

Нефтехимия, 2020, T. 60, № 5, стр. 586-599
Окислительное обессеривание углеводородного сырья с использованием кислорода как окислителя (обзор)
Е. А. Есева 1, *, А. В. Акопян 1, А. В. Анисимов 1, А. Л. Максимов 2
1 Московский государственный университет имени М.В. Ломоносова, Химический факультет
119991 Москва, Россия
2 Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Россия
* E-mail: esevakatya@mail.ru
Поступила в редакцию 27.04.2020
После доработки 08.05.2020
Принята к публикации 12.05.2020
Аннотация
В работе представлен обзор научных исследований в области окислительного обессеривания нефтяных фракций кислородом воздуха (в качестве окислителя), опубликованных за последние 15–20 лет. Проведен сравнительный анализ различных каталитических систем в данной области. Определены перспективные направления для разработки эффективной каталитической системы процессов окислительного обессеривания.
Природная нефть – сложная смесь углеводородов (УВ), серо-, азот- и других гетероатомных, в том числе и металлсодержащих соединений. При первичной разгонке фракции нефти содержат значительные количества сераорганических соединений, вызывающих коррозию оборудования и отравление гетерогенных катализаторов вторичной переработки, а при эксплуатации получаемых из них топлив в атмосферу выделяются оксиды серы и азота [1]. В связи с этим обессеривание является одним из важных процессов переработки нефтяных фракций и направлено на получение продуктов с низким содержанием серы для использования в качестве топлива или сырья для нефтехимических предприятий. Поэтому допустимые значения содержания серы и установленные на них стандарты в различных видах топлив в промышленно-развитых странах строго регламентированы, при этом наблюдается тенденция к их ужесточению. На данный момент в Европейских странах существуют экологические стандарты на ограничение содержания серы не более 10 ppm в бензиновом и дизельном топливе [2].
Для реализации данных требований в нефтепереработке применяют процесс гидроочистки, в основе которого лежит удаление сера- и азотсодержащих соединений под давлением водорода и при высокой температуре в присутствии CoMo- или NiMo-катализаторов [3]. При усложнении состава углеводородного сырья при переходе от бензиновой фракции к вакуумному газойлю ужесточаются условия проведения процесса: увеличивается давление от 30 до 170 атм, температура от 300 до 420°С, а также появляется необходимость построения установки рециклизации водородсодержащего газа из-за избыточного его использования. В процессе гидроочистки происходит удаление меркаптанов, сульфидов и дисульфидов, а производные бензотиофена и дибензотиофена удаляются лишь частично, поскольку являются наименее реакционноспособными [4]. В ходе гидроочистки происходит также восстановление олефинов и ароматических соединений до нафтенов и парафинов, расходуется большой избыток водорода, что приводит к росту капитальных и энергетических затрат. В результате указанных факторов в настоящее время усиливается тенденция к использованию альтернативных процессов обессеривания, обеспечивающих высокую степень сероочистки. К альтернативным методам относятся экстракция органическими растворителями и ионными жидкостями [5–7], использование селективных сорбентов, биообессеривание и окислительное обессеривание [1, 3, 8, 9].
Метод окислительного обессеривания основан на окислении серосодержащих соединений до соответствующих сульфоксидов и сульфонов с последующим извлечением окисленных продуктов адсорбционными и экстракционными методами. Основные преимущества метода окислительного обессеривания заключаются в мягких условиях процесса и в отсутствии необходимости использования дорогого водорода. Используемый в качестве окислителя пероксид водорода при окислительном обессеривании образует воду как единственный побочный продукт реакции, но его основной недостаток связан с высокой степенью взрывоопасности. Кроме того, для процесса окислительного обессеривания применяются и другие окислители – органические пероксиды и гидропероксиды, неорганические окислители и озон, которые не могут быть использованы в переработке углеводородного сырья из-за их высокой стоимости и низкой селективности. Молекулярный кислород является экологически безопасным и дешевым окислителем, однако его использование ограничено значительным индукционным периодом, делающим его довольно инертным.
Решением этой проблемы является проведение процесса окисления чистым молекулярным кислородом или воздухом при высоких температурах и давлениях, а также с использованием различных катализаторов, среди которых получили распространение полиоксометаллаты, проявляющие активность при жидкофазном окислении различных органических соединений.
ОКИСЛИТЕЛЬНОЕ ОБЕССЕРИВАНИЕ В ПРИСУТСТВИИ ПОЛИОКСОМЕТАЛЛАТОВ
Известно окисление сернистых соединений воздухом при температурах 80–180°С в присутствии некислотных катализаторов, промотированных переходными металлами. Металлсодержащие каталитические системы катализируют разложение гидропероксидов, являющиеся интермедиатами процесса автоокисления, вследствие чего удается существенно снизить температуру реакции и энергетический барьер окисления [8]. Автоокисление заключается в образовании кислородсодержащих радикалов, генерирующихся из УВ под действием кислорода внутри реакционной системы. Гидропероксид – ключевой интермедиат процесса автоокисления. Механизм процесса окисления – свободно-радикальный, затрагивающий наиболее слабую связь С–Н для образования радикалов. Стабильный гидропероксид инициирует радикально-цепную реакцию окисления, неустойчивый – радикально-цепной процесс с вырожденным разветвлением [10].
Кислород – парамагнитная и бирадикальная частица, поэтому прямое его введение в реакции с устойчивыми диамагнитными органическими субстратами запрещено и требует особой активации окислителя. Электронно-насыщенный супероксидный радикал ${\text{О}}_{2}^{\centerdot },$ образующийся при химической активации в результате переноса одного электрона, легко окисляет спирты, амины, но не реагирует с нуклеофилами – сульфидами. Для протекания реакции по электрофильному механизму необходима генерация гидропероксильных радикалов (HOO•); в этом случае введение доноров электронов облегчает образование пероксидных радикалов (галогенид-ионы, аммиак, вода), но такой подход является энергоемким и связан с образованием значительных количеств побочных продуктов.
Альтернативный подход в активации кислорода основан на использовании катализатора, выполняющего роль переносчика электронов [11]. Химия полиоксометаллатов, представленная структурами Кеггина, Уэллс–Доусона, Линдквиста, Андресона и др., привлекает свое особое внимание за счет многофункционального активного центра, содержащего протоны, атомы кислорода и металлов. Преимущество взаимодействия ионов металлов и координированных мостиковых и концевых групп заключается в способности таких структур активировать кислород для окисления серосодержащих соединений.
Впервые применение полиоксометаллатов в окислительном обессеривании было связано со специфической активацией молекулярного кислорода в относительно мягких условиях в реакциях окисления УВ. Авторами работы [12] было замечено, что полиоксометаллат типа [PVxMo12 –xO40](3 +x) в присутствии диметилсульфоксида (ДМСО) как растворителя повышает селективность окисления бензилового спирта до альдегида. Рост селективности непосредственно связан с участием ДМСО в окислении спирта, фактически он является окислителем в реакциях, катализируемых фосфованадомолибдатами. Механизм реакции, представленный на рис. 1, включает активацию сульфоксида путем комплексообразования с полиоксометаллатом и перенос кислорода от активированного сульфоксида. Комплекс сульфоксид–полиоксометаллат проявляет высокую эффективность в окислении алкилароматических субстратов. Следует заметить, что восстановление катализатора происходило за счет избыточного количества диметилсульфоксида с получением регенерированного полиоксометаллата, сульфида и воды. С появлением этого исследования проявился интерес к обратному процессу окисления сульфидов до сульфоксидов с участием полиоксометаллатов.
Рис. 1.
Механизм окисления бензилового спирта в ДМСО в присутствии фосфованадомолибдата (по данным [12]).
