

Нейрохимия, 2019, T. 36, № 1, стр. 56-64
Афобазол восстанавливает содержание дофамина при моделировании болезни паркинсона 6-гидроксидофамином
М. В. Воронин 1, И. А. Кадников 1, С. Б. Середенин 1
1 Федеральное государственное бюджетное научное учреждение
“Научно-исследовательский институт фармакологии имени В.В. Закусова”
Москва, Россия
Поступила в редакцию 19.04.2018
После доработки 18.06.2018
Принята к публикации 08.06.2018
Аннотация
Изучено содержание моноаминов стриатума мышей при введении афобазола в дозе 2.5 мг/кг в/б в течение 14 дней с началом курса через 30 мин после унилатерального интрастриатного введения 5 мкг 6-гидроксидофамина (6-OHDA). Афобазол предотвращал падение содержания дофамина в поврежденном 6-OHDA стриатуме опытных животных и не влиял на уровень норадреналина, серотонина и его метаболита в стриатуме как контрольных, так и получавших 6-OHDA мышей.
ВВЕДЕНИЕ
Эпидемиологические данные по болезни Паркинсона и отсутствие достаточно эффективной фармакотерапии заболевания определяют актуальность поиска новых фармакологических средств, обладающих нейропротекторным действием [1]. Значимым фактором патогенеза нейродегенеративных болезней, включая болезнь Паркинсона, рассматривается ошибочный фолдинг белков (protein misfolding disorders, conformational diseases), что обуславливает запуск сигнальных каскадов ЭПР стресса, повреждение митохондрий и избыточную генерацию активных форм кислорода (АФК) [2, 3]. Поэтому аргументированной представляется нейропротекторная стратегия, основанная на регуляции функций шаперонов [4–6].
Моделирование in vivo болезни Паркинсона традиционно основано на индукции избирательной гибели дофаминергических нейронов черной субстанции (Substantia nigra), опосредованной как минимум тремя патологическими процессами: дисфункцией митохондрий, окислительным стрессом и воспалением [4, 7–9]. Однако исследования последних лет внесли в эту триаду важный связующий компонент – нарушение фолдинга и деградации белков [5, 10].
Одним из наиболее распространенных способов моделирования болезни Паркинсона служит ретроградное токсическое воздействие 6-гидроксидофамина (6-OHDA) на нейроны черной субстанции при интрастриатном введении [11–13]. Запущенный 6‑OHDA окислительный стресс усиливает дофаминергическую токсичность, что способствует накоплению в полости ЭПР агрегатов белков с нарушенной конформацией [14–17]. В условиях сниженной активности энергозависимых процессов протеосомальной деградации нейроны переходят в состояние ЭПР стресса [18, 19], что вызывает универсальный клеточный ответ в виде активации специфичной сигнальной сети (UPR, unfolded protein response) [14, 15]. В адаптивной фазе ответа ингибируется трансляция и активируется экспрессия генов, отвечающих за синтез шаперонов, фолдаз, компонентов системы контроля качества и деградации незрелых белков [20]. Процессы UPR инициируются активацией белковых сенсоров ЭПР стресса (протеинкиназами IRE1α, PERK и фактором транскрипции ATF6) [19]. Шаперон Sigma1R, регулируя активность сенсоров ЭПР стресса, основного шаперона ЭПР BiP (Binding-immunoglobulin protein, GRP78), кальциевых каналов в области контакта ЭПР и митохондрий, играет важную роль в адаптации клеток нервной системы к стрессовым воздействиям [21, 22]. Известно, что при лигандной активации Sigma1R в составе специфических липидных доменов способен к внутриклеточному перераспределению и шаперонной активности в отношении ионных каналов, рецепторов и ферментов цитоплазматической мембраны, что оказывает защитное действие на нейроны [23, 24].
В результате ферментативного катализа и автоокисления 6‑OHDA в нейроне образуются хинонные производные ДА (DAQ) [25], способствующие развитию окислительного стресса [4, 11]. Активация свободнорадикальных процессов инициирует системное нарушение работы митохондрий [26, 27], снижение активности и окислительную модификацию ключевых белков дофаминергической системы, что вызывает нарушение обмена и депонирования ДА, триггерное усиление продукции DAQ [25, 27–30]. Восстановление DAQ NQO2 влечет за собой возобновление цитозольного окисления ДА с образованием супероксид анион радикала [31, 32], поэтому ингибирование NQO2 может приводить к снижению продукции АФК и радикалов o-семихинона [33].
Настоящая работа посвящена анализу нейропротекторного действия препарата афобазол на экспериментальной модели болезни Паркинсона с интрастриатным введением 6-OHDA. Афобазол (5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид) разработан и фармакологически изучен в ФГБНУ “НИИ фармакологии имени В.В. Закусова” [34]. Среди мишеней препарата установлены шаперон SigmaR1 (sigma-1 рецептор; IC50 = 7.1 × 10–6 M) и мелатонинзависимый регуляторный сайт оксидоредуктазы NQO2 (МТ3 рецептор; IC50 = 9.9 × 10–7 M) [35]. В наших предыдущих исследованиях показано, что афобазол вызывает внутриклеточное перераспределение SigmaR1 [36, 37], соответствующее агонистическому влиянию [38]. В бесклеточной системе in vitro выявлено ингибирующее действие афобазола на NQO2 человека [39]. В различных экспериментальных моделях установлены нейропротекторные и цитопротекторные свойства афобазола, а применение селективных лигандов позволило in vitro установить вклад SigmaR1 и NQO2 в реализацию его защитного действия [40–42]. SigmaR1 зависимые механизмы развития протекции систематизированы в обзоре С.Б. Середенина с соавторами [43]. Таким образом, имеются теоретические и экспериментальные предпосылки для гипотезы о том, что совместная регуляция SigmaR1 и NQO2 может быть эффективной для терапии болезни Паркинсона, об эффективности которой можно судить по восстановлению содержания дофамина нигро-стриатной области [44]. Поэтому целью настоящей работы стало изучение влияния афобазола на содержание катехоламинов стриатума головного мозга мышей на модели болезни Паркинсона, вызванной унилатеральным введением 6‑OHDA.
МЕТОДЫ ИССЛЕДОВАНИЯ
Реактивы. При выполнении данной работы были использованы следующие реактивы: афобазол (5-этокси-2-[2-(морфолино)-этилтио] бензимидазола дигидрохлорид; ФГБНУ “НИИ фармакологии имени В.В. Закусова”), 6-гидроксидофамина гидрохлорид (6‑OHDA), аскорбиновая кислота, NaCl, HClO4, сахароза (Sigma-Aldrich), хлоралгидрат (Serva), 3,4‑дигидроксибензиламин (DHBA), дофамин (ДА), дигидроксифенилуксусная кислота (ДОФУК), норадреналин (НА), серотонин (5-ОТ), гомованилиновая кислота (ГВК), 5-гидроксииндолуксусная кислота (5-ОИУК), KH2PO4, H3PO4, лимонная кислота, EDTA-Na2, октансульфоновая кислота, ацетонитрил (Fluka).
Животные. В работе были использованы мыши-самцы аутбредной линии CD-1 весом 25–30 г (n = 31). Животные были получены из НПП Питомника лабораторных животных ФИБХ. Мышей содержали в условиях вивария (20–22°С, относительная влажность 30–70%, 12‑ч световой цикл) в пластиковых клетках со свободным доступом к пище и воде по 5–7 особей.
Все процедуры с животными в исследовании были рассмотрены и утверждены комиссией Института по уходу и использованию животных (IACUC) на предмет соответствия этическим принципам обращения с животными.
Моделирование болезни Паркинсона интрастриатным введением 6‑OHDA. За 30 мин до операции животное анестезировали хлоралгидратом (400 мг/кг, внутрибрюшинно). Анестезированное животное помещали в стереотаксические рамки (Stoelting Motorized Stereotaxis, Stoelting Co., Великобритания), где 6-OHDA однократно вводили в правый стриатум в координатах A = 0.4; L = 1.8; V = –3.5, относительно брегмы [12]. Концентрация 6-OHDA составляла 5 мкг на 1 мкл раствора, содержащего 0.9% NaCl и 0.02% аскорбиновой кислоты. Опытным животным вводили 1 мкл раствора 6-OHDA со скоростью 0.5 мкл/мин гамильтоновским шприцом с иглой из нержавеющей стали (30 gauge), спустя 2 мин после инъекции иглу извлекали. Ложно-оперированным животным вводили 1 мкл контрольного раствора, содержащего 0.9% NaCl и 0.02% аскорбиновой кислоты, в тех же координатах.
Схема введения афобазола. Афобазол в дозе 2.5 мг/кг в/б и плацебо (вода для инъекций) вводили ежедневно на протяжении 14 сут с началом курса через 30 минут после интрастриатной инъекции 6‑OHDA или контрольного раствора. Животные были разделены на 4 группы: ложно-оперированные, получавшие плацебо (n = 7); ложно-оперированные, получавшие афобазол (n = 5); опытные, получавшие плацебо (n = 12); опытные, получавшие афобазол (n = 7).
Метод ВЭЖХ-ЭД. Моноамины и их метаболиты определяли в поврежденном и интактном стриатумах головного мозга мышей методом высокоэффективной жидкостной хроматографии с электрохимической детекцией (ВЭЖХ-ЭД). Через 14 сут после введения 6‑OHDA мышей декапитировали и извлекали головной мозг. Левый и правый стриатумы выделяли при температуре тающего льда (0–4°С) на фильтровальной бумаге, смоченной 0.32 М раствором сахарозы. Каждый выделенный стриатум замораживали в жидком азоте (–196°С), взвешивали и хранили при температуре –80°C. Для определения содержания моноаминов и их метаболитов выделенный стриатум гомогенизировали в 0.1 M НClO4 с добавлением в качестве внутреннего стандарта DHBA в концентрации 0.25 нмоль/мл в шаровом гомогенизаторе Tissue LyserLT (Quiagen, Германия) с частотой 45 ударов в минуту в течение 5 мин. Пробы центрифугировали при 10000 g в течение 10 мин при 4°C. Надосадочную жидкость в объеме 20 мкл наносили на аналитическую колонку Kromasil C-18 4.6 × × 150 (Dr.Maisch, Германия) с помощью автосемплера SIL-20 ACHT (Shimadzu, Япония). Моноамины и их метаболиты разделяли на колонке с использованием в качестве подвижной фазы 0.1 М цитратно-фосфатного буфера, содержащего 0.3 мМ октансульфоната натрия, 0.1 мМ ЭДТА и 8% ацетонитрила (рН 3.0). Определение моноаминов и их метаболитов осуществляли на хроматографической станции Shimadzu LC-20 Prominence (Shimadzu, Япония) с использованием электрохимической ячейки ESA 5011 (Е1 = –175; Е2 = +250) и электрохимического детектора Coulochem III (ESA, США). Скорость потока подвижной фазы составляла 1 мл/мин. Полученные результаты обрабатывали на ПК с использованием программно-аппаратного комплекса “Мультихром 1.5" (Ampersand, Россия).
Математическая обработка экспериментальных данных. Поскольку в биодеградации дофамина и серотонина принимают участие ферменты МАО и катехол-О-метилтрансфераза (КОМТ), были использованы метаболические соотношения 5‑ОИУК/5-ОТ и ДОФУК/ДА, отражающие активность МАО, и ГВК/ДА, отражающее активность КОМТ, а также результирующее (ДОФУК + + ГВК)/ДА [45]. Табличные данные представлены в виде медиана (25–75 квартиль) (Mdn (q25–75)). Данные на рисунках представлены в виде медиана (минимум – максимум) (Mdn (min-max). Оценку статистической значимости полученных результатов проводили с применением критерия Вилкоксона и одностороннего дисперсионного анализа Краскела–Уоллиса (Kruskal–Wallis test, Dunn’s post hoc). Статистическую обработку и визуализацию полученных данных осуществляли с помощью программного пакета GraphPad 5.0.2 (GraphPad Software, San Diego California USA, (www.graphpad.com).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Через 14 сут после однократного введения контрольного раствора в правый стриатум ложно-оперированным мышам содержание моноаминов и их метаболитов количественно соответствовало интактному левому стриатуму этих животных (табл. 1, рис. 1–3). Однократное введение 6‑OHDA в группе плацебо через аналогичный временной интервал привело к статистически значимому снижению медианы содержания ДА в поврежденном стриатуме до 35.3 нмоль/мг ткани по сравнению с поврежденным стриатумом ложно‑оперированных (74.7 нмоль/мг ткани) и интактным стриатумом опытных животных (79.4 нмоль/мг ткани) (рис. 1). Содержание основных метаболитов ДА – ДОФУК и ГВК в поврежденном стриатуме опытных животных уменьшилось до 2.8 и 5.3 нмоль/мг ткани соответственно в сравнении с поврежденным стриатумом ложно-оперированных мышей (5.5 и 10.0 нмоль/мг ткани) (рис. 2, 3). Внутригрупповое сравнение с интактным стриатумом опытных животных показало, что уровень ДОФУК снизился с 5.7 до 2.8 нмоль/мг ткани, а ГВК – с 9.7 до 5.3 нмоль/мг ткани (рис. 2, 3). В то же время отмечалось увеличение метаболических отношений ГВК/ДА на 30.1% и (ДОФУК + ГВК)/ДА на 33% (табл. 1). Таким образом, в группе плацебо однократное интрастриатное введение 6-OHDA привело к статистически значимому снижению содержания ДА и его метаболитов ДОФУК и ГВК в поврежденном стриатуме и не влияло на содержание ДА и его метаболитов в интактном стриатуме.
Таблица 1.
Влияние 14-дневного введения афобазола в дозе 2.5 мг/кг на содержание моноаминов и их метаболизм в интактном и поврежденном стриатуме мышей линии CD-1
| Группа | Плацебо | Афобазол | ||||||
|---|---|---|---|---|---|---|---|---|
| Л.О. n = 7 | 6-OHDA n = 12 | Л.О. n = 5 | 6-OHDA n = 7 | |||||
| И | П | И | П | И | П | И | П | |
| НА (нмоль/г ткани) | 1.24 (1.12–2.56) |
1.04 (0.66–1.32) |
0.83 (0.31–1.25) |
0.29 (0.27–1.16) |
1.04 (0.85–1.31) |
1.12 (0.81–1.33) |
0.94 (0.56–1.98) |
0.68 (0.4–1.12) |
| 5-ОТ (нмоль/г ткани) | 6.44 (6.14–8.72) |
5.16 (4.58–5.82) |
4.88 (4.36–7.38) |
4.75 (2.81–5.7) |
6.4 (4.35–9.22) |
5.64 (3.4–6.59) |
4.08 (2.88–5.36) @ p = 0.04 |
4.58 (3.36–5.48) |
| 5-ОИУК (нмоль/г ткани) | 3.22 (3.06–4.26) |
3.06 (2.08–3.46) |
2.65 (2.2–3.21) |
2.46 (1.9–2.73) |
2.42 (2.08–3.03) |
2.0 (1.75–2.85) |
2.06 (1.36–2.48) @ р = 0.011 |
2.04 (1.78–2.46) |
| 5-ОИУК/5-ОТ | 0.51 (0.39–0.57) |
0.67 (0.47–0.7) |
0.56 (0.39–0.58) |
0.58 (0.5–0.67) |
0.38 (0.3–0.59) |
0.42 (0.32–0.85) |
0.44 (0.4–0.55) |
0.48 (0.43–0.65) |
| ДОФУК/ДА | 0.078 (0.069–0.091) |
0.074 (0.062–0.1) |
0.076 (0.062–0.088) |
0.09 (0.073–0.13) |
0.056 (0.05–0.082) |
0.052 (0.044–0.062) |
0.072 (0.062–0.072) |
0.06 (0.045–0.064) * p = 0.026 |
| ГВК/ДА | 0.15 (0.13–0.15) |
0.13 (0.12–0.14) |
0.13 (0.11–0.15) |
0.17 (0.13–0.31) ^ р= 0.027 |
0.12 (0.09–0.17) |
0.1 (0.08–0.12) |
0.11 (0.11–0.13) |
0.11 (0.09–0.12) * p = 0.025 |
| (ДОФУК + ГВК)/ДА | 0.22 (0.2–0.25) |
0.22 (0.18–0.23) |
0.21 (0.18–0.23) |
0.28 (0.22–0.46) ^ р= 0.012 |
0.17 (0.14–0.25) |
0.15 (0.13–0.18) |
0.18 (0.17–0.21) |
0.17 (0.14–0.18) ** p = 0.0027 |
Данные представлены в виде Mdn (q25–75). n – количество животных в группе. Л.О. – ложно-оперированные животные. 6-OHDA – опытные животные. И – интактный стриатум (левый), П – поврежденный стриатум (правый). * Статистически значимые различия по сравнению с поврежденным стриатумом опытных животных, получавших плацебо (p < 0.05, Kruskal–Wallis test, Dunn’s post hoc). ** Статистически значимые различия по сравнению с поврежденным стриатумом опытных животных, получавших плацебо (p < 0.01, Kruskal–Wallis test, Dunn’s post hoc). ^ Статистически значимые различия по сравнению с интактным стриатумом опытных животных, получавших плацебо (p < 0.05, Wilcoxon test). @ Статистически значимые различия по сравнению с интактным стриатумом ложно оперированных животных, получавших плацебо (p < 0.05, Kruskal–Wallis test, Dunn’s post hoc).
Рис. 1.
Влияние 14-дневного введения афобазола в дозе 2,5 мг/кг на содержание дофамина в интактном и поврежденном 6-OHDA стриатуме мышей линии CD-1. Данные представлены в виде Mdn (min–max). + Среднее значение. Л.О. – ложно-оперированные животные. 6-OHDA – опытные животные. ** Статистически значимые различия по сравнению с поврежденным стриатумом опытных животных, получавших плацебо (p < 0.01, Kruskal–Wallis test, Dunn’s post hoc). ### Статистически значимые различия по сравнению с поврежденным стриатумом ложно-оперированных животных, получавших плацебо (p < 0.001, Kruskal–Wallis test, Dunn’s post hoc). ^^^ Статистически значимые различия по сравнению с интактным стриатумом опытных животных, получавших плацебо (p < < 0.001, Wilcoxon test). ⚪ Интактный стриатум, ⚫ поврежденный стриатум.
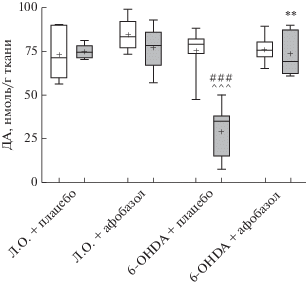
Рис. 2.
Влияние 14-дневного введения афобазола в дозе 2.5 мг/кг на содержание дигидроксифенилуксуной кислоты в интактном и поврежденном 6-OHDA стриатуме мышей линии CD-1. Обозначения как на рис. 1.
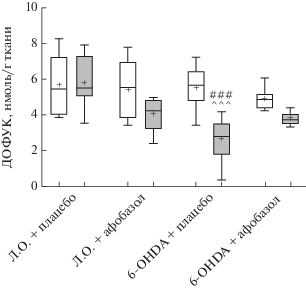
Рис. 3.
Влияние 14-дневного введения афобазола в дозе 2.5 мг/кг на содержание гомованилиновой кислоