средах. М.: Химия, 1982. 320 с.
Миттова И.Я., Самсонов А.А., Сладкопевцев Б.В. Основные понятия и механизмы каталитических и сопряженных реакций. Неравновесные процессы в катализе (Классические системы). Воронеж: Издательско-полиграфический центр Воронежского государственного университета, 2012 . 98 с.
Яндиева Ф.А., Цодиков М.В., Чистяков А.В., Кугель В.Я., Зубавичус Я.В., Велигжанин А.А., Китаев Л.Е., Ющенко В.В., Гехман А.Е., Моисеев И.И. // Кинетика и катализ. 2010. Т. 51. № 4. С. 572.
Дополнительные материалы отсутствуют.