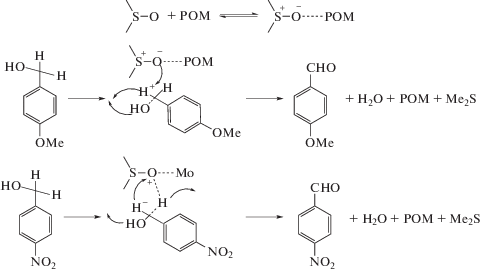
В работе [13] было синтезировано 78 типов фосфованадомолибденовых катализаторов общей формулы H(3 +x)[PVxM12 −хO40], где М = Мо, W и была протестирована их каталитическая активность в реакциях окислительного обессеривания под давлением кислорода при температуре 135°С в течение 140 мин. Было установлено, что наибольшей активностью в окислении модельной смеси, содержащей тиофен, бензотиофен (БТ) и дибензотиофен (ДБТ), обладает полиоксометаллат состава H5PV2W10O40. В оптимальных условиях (объемное соотношение катализатор/ацетонитрил/топливо = 2/9/15, ${{P}_{{{{{\text{O}}}_{2}}}}}$ = 20 атм, t = 2 ч, Т = 135°С) была достигнута 100%-ная конверсия ДБТ, а через 140 мин – полное окисление тиофена до сульфона.
Возникающие межфазные ограничения, обусловленные локализацией органического субстрата и катализатора в разных фазах, можно преодолеть введением четвертичных аммонийных солей в катион полиоксометаллата. Например, в работе [14] приведен катализатор, содержащий катион аммония состава (NH4)5H6PV8Mo4O40. В сравнении с широко известными катализаторами на основе солей переходных металлов (NaVO3, Na2MoO4), предложенный авторами полиоксометаллат обладает высокой каталитической активностью в сочетании с кислородом. Достигается 100%-ная конверсия ДБТ в течение 6 ч при 100°C. Для дизельного топлива с содержанием общей серы 1000 ppm через 6 ч было достигнуто снижение до 9 ppm. Авторами работ [15, 16] было изучено влияние изополиоксованадатов и тетрадекаванадофосфатов на окисление ДБТ в мягких условиях. Катализаторы состава [C8H17N(CH3)3]3H3V10O28 и [C8H17N(CH3)3]5H4PV14O42 взаимодействуют с молекулярным кислородом, образуя активные оксованадиевые комплексы, окисляющие ДБТ в модельной смеси в соответствующий сульфон с высокой селективностью. Этой же группой ученых [17] был синтезирован амфифильный катализатор состава [(C18H37)2N(CH3)2]5IMo6O24. Отмечено полное окисление сернистых субстратов при достаточно мягких условиях (80°С, атмосферное давление). Обнаружено, что ацетонитрил и вода оказывают негативное влияние на окисление в данной системе. Предполагается, что происходит блокировка активных центров катализатора в результате сильного взаимодействия между полиоксометаллатом и ацетонитрилом или водой. Было выявлено, что каталитическая активность катализатора зависит от строения катиона четвертичной аммонийной соли, а реакционная способность сернистых соединений снижается в ряду 4,6-ДМДБТ > > ДБТ > БТ. Ли и др. [18] сообщили, что ванадатопериодат структуры [C8H17N(CH3)3]3HIV9O28 обладает высокой каталитической активностью в реакциях окисления сернистых соединений за счет формирования супероксованадиевого комплекса внутри реакционной системы.
Другой группой исследователей [19] был также получен амфифильный полиоксометаллат, содержащий в своем составе пероксованадиевый комплекс. Установлено, что наличие пероксогруппы повышает эффективность окисления ДБТ в мягких условиях (70°С, атмосферное давление). Предложенный авторами работы механизм заключается в окислении субстрата до сульфоксида с последующей регенерацией пероксогруппы за счет непрерывного введения кислорода в реакционную систему и, в заключение, полного окисления до соответствующего сульфона (рис. 2). Изучено влияние природы четвертичного аммонийного катиона, наибольшей активностью обладает катализатор [C16H33N(CH3)3]5[PMo10V2O38(O2)2].
Рис. 2.
Предложенный механизм окисления ДБТ в присутствии пероксованадия полиоксометаллата (по данным [19]).
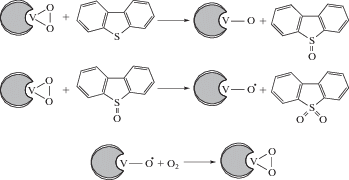
Для увеличения скорости реакции окисления сернистых соединений за счет снижения ограничений, связанных с диффузией и массопереносом, используют мицеллярные среды. Четвертичные аммониевые соли, состоящие из ядра полиоксометаллата, окруженного поверхностно-активным веществом, образуют супрамолекулярные обращенно-фазовые агрегаты в неполярных растворителях, позволяющие снижать содержание серы в дизельном топливе от 500 до 1 ppm [20].
В работе [21] было продемонстрировано применение кластерных полиоксометаллатных агрегатов в окислении органических сульфидов. Идея авторов заключалась в создании мицелл на основе додецилсульфата цезия с последующим осаждением на их поверхности наночастиц полиоксометаллата структуры Кеггина. Контроль размера, формы и структуры наночастиц, а также возможность их корреляции в таких системах с ядром поверхностно-активного соединения и поверхностью полианиона обеспечивает повышение активности катализатора в окислении сульфидов в модельной смеси.
Полиоксометаллаты типа Андерсона со смешанной валентностью центрального гетероатома проявляют высокую активность в мягких условиях (80°С) в реакциях окислительного обессеривания модельных смесей [22]. Сообщено, что в присутствии [(C18H37)2N(CH3)2]3Co(OH)6Mo6O18 достигается полное удаление ДБТ в течение 7 ч.
Новый подход к окислению серосодержащих соединений в углеводородном сырье продемонстрирован в работе [23]. Сущность данного метода заключается в окислении ДБТ до водорастворимых сульфатов под давлением кислорода в присутствии H8PV5Mo7O40. Такой подход позволяет исключить стадию удаления окисленных форм серы путем экстракции или адсорбции и позволяет достигнуть 99%-ного удаления ДБТ из модельного топлива при 120°С и давлении 20 атм.
Мицеллярный катализ имеет свои недостатки. В результате метастабильности эмульсий каталитическая система разрушается, что приводит к выделению полиоксометаллата и разделению фаз, что напрямую сказывается на снижении каталитической активности. Для решения данной проблемы авторы работ [24–26] предложили использование высокопористых металлоорганических каркасов (MOF). Отмечено, что благодаря гидрофобным взаимодействиям с участием металлорганических каркасов происходит диффузия сернистого компонента и его концентрирование в порах структуры, где расположены каталитически активные центры полиоксометаллата. За счет создания такой системы усиливаются субстрат-каталитические взаимодействия в области каркаса и улучшается кинетика реакции окисления. Например, было достигнуто полное окисление ДБТ молекулярным кислородом в присутствии металлоорганических структур, модифицированных полиоксометаллатами типа Кеггина и нанесенных на мезопористые материалы типа МСМ-41 [25]. Применение подобных каталитических систем позволяет увеличить стабильность и срок службы катализатора без потери активности как минимум в течение пяти каталитических циклов. Также изучено применение катализаторов на основе гетерополикислот, нанесенных на макропористую поверхность [27].
Металлоорганические каркасы привлекают внимание не только большой площадью поверхности и пористостью структуры, но и возможностью регулировки химического состава и структуры пор. Включение гетерополикислот в структуру пор металлоорганических каркасов позволяет получить высокоэффективный гетерогенный катализатор для процесса окислительного обессеривания. В работе [28] продемонстрировано влияние природы гетерополикислот в металлоорганических каркасах. Обнаружено, что HPW@MOF (где HPW – фосфорновольфрамовая кислота) проявляет наибольшую активность; в ее присутствии достигается 90% удаления ДБТ при 90°С в течение 4 ч, что хорошо согласуется с литературными данными, поскольку HPW является самой сильной кислотой Бренстеда в ряду Кеггина [29].
При гидрофобизации катализатора возникает проблема его отделения от реакционной среды, переработки и утилизации. Решением проблемы является нанесение полиоксометаллатов на инертные материалы, позволяющее существенно снизить механические потери катализатора и его выщелачивание в ходе реакции. Синтезированный методом пропитки гетерогенный катализатор был эффективно использован для удаления конденсированных производных тиофена из модельного топлива [30, 31]. Результаты показывают, что в присутствии гетерогенного катализатора H5PV2Mo10O40/SiO2 достигается 70%-ная конверсия ДБТ в течение 6 ч, в том время как, применение ультразвукового воздействия на модельную смесь в присутствии аналогичной каталитической системы позволяет повысить эффективность процесса обессеривания и достичь 90%-ной конверсии за 3.5 ч. Интенсивное перемешивание предотвращает осаждение продуктов конденсации, образующихся при взаимодействии сернистых соединений в присутствии катализатора и, соответственно, блокирующих поступающий окислитель, а также позволяет создать гомогенный раствор для равномерного распределения кислорода в реакционной системе.
Путем хемосорбции полиоксометаллат ион [Co(OH)6Mo6O18]–3 наносили на нанокристаллическую поверхность целлюлозы (CNC) с использованием полиионной жидкости (PIL) – бромида винил-3-додецилимидазолия [32]. Полученный катализатор состава CNC@PIL@POM показал высокую активность по отношению к конденсированным производным тиофена при их окислении в мягких условиях кислородом воздуха (97% удаления ДБТ при 80°С в течение 6 ч). Благодаря липофильности полимерной ионной жидкости, катализатор создает высокодисперсную реакционную систему, повышая возможность контакта активных центров с молекулой субстрата.
В работе [33] была изучена возможность нанесения полиоксометаллата типа Доусона H6 +nP2Mo18 –nVnO62 · mH2O на углеродные материалы. Авторы использовали гибридный материал CNT@MOF-199, который имеет подходящую удельную площадь поверхности для процесса десульфуризации (640 м2/г), обладает термической и химической стабильностью. В присутствии синтезированного катализатора было достигнуто 98% удаления ДБТ при оптимальных условиях проведения процесса (80°С, 3 ч). Липофильный ванадат, нанесенный на нитрид углерода ([(C8H17)3NCH3]3H3V10O28/3Dg-C3N4) за счет четвертичного аммонийного катиона и гидрофобного материала носителя, интенсифицирует межфазный перенос, а значит, повышает эффективность окисления серосодержащих соединений [34]. Носитель имеет трехмерную пористую структуру, в присутствии которой достигается высокая дисперсность системы. Материал носителя по данным авторов хорошо адсорбирует сернистые соединения, создавая локально высокие концентрации серы, что позволяет эффективно проводить реакции окислительного обессеривания.
Большинство работ по окислению включают генерацию надкислот внутри реакционной системы добавлением различных соокислителей. В связи с высоким энергетическим барьером активации кислорода процесс образования активных частиц в мягких условиях под действием молекулярного кислорода затруднен. В работе [35] рассматривается возможность активации кислорода с использованием медиаторов переноса электронов. Такой подход называется биомимическим и является на сегодняшний день одним из перспективных решений проблемы окисления сернистых соединений. В присутствии каталитической системы, состоящей из полиоксометаллата типа Андерсона (NH4)3Co(OH)6Mo6O18 и п-толуолсульфокислоты на основе эвтектического растворителя, происходит селективное окисление сульфидов до сульфонов подобно тому, как это имеет место при ферментативном катализе. Сообщено об синергетическом эффекте сочетания катализатора и сокатализатора в приведенной системе.
Полимерные ионные жидкости (PIL) имеют ряд преимуществ: низкое давление насыщенных паров, химическая стабильность, простота синтеза и возможность модификации катиона и аниона. Авторами работы [36] разработан супергидрофобный магнитный углеродсодержащий композитный катализатор MoOx/MC-600. Синтезированный катализатор представляет собой полиоксометаллат типа Кеггина, привитый на полимерной ионной жидкости и иммобилизированный на поверхности силикагеля, модифицированного 3-метакрилоксипропилтриметоксисиланом. Механизм реакции окисления ДБТ представляет собой свободно-радикальный процесс, доказанный введением в реакционную систему радикальных ловушек – трет-бутанола и п-бензохинона. Генерирование на поверхности катализатора супероксидных частиц приводит к образованию на ней пероксокомплекса молибдена, способствующему окислению адсорбированного ДБТ до ДБТО2 (рис. 3). Катализатор обладает магнитным эффектом, благодаря чему может быть легко отделен из реакционной системы и повторно использован без снижения каталитической активности на протяжении семи циклов. Аналогичный подход продемонстрирован в работе [37], где катализатор структуры Кеггина наносили на магнитный мезопористый материал.

Использование экстрагентов в процессе окислительного обессеривания сопряжено с рядом недостатков, в первую очередь ввиду их летучести и токсичности. Экологически безопасными растворителями и экстрагентами являются ионные жидкости, однако высокая стоимость ограничивает их применение в промышленности. Обнаружено, что кислотные эвтектические растворители (DES) обладают функциями, как растворителя, так и катализатора, и способны в одну стадию окислять субстраты и экстрагировать окисленные продукты. Экологически чистые растворители DES формируются в результате образования водородной связи между донором и акцептором водорода. Разработанный катализатор состава [(CH3)3NCH2CH2OH]xNa5 –xIMo6O24 эффективно работает в реакциях окисления сернистых соединений, является химически стабильным и механически прочным [38]. Сернистые соединения, такие как ДБТ, БТ и 4,6-ДМДБТ могут быть полностью окислены молекулярным кислородом с высокой селективностью согласно механизму, реализуемому при использовании эвтектического растворителя.
Авторами работы [39] был синтезирован новый вид катализатора сэндвичного типа {K8[(FeIII(OH2)2)3(A-α-PW9O34)2]}58(Si/AlO2) на катионите для селективного окисления воздухом альдегидов и сульфидов в мягких условиях. В качестве сернистого субстрата использовали тетрагидротиофен, конверсия которого составила 28% при 75°С и 120 ч с высоким TON (число оборотов). Этой же группой ученых была испытана каталитическая система, состоящая из солей меди и полиоксометаллата вышеуказанной структуры, для окисления полуиприта (2-хлорэтилэтилсульфида) [40]. Благодаря такому сочетанию катализаторов была достигнута 94%-ная конверсия при окислении горчичного газа. Позже группа ученых под руководством Окуна предложили новый тип катализатора для окисления горчичного газа при мягких условиях процесса (1 атм, 25°С) [41]. Катализатор, состоящий из гетерополивольфрамата типа Кеггина с динитратной функциональной группой, состава Fe(III)[H(ONO2)2]PW11${\text{O}}_{{{\text{39}}}}^{{ - 5}},$ проявляет высокие каталитические свойства по отношению к 2-хлорэтилэтил сульфиду.
Благодаря высокой каталитической активности в реакциях окислительного обессеривания под действием кислорода, полиоксометаллаты относятся к классу универсальных катализаторов-переносчиков электронов. Основное количество работ по окислению сернистых соединений в присутствии полиоксометаллатов описывают радикальный механизм реакции окисления. Металлы, входящие в состав полиоксометаллатов, облегчают и ускоряют разложение гидропероксидов, выступающих в качестве окислителей серосодержащих соединений в модельной смеси. При переходе к реальному углеводородному сырью возникает проблема селективности окисления, в ходе которого по свободно-радикальному механизму окисляются углеводородные компоненты, что приводит к повышению содержания кислородсодержащих соединений и, соответственно, к росту вязкости углеводородной среды. Эта проблема ограничивает их распространение как катализаторов окисления.
Окисление в присутствии полиоксометаллатов через образование пероксокомплексов является наиболее желательным. Функционализация такого полиоксометаллата с органическими катионами позволяет улучшить дисперсность системы и повысить адсорбцию субстрата. Пропитка или иммобилизация на инертный носитель дает возможность легко отделять катализатор от реакционной системы и повторно использовать, а также предотвратить потери.
ОКИСЛИТЕЛЬНОЕ ОБЕССЕРИВАНИЕ В ПРИСУТСТВИИ МЕТАЛЛСОДЕРЖАЩИХ КОМПЛЕКСОВ
Активировать кислород можно добавлением в реакционную систему комплексов металлов, содержащих в качестве лигандов порфирины, фталоцианины и имины различного строения (сален-лиганды). В связи с простотой синтеза, такие сален-комплексы являются одними из наиболее популярных, но в связи со сложностью отделения таких катализаторов от реакционной системы существуют ограничения по их использованию в промышленном масштабе.
Авторами работы [42] в качестве катализатора окисления был синтезирован смешанный биметаллический сален-комплекс (Сo-Cu(salen)), внедренный на цеолит NaY (где salen = N,N'-бис(салицилиден)этилендиамин). Наблюдаемый синергетический эффект между Co+2 и Сu+2 приводит к повышению активности комплекса в окислении ДБТ под действием кислорода, а 95%-ная конверсия при 100°С сохраняется до 5 каталитических циклов.
Металлсодержащие фталоцианины и их производные проявляют высокие селективность и реакционную активность по отношению к сульфидной и меркаптановой сере. Поэтому модифицированные фталоцианины являются интересной областью исследования для процесса окислительного обессеривания. Известно применение различных фталоцианинов в качестве катализаторов окисления сернистых соединений кислородом воздуха. Авторы [43] использовали модифицированные фталоцианины железа и показали высокую эффективность окисления ДБТ при 3 атм и 100°С. Было исследовано влияние функциональной группы катализатора на его активность, которая снижается в следующем ряду при переходе от нитро- к аминозамещенному фталоцианину FePc(NO2)4 > FePc(NO2)3NH2 > FePc(NH2)4. Пришитые фталоцианины железа на углеродные волокна были изучены в работе [44], авторами был предложен механизм окисления ДБТ, включающий образование радикалов пятикоординационного оксожелеза. Предложенная каталитическая система обладает высокой активностью, а конверсия ДБТ достигает 93% при 130°С и 2 атм. Свободно-радикальный механизм процесса представленна рис. 4.
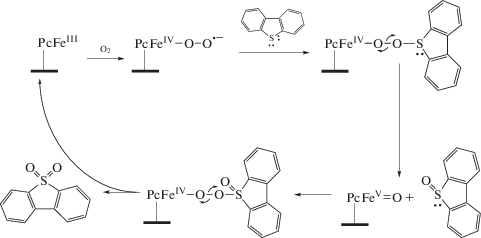
Чжао и др. использовали катализаторы на основе фталоцианина меди, пришитые к цеолитам: Cu2(PcAN)2–W-HZSM-5 и Cu2(PcTN)2/ W-HZSM-5 [45]. Пришитые тетразамещенные нитрофталоцианины редкоземельных металлов на цеолиты типа ZSM-5 были изучены в работе [46]. Показано, что сочетание фталоцианина и цеолитных материалов позволяет достигнуть высокой каталитической активности в реакциях окисления, при которой 99% тиофена удаляется уже при комнатной температуре.
Окисление ДБТ кислородом воздуха в присутствии хлорзамещенного фталоцианина кобальта, как катализатора, и ионной жидкости, как экстрагента, исследовано в работе [47]. По данным авторов природа заместителя в молекуле фталоцианина влияет на каталитическую активность. Обнаружено, что электроноакцепторные группы оказывают положительный эффект на каталитические свойства [48]. В присутствии полностью замещенного хлором фталоцианина кобальта достигается степень удаления ДБТ 90% уже при комнатной температуре в течение 2 ч. Также изучено применение нефтерастворимых катализаторов на основе хлорида меди и моноэтаноламина, обладающих противокоррозионной стойкостью, в присутствии которых происходит полная демеркаптанизация углеводородных смесей под действием кислорода [49].
ОКИСЛИТЕЛЬНОЕ ОБЕССЕРИВАНИЕ В ПРИСУТСТВИИ НАНЕСЕННЫХ ОКСИДОВ МЕТАЛЛОВ
Окислительно-адсорбционный подход включает использование специфических адсорбентов, имеющих высокое сродство к серосодержащим соединениям, промотированных оксидами переходных, в том числе редкоземельных металлов. Окисление модельного топлива с использованием гетерогенных каталитических систем обычно включает применение трехфазных реакторов с неподвижным слоем катализатора, широко известных в нефтехимической области, что облегчает распространение и внедрение данного подхода в химическую промышленность. К настоящему времени имеются многочисленные данные по процессу прямого окисления сернистых соединений под действием кислорода воздуха в присутствии гетерогенно-каталитических систем. В табл. 1 приведены сведения о каталитических системах для удаления серосодержащих соединений из углеводородного сырья.
Таблица 1.
Гетерогенно-каталитические системы в окислительном обессеривании
| Катализатор | Растворитель | Субстрат | Окислитель | Температура, °С | Время, ч | Объемная скорость подачи, ч–1 | Конверсия, % | Ссылка |
|---|---|---|---|---|---|---|---|---|
| CuO/ZnO/Al2O3 | Толуол | Тиофен | Кислород | 430 | 18 | – | 25 | [51] |
| 1% B, 5% MoO3 | ДБТ | 405 | 13 | 60 | ||||
| CeO2/MoO3 | Декалин | БТ ДБТ 4,6-ДМДБТ |
Кислород | 100 | 6 | – | 80 99 83 |
[50] |
| Cu/CeO2 | Изооктан | ДБТ | Воздух | 300 | 20 | – | 99 | [52] |
| 1.5% Pt/CeO2 CuO/ZnO/Al2O3 |
Тиофен | 18 | 93 | |||||
| CuO/ZnO/Al2O3 | Изооктан | Тиофен | Воздух | 300 | – | 6 | 96 | [53] |
| 37% Cu, 15% Zn, 48% Al | БТ | 18 | 95 | |||||
| ДБТ | 10 | 96 | ||||||
| 2% Co3O4/γ-Al2O3 | Легкий газойль | ДБТ | Воздух | 200 | – | 1 | 78 | [54] |
| MnO2/γ-Co3O4/γ-Al2O3 | Тетрадекан | ДБТ + алкил производные | Воздух | 130–200 | 24 | – | 65–75 | [55] |
| 2% Co3O4/γ-Al2O3; 13% MnO2/γ-Al2O3 |
Легкий газойль | ДБТ | Воздух | 200 | – | 1 | 82 78 |
[56] |
| Оксиды Mo, V, Ti, Cr | Бензол | Тиофен | Воздух | 310 | – | 0.7–0.9 | 100 | [57] |
| MoO3 ⋅ V2O5 | Бензол | Тиофен | Воздух | 320 | 9 | – | 94 | [58] |
| V2O5 ⋅ МоО3 | Додекан | Тиофен | Воздух | 300 | 2 | – | 82 | [59] |
| ${{{\text{SO}}_{{\text{4}}}^{{{\text{2}} - }}} \mathord{\left/ {\vphantom {{{\text{SO}}_{{\text{4}}}^{{{\text{2}} - }}} {{\text{Zr}}{{{\text{O}}}_{{\text{2}}}}}}} \right. \kern-0em} {{\text{Zr}}{{{\text{O}}}_{{\text{2}}}}}}$ | Гексадекан | Тиофен | Воздух | 25 | 1.5 | – | 100 | [60] |
| 5% Pd/C Mo2C/C |
Дизельное топливо | ДБТ | Кислород | 50–90 | 1–4 | – | 30 | [61] |
| Co–Mo–O | ДБТ | Воздух | 100 | 4 | – | 100 | [62] | |
| Fe(NO3)3–FeBr3 Fe(NO3)3–FeBr2 Cu(NO3)2–CuBr2 |
Ацетонитрил | MeSPh | Воздух | 25 | 1 | – | 98 | [63] |
| Fe(NO3)3–FeBr3/ACMB | Декан Тетрадекан |
БТ + 2-МБТ + 5-МБТ + ДБТ | Кислород | 25 | 2 | – | 99 | [64] |
В работах [50, 51] было предложено использовать оксиды металлов для аэробного окисления сернистых соединений в углеводородном сырье без добавок жертвенных реагентов (соокислителей). Обнаружено, что модифицированные оксиды металлов меди, цинка и алюминия с добавками 1% бора и 5% оксида молибдена позволяют удалить до 80% конденсированных производных тиофена [50]. Показано, что при температуре выше 375°С реакционная способность субстратов увеличивается в ряду тиофен < ДБТ < 4,6-ДМДБТ. Окисление сераорганических соединений с последующим термическим разложением окисленных форм – сульфоксидов и сульфонов до углеводородов и оксида серы (SO2) рассматривается в работах [51–53, 57–59].
Окисление сернистых соединений с применением экстракционного метода для удаления окисленных форм продемонстрировано в работе [55]. Обнаружено, что 1-метил-2-пирролидон является самым эффективным экстрагентом в удалении сернистых соединений из окисленного углеводородного сырья. В работе [60] были рассмотрены различные окислительные системы (без окислителя, в присутствии воздуха, озона) в среде с высоким содержанием тиофена в присутствии сульфатированного оксида циркония. Обнаружено, что наибольшую эффективность в окислении тиофена проявляет озон, достигая 100% конверсии за 0.5 ч. Предложенный авторами механизм заключается в формировании суперактивных интермедиатов – озонидов, образующихся в результате присоединения кислорода к кислотным Льюсовским центрам, с последующим циклоприсоедиением озона к активированному тиофену по положению 2 и 5.
Авторы исследовали 3 класса катализаторов, включая нанесенные благородные металлы на активированный углерод или оксид алюминия (Pd/C, Pd/Al2O3), нанесенные карбиды металлов (MoC2 и WC) и мезопористые титаносиликаты, в реакциях окислительного обессеривания под действием кислорода как окислителя [61]. Показано, что низкая конверсия достигается в результате протекания побочных реакций окисления УВ и олигомеризации. Авторы считают, что ключевая стадия в окислении серосодержащих соединений заключается в генерации гидропероксидов из алкилароматических углеводородов и азотсодержащих соединений, находящихся в дизельном топливе.
Авторы работы [62] наблюдали синергетический эффект между кобальтом и молибденом. Нанолисты Co–Mo–O показывают высокую активность в реакциях окисления ДБТ воздухом в мягких условиях процесса. В работе приведено детальное исследование синтезированного катализатора.
Окислительное обессеривание модельного топлива кислородом воздуха в присутствии солей железа и меди предложили в работе [63]. Наибольшую активность показала каталитическая бинарная система на основе железа(III), в присутствии которой достигнуто 98%-ная конверсии тиоанизола. Обнаружено, что бромид-ион является переносчиком электронов и выступает в качестве медиатора окислительно-восстановительного процесса превращения сульфидов. Была также установлена роль азотной кислоты, окисляющей бромид-ион до брома, что запускает процесс активации и окисления сульфидов на координированном металлическом центре. Аналогичный адсорбционно-окислительный подход изучался в работе [64]. Окисление конденсированных производных тиофена проводили в присутствии смеси нитрата и бромида железа(III) в мягких условиях процесса под действием молекулярного кислорода. Нанесение смеси солей железа на активированный углерод позволяет увеличить адсорбционную емкость с результатом до 99%-ной сероочистки, что обусловлено большим сродством углеродного материала к сульфонам и сульфоксидам. В данной работе авторами предлагается двухступенчатый метод удаления серы из реального углеводородного сырья, который по их данным является наиболее экономически эффективным, рентабельным и экологически безопасным. Первая стадия включает окислительное обессеривание кислородом в мягких условиях, второй шаг основан на адсорбции окисленных форм серы углеродистыми материалами.
В работе [65] рассматривают окислительно-адсорбционный подход обессеривания в присутствии воздуха путем образования плазмы, создаваемой диэлектрическим барьерным разрядом (DBD). Каталитическая система на основе FeCl3–SiO2 сочетает в себе функции катализатора и адсорбента, что эффективно сказывается на десульфуризации даже при комнатной температуре. Диэлектрическая плазма позволяет генерировать радикалы из воздуха, в том числе активный кислород. Такие высокоактивные частицы являются идеальными окислителями для обессеривания и способны снизить содержание ДБТ в модельной смеси с 200 до 3 ppm, что дает 98%-ную конверсию при комнатной температуре в течение 30 мин. Группой ученых был разработан аналогичный способ окислительной десульфуризации в присутствии MnO2 как катализатора и [BMIM]CH3COO как экстрагента [66]. Авторами была изучена каталитическая активность различной модификации оксида марганца (α-, β-, и γ-MnO2) и влияние температуры синтеза на конверсию ДБТ. Обнаружено, что наибольшей активностью обладают кристаллические фазы α- и β-MnO2 (более 94% удаления серы). Показано, что окислительная активность сернистых соединений под действием диэлектрического разряда снижается в следующем ряду: тиофен > БТ > ДБТ > 4,6-ДМДБТ.
Данные о каталитических системах на основе оксидов металлов или их простых солей показывают, что их использование нецелесообразно и нерентабельно в реакциях окислительного обессеривания. Приведенные системы требуют высокой температуры реакции и продолжительности окисления более 10 ч, что может вызвать сопутствующее окисление углеводородных компонентов вплоть до оксидов углерода. Использование каталитических систем с активными центрами на основе редкоземельных или благородных металлов резко увеличивает стоимость процесса, а применение адсорбентов на их основе также имеет ряд недостатков, обусловленных энергоемкой регенерацией.
ОБЕССЕРИВАНИЕ С ИСПОЛЬЗОВАНИЕМ ГРАФЕНОПОДОБНЫХ СИСТЕМ
В последнее десятилетие особое внимание привлекают углеродные материалы в качестве основы для создания катализаторов окисления различных субстратов, что обусловлено их большой площадью поверхности, высокой прочностью, возможностями функционализации поверхности и наличием дефектов на поверхности. Материалы на основе углерода проявляют высокую каталитическую активность для большинства органических реакций, в том числе реакции кросс-сочетания, кислотно-основные реакции и окислительно-восстановительные процессы. Обнаружено, что материалы на основе графена способны активировать молекулы кислорода при использовании его в качестве окислителя [67].
Авторы работы [68] впервые применили восстановленный оксид графена в реакции окислительного обессеривания с результатом 100%-ной конверсии ДБТ при температуре 150°С в течение 2 ч при дозировке катализатора 8.7 мас. %. Авторы исследовали каталитическую активность различных материалов на основе этой модификации углерода в процессе окислительного обессеривания; результаты приведены в табл. 2. Доказано, что дефекты на поверхности графена играют важную роль в окислительном процессе, а также было обнаружено образование карбонильных групп на дефектах структуры, обеспечивающих активацию кислорода и улучшение адсорбционной способности. Авторами было показано эффективное применение материалов на основе углерода и показана высокая каталитическая активность по отношению к сераорганическим соединениям.
Таблица 2.
Каталитическое окисление дибензотиофена в присутствии различных углеродсодержащих катализаторов
| Катализатор | Площадь поверхности по БЭТ, м2/г | Время, ч | Температура, °С | Конверсия, % |
|---|---|---|---|---|
| Без добавок | – | 2 | 140 | 14.6 |
| Восстановленный оксид графена (rGO) | 492 | 2 | 140 | 65.3 |
| Оксид графита (GO) | 46 | 2 | 140 | 36.5 |
| Нанотрубки углерода (CNT) | 219 | 2 | 140 | 51.3 |
| Фуллеренподобные структуры углерода (OLG) | 463 | 2 | 140 | 21.5 |
| Высокоориентированный пиролитический графит (HOPG) | – | 2 | 140 | 25.8 |
| Коммерческое высокотемпературное наноуглеродистое волокно с высоко графитизированной структурой (HHT) | 35 | 2 | 140 | 34.3 |
Исследовано использование углеродных нанотрубок в качестве катализатора окисления модельной смеси на основе ДБТ [69]. Приведенные данные указывают, что важную роль в каталитической активности в реакциях окислительного обессеривания играет степень графитизации нанотрубок углерода. Также отмечено, что с увеличением графитизации повышается эффективность окисления ДБТ в сульфон и достигнуто полное его удаление в течение 40 мин при 150°С. Кроме этого катализатор может быть легко восстановлен в токе аргона при 900°С.
Группа исследователей [70, 71] обнаружила эффективное использование графеноподобных структур на основе нитрида бора в качестве катализатора аэробного окисления. Такие структуры обладают высокой площадью поверхности, низкой стоимостью и способностью введения на поверхность слоя различных металлов для повышения активности. Сообщено, что возможно полное удаление алкилпроизводных ДБТ в декалине под действием воздуха при 120°С в течение 8 ч [70]. В качестве катализатора использовали нитрид бора с включениями наночастиц меди, обладающих высокой электронной подвижностью.
ОКИСЛИТЕЛЬНОЕ ОБЕССЕРИВАНИЕ С ИСПОЛЬЗОВАНИЕМ СООКИСЛИТЕЛЕЙ
Использование дополнительных жертвенных агентов (соокислителей) с кислородом воздуха основано на генерировании активных частиц, выступающих в роли окислителя, в процессе автоокисления УВ. К таким частицам относятся пероксикислоты, гидропероксиды и различные пероксидные радикалы, инициирующие процесс окисления. Например, Рао и другие [72] впервые исследовали окислительное обессеривание с использованием комбинации альдегида и молекулярного кислорода с целью образования ацильных радикалов для инициирования реакций автоокисления УВ (рис. 5). При дальнейшем введении кислорода в реакционную систему формируется пероксильный радикал, который отщепляет атом водорода от другой молекулы альдегида, образуя пероксикислоты и другой ацильный радикал на стадии роста цепи.

Указанной выше идеей генерирования внутри системы надкислоты воспользовались авторы следующей работы для окисления 2-хлордиэтилсульфида в присутствии нанесенного оксида ванадия на мезопористые материалы [73]. Влияние природы переходного металла и длины углеродной цепи алифатических альдегидов на окисление сернистых соединений показано в работах [74, 75]. В присутствии солей переходных металлов (ацетаты кобальта, никеля и марганца) под действием молекулярного кислорода происходит окисление альдегидов до пероксикислот, которые являются дополнительным реагентом-соокислителем в данных системах. Результаты указывают, что самыми эффективными катализаторами окисления являются ацетат и хлорид кобальта(II) и альдегиды с длиной углеводородной цепи 6–10 атомов углерода.
Авторы работы [76] разработали эффективный и простой способ окислительного обессеривания с использованием трехкомпонентной системы, состоящей из воздуха, трет-бутилгидропероксида и изобутилового альдегида. В мягких условиях окисления было достигнуто снижение общего содержания серы в дизельном топливе с 400 до 13 ppm и показано, что добавление трет-бутилгидропероксида как инициатора улучшает обессеривание в результате роста скорости образования карбонильных радикалов.
Анионы на основе гетерополикислот были успешно применены в реакциях окислительного обессеривания в присутствии сооокислителей [77, 78]. Как было замечено ранее, преимущество использования гетерополикислот как многофункциональных катализаторов заключается в мягких условиях проведения процесса [79]. Это подтверждается сообщением о применении полиоксометаллата состава [C18H37N(CH3)3]5[PV2Mo10O40] для окисления ДБТ в модельной смеси под действием кислорода в присутствии альдегидов в мягких условиях [77]. Предложенный авторами катализатор играет роль межфазного переносчика за счет длинного углеводородного хвоста и может приводить к образованию эмульсионной системы, позволяющей снизить масс-диффузионные ограничения и увеличить контакт двух фаз. Окисление сераорганических соединений в присутствии полиоксометаллата типа Кеггина [C16H33N(CH3)3]3[PW9Mo3O40] с добавлением муравьиной кислоты, пероксида водорода и воздуха продемонстрировано в работе [78]. Результаты показывают, что основное снижение содержания ДБТ происходит за счет окисления надмуравьиной кислотой, формирующейся на этапе введения муравьиной кислоты и пероксида водорода, и составляет 79%. Воздух в данной системе играет роль соокислителя, генерируя образование пероксокислоты в ходе реакции.
Авторами работы [80] было изучено применение диоксидованадиевого комплекса на основе никеля состава [Ni2(C2O4)(dpa)4][(C4H6O4)(VO2)]2 ⋅ ⋅ 2[CH3OH] (где dpa = 2,2'-дипиридинамин) в реакциях окислительного обессеривания. В присутствии данного катализатора под действием молекулярного кислорода и изобутилальдегида как соокислителя достигается полное окисление ДБТ в течение 4 ч, а в случае 4,6-ДМДБТ 100%-ная конверсия за 5 ч. Катализатор может быть использован в течение трех циклов без потери активности.
Широко распространен экстракционно-каталитический метод окислительного обессеривания [81–83]. В данном методе обычно используются ионные жидкости как катализаторы окисления сернистых соединений в сульфоны и как экстрагенты для их извлечения. Однако из-за высокой стоимости и трудности регенерации применение ионных жидкостей ограничено. Вышеуказанные недостатки отсутствуют в ионных жидкостях на основе металлов (так называемые MIL), обладающие большой экстракционной емкостью, что привлекает их применение в качестве экстрагентов для процесса окислительного обессеривания. Такой подход использовали авторы [84, 85], в работах которых представлены катализаторы на основе ионных жидкостей состава (Cr3F(H2O)2O[(O2C)–C6H4–(CO2)]3 · nH2O) (n ≤ ≤ 25) [84] и [Et3NH]FeCl4 в фотохимической реакции [85]. Полученные катализаторы являются эффективными и стабильными, и могут быть использованы повторно без снижения активности. С помощью методов ЭПР и радикальных ловушек авторы [84] доказали радикальный механизм реакции и образование радикалов О• и НОО•, инициирующих автоокисление УВ.
В работе [86] был рассмотрен адсорбционно-окислительный метод обессеривания, где в качестве сорбционного материала применяли оксид графена (GO) и модифицированный азотной кислотой технический углерод (mCB), а также систему октаналь–воздух как окислительную добавку при мягких условиях (60°С) процесса с последующим извлечением окисленных соединений ацетонитрилом.
Предложен некаталитический экстракционно-окислительный метод окисления воздухом сернистых соединений [87]. В качестве растворителя для извлечения окисленных форм серы использовали γ-бутиролактон. Сообщено, что указанный растворитель может выступать в качестве окислителя, генерируя активные частицы под действием кислорода воздуха. Кроме этого растворитель может повышать растворимость кислорода в углеводородной среде, что увеличивает возможность контакта субстрата и окислителя. Авторы привели доказательства радикального механизма реакции окисления в среде γ-бутиролактона.
Сравнение различных каталитических систем для окисления ДБТ кислородом воздуха в присутствии октаналя приведено в работе [88]. Испытан широкий спектр разнообразных гомогенных (ацетаты) и гетерогенных катализаторов – оксиды (ненанесенные и нанесенные на оксид алюминия) на основе переходных металлов: кобальта, марганца и никеля, среди которых наибольшую активность проявили системы на основе кобальта и марганца.
О применении метода фракционной дистилляции было сообщено в работе [89]. Исследователи показали возможность некаталитического окисления прямогонного керосина атмосферным кислородом при температуре 200–220°С и высоком давлении в реакторе барботирующего типа в присутствии воды с получением топлива с содержанием серы менее 0.05%. Изучено влияние температуры, времени и интенсивности перемешивания при дистилляции на степень сероочистки.
Рассмотренные системы предполагают добавление жертвенных реагентов в ходе процесса окисления, что отражается на существенном повышении стоимости такой технологии. Кроме этого, стоит отметить об образовании кислот как побочных продуктов, являющихся коррозионно-активными для оборудования. Основным недостатком в приведенных выше работах является невозможность регенерации каталитических систем.
ЗАКЛЮЧЕНИЕ
Идеальными условиями для проведения химических процессов окисления являются использование воздуха как окислителя, наличие регенерируемого катализатора и нетоксичного и регенерируемого растворителя, а также мягких параметров проведения процесса. Однако анализ научной литературы по окислительному обессериванию кислородом воздуха показал, что эффективные каталитические системы в этой области еще не разработаны. В результате приведенного материала отмечено несколько вариантов окислительного обессеривания: окисление чистым кислородом или кислородом воздуха в присутствии полиоксометаллатов; окисление в присутствии металлокомплексных систем; окисление в присутствии оксидов переходных металлов; окисление в присутствии графеноподобных материалов и соокислителей.
Обсуждение в обзоре механизма окислительного обессеривания сераорганических соединений кислородом воздуха показало, что полиоксометаллаты являются наиболее интересным и перспективным классом металлсодержащих соединений, применяющихся в области каталитической химии. Полиоксометаллаты позволяют вести окисление серосодержащих соединений не только чистым кислородом, но и воздухом, что является наиболее экономически выгодным ввиду отсутствия ограничений по исходному содержанию общей серы в нефтяном сырье.
Список литературы
Srivastava V.C. // RSC Advances. 2012. V. 2. P. 759.
Bhutto A.W., Abro R., Gao S.R., Abbas T., Chen X.C., Yu G.R. // J. Taiwan Inst. Chem. Eng. 2016. V. 62. P. 84.
Houda S., Lancelot C., Blanchard P., Poinel L., Lamonier C. // Catalysts. 2018. V. 8. P. 344.
Yashnik S.A., Salnikov A.V., Kerzhentsev M.A., Saraev A.A., Kaichev V.V., Khitsova L.M., Ismagilov Z.R., Yamin J., Koseoglu O.R. // Kinetics and Catalysis. 2017. V. 58. № 1. P. 58.
Zhao D., Ren H., Wang J., Yang Y., Zhao Y. // Energy and Fuels. 2007. V. 21. P. 2543.
Lu L., Cheng Sh., Gao J., Gao G., He M. // Energy Fuels. 2007. V. 21. P. 383.
Kulkarni P.S., Afonso C.A.M. // Green Chem. 2010. V. 12. P. 1139.
Javadli R., Klerk A. // Appl. Petrochem. Res. 2012. V. 1. P. 3.
Ismagilov Z., Yashnik S., Kerzhentsev M., Parmon V., Bourane A., Al-Shahrani F.M., Hajji A.A., Koseoglu O.R. // Catalysis Rev. Science and Engineering. 2011. V. 53. P. 199.
Фомин В.М. Радикально-цепное окисление органических соединений и его торможение ингибиторами фенольного типа. Электронное учебное пособие. Нижегородский госуниверситет, 2010. С. 5.
Neumann R. // Inorg. Chemistry. 2010. V. 49. № 8. P. 3594.
Neumann R., Khenkin A. // Chem. Commun. 2006. V. 24. P. 2529.
Yu F., Liu C., Yuan B., Xie C., Yu S. // Catal. Commun. 2015. V. 68. P. 49.
Shi M., Zhang D., Yu X., Li Y., Wang X., Yang W. // Fuel Processing Technology. 2017. V. 160. P. 136.
Tang N., Zhang Y., Lin F., Lu H., Jiang Z., Li C. // Chem. Commun. 2012. V. 48. P. 11 647.
Tang N., Zhao X., Jiang Z., Li C. // Chin. J. Catal. 2014. V. 35. № 9. P. 1433.
Lu H., Zhang Y., Jiang Z., Li C. // Green Chem. 2010. V. 2. P. 1954.
Lu H., Zhang Y., Jiang Z., Li C. // Green Chem. 2015. V. 17. P. 817.
Tao M., Zheng H., Shi J., Wang S., Wang X., Huang G. // Catal. Surv. Asia. 2015. V. 9. P. 257.
Jiang C., Wang J., Wang S., Guan H., Wang X., Huo M. // Appl. Catalysis. B: Environmental. 2011. V. 106. P. 343.
Maayan G., Popovitz-Biro R., Neumann R. // J. Am. Chem. Soc. 2006. V. 28. P. 4968.
Lu H., Ren W., Liao W., Chen W., Li Y., Suo Z. // Appl. Catalysis. B: Environmental. 2013. V. 138. P. 79.
Bertleff B., Claußnitzer J., Korth W., Wasserscheid P., Jess A., Albert J. // ACS Sustainable Chem. Eng. 2017. V. 5. P. 4110.
Liu Y., Liu S., Liu S., Liang D., Li S., Tang Q., Wang X., Miao J., Shi Z., Zheng Z. // ChemCatChem. 2013. V. 5. P. 3086.
Li S.W., Li J.R., Gao Y., Liang L.L., Zhang R.L., Zhao J. // Fuel. 2017. V. 197. P. 551.
Li S.W., Gao R.M., Zhang R.L., Zhao J. // Fuel. 2016. V. 184. P. 18.
Li S.W., Gao R., Zhang W., Zhang Y., Zhao J. // Fuel. 2018. V. 221. P. 1.
Ding J.W., Wang R. // Chinese Chemical Letters. 2016. V. 27. P. 655.
Kozhevnikov I.V. // Chem. Rev. 1998. V. 98. P. 171.
Khenkin A.M., Neumann R. // ChemSusChem. 2011. V. 4. P. 346.
Mansourian S.H., Shahhosseini S., Maleki A. // J. of Industrial and Engineering Chemistry. 2019. V. 80. P. 576.
Yang H., Zhang Q., Zhang J., Yang L., Ma Z., Wang L., Li H., Bai L., Wei D., Wang W., Chen H. // J. of Colloid and Interface Science. 2019. V. 554. P. 572.
Gao Y., Lv Z., Gao R., Zhang G., Zheng Y., Zhao J. // J. Hazardous Materials. 2018. V. 359. P. 258.
Gu J., Liu M., Xun S., He M., Wu L., Zhu L., Wu X., Zhu W., Li H. // Molecular Catalysis. 2020. V. 483. P. 110 709.
Sun L., Su T., Xu J., Hao D., Liao W., Zhao Y., Ren W., Deng C., Lü H. // Green Chem. 2019. V. 21. P. 2629.
Jiang W., Xiao J., Dong L., Wang C., Li H., Luo Y., Zhu W., Li H. // ACS Sustainable Chem. Eng. 2019. V. 7. № 8. P. 15 755.
Xun S., Jiang W., Guo T., He M., Ma R., Zhang M., Zhu W., Li H. // J. of Colloid and Interface Science. 2019. V. 534. P. 239.
Yu X., Shi M., Yan S., Wang H., Wang X., Yang W. // Fuel. 2017. V. 207. P. 13.
Okun N.M., Anderson T.M., Hill C.L. // J. Am. Chem. Soc. 2003. V. 125. P. 3194.
Okun N.M., Anderson T.M., Hill C.L. // J. Mol. Catal. A: Chemica. 2003. V. 197. P. 283.
Okun N.M., Tarr J.C., Hilleshiem D.A., Zhang L., Hardcastle K.I., Hill C.L. // J. Mol. Catal. A: Chemica. 2006. V. 246. P. 11.
Yu J., Zhu Z., Ding Q., Zhang Y., Wu X., Sun L., Du J. // Catal. Today. 2020. V. 339. P. 105.
Zhou X., Li J., Wang X., Jin K., Ma W. // Fuel Processing Technology. 2009. V. 90. P. 317.
Chen S., Lu W., Yao Y., Chen H., Chen W. // Reac. Kinet. Mech. Cat. 2014. V. 111. P. 535.
Zhao N., Li S., Wang J., Zhang R., Gao R., Zhao J., Wang J. // J. Solid-State Chem. 2015. V. 225. P. 347.
Zhang Y., Wang D., Zhang R., Zhao J., Zheng Y. // Communications. 2012. V. 29. P. 21.
Zhang J., Li J., Ren T., Hu Y., Ge J., Zhao D. // RSC Advances. 2014. V. 4. P. 3206.
Buck T., Bohlen H., Wohrle D. // J. Mol. Catal. 1993. V. 80. P. 253.
Плетнева И.В., Гаврилов Ю.А. // Химия и хим. технология. 2017. V. 60. № 8. С. 71.
Shi Y., Liu G., Zhang B., Zhang X. // Green Chem. 2016. V. 18. P. 5273.
Yashnik S.A., Salnikov A.V., Kerzhentsev M.A., Saraev A.A, Kaichev V.V., Khitsova L.M., Ismagilov Z.R., Yamin J., Koseoglu O.R. // Kinetics and Catalysis. 2017. V. 58. № 1. P. 58.
Lu Y., Wang Y., Gao L., Chen J., Mao J., Xue Q., Liu Y., Wu H., Gao G., He M. // ChemSusChem. 2008. V. 1. P. 302.
Gao L., Tang Y., Xue Q., Liu Y., Lu Y. // Energy Fuels. 2009. V. 23. P. 624.
Nawaf A.T., Jarullah A.T., Gheni S.A. // Ind. Eng. Chem. Res. 2015. V. 54. P. 12 503.
Sampanthar J.T., Xiao H., Dou J., Nah T.Y., Rong X., Kwan W.P. // Applied Catalysis B: Environmental. 2006. V. 63. P. 85.
Nawaf A.T., Gheni S.A., Jarullah A.T., Mujtaba I.M. // Fuel Processing Technology. 2015. V. 138. P. 337.
Boikov E.V., Vakhrushin P.A., Vishnetskaya M.V. // Chemistry and Technology of Fuels and Oils. 2008. V. 44. № 4. P. 271.
Бойков Е.В., Вишнецкая М.В. // Журнал физической химии. 2013. Т. 87. № 2. С. 196.
Томский И.С., Вишнецкая М.В., Вахрушин П.А., Томская Л.А. // Нефтехимия. 2017. Т. 57. № 5. С. 586 [Petrol. Chemistry. 2017. V. 57. P. 908].
Wang B., Zhu J., Ma H. // J. of Hazardous Materials. 2009. V. 164. P. 256.
Dooley K.M., Liu D., Madrid A.M., Knopf F.C. // Applied Catalysis A: General. 2013. V. 468. P. 143.
Dong Y., Zhang J., Ma Z., Xu H., Yang H., Yang L., Bai L., Wei D., Wang W., Chen H. // Chem. Commun. 2019. V. 55. P. 13 995.
Martin S.E., Rossi L.I. // Tetrahedron Lett. 2001. V. 42. P. 7147.
Ma X., Zhou A., Song C. // Catal. Today. 2007. V. 123. P. 276.
Ban L., Liu P., Ma C., Dai B. // Catal. Today. 2013. V. 211. P. 78.
Ma C., Dai B., Xu C., Liu P., Qi L., Ban L. // Catal. Today. 2013. V. 211. P. 84.
Long J., Xie X., Xu J., Gu Q., Chen L., Wang X. // ACS Catal. 2012. V. 2. P. 622.
Gu Q., Wen G., Ding Y., Wu K.H., Chen C., Su D. // Green Chem. 2017. V. 19. P. 1175.
Zhang W., Zhang H., Xiao J., Zhao Z., Yuand M., Li Z. // Green Chem. 2014. V. 16. P. 211.
Wu P., Zhu W., Dai B., Cha Y., Li C., Li H., Zhang M., Jiang W., Li H. // Chem. Engineering J. 2016. V. 301. P. 123.
Wu P., Zhu W., Chao Y., Zhang J., Zhang P., Zhu H., Li C., Chen Z., Li H., Dai S. // Chem. Commun. 2016. V. 52. P. 144.
Rao T.V., Sain B., Kafola S., Nautiyal B.R., Sharma Y.K., Nanoti S.M., Garg M.O. // Energy & Fuels. 2007. V. 21. P. 3420.
Livingston S.R., Landry C.C. // J. Am. Chem. Soc. 2008. V. 130. P. 13214.
Murata S., Murata K., Kidena K., Nomura M. // Prepr. Pap.-Am. Chem. Soc. Div. Fuel Chem. 2003. V. 48. № 2. P. 531.
Murata S., Murata K., Kidena K., Nomura M. // Energy & Fuels. 2004. V. 18. P. 116.
Guo W., Wang C., Lin P., Lu X. // Appl. Energy. 2011. V. 88. P. 175.
Lu H., Gao J., Jiang Z., Wang Y., Song B., Li C. // Chem. Commun. 2007. V. 2. P. 150.
Imtiaz A., Waqas A., Muhammad I. // Chinese J. of Catalysis. 2013. V. 34. P. 1839.
da Silva M.J., Teixeira M.G. // RSC Adv. 2017. V. 7. P. 8192.
Li J.K., Xu Y.Q., Hu C.W. // Inorg. Chem. Commun. 2015. V. 60. P. 12.
Ribeiro S., Julião D., Cunha-Silva L., Domingues V., Valença R., Ribeiro J., de Castro B., Balula S. // Fuel. 2016. V. 166. P. 268.
Mirhoseini H., Taghdiri M. // Fuel. 2016. V. 167. P. 60.
Jiang W., Li H., Wang C., Liu W., Guo T., Liu H., Zhu W., Li H. // Energy Fuels. 2016. V. 30. № 10. P. 8164.
Gómez-Paricio A., Santiago-Portillo A., Navalón S., Concepción P., Alvaroa M., Garcia H. // Green Chem. 2016. V. 18. P. 508.
Wang C., Chen Z., Zhu W., Wu P., Jiang W., Zhang M., Li H., Zhu W., Li H. // Energy Fuels, 2017. V. 31. P. 1376.
Zhang Y., Wang R. // Diamond & Related Materials. 2017. V. 73. P. 161.
Xu X., Moulijn J.A., Ito E., Wagemans R., Makkee M. // Chem. Sus. Chem. 2008. V. 1. P. 817.
Dumont V., Oliviero L., Mauge F., Houalla M. // Catalysis Today. 2008. V. 130. P. 195.
Paniv P.M., Pysh’ev S.V., Gaivanovich V.I., Lazorko O.I. // Chemistry and Technology of Fuels and Oils. 2006. V. 42. № 3. P. 159.
Дополнительные материалы отсутствуют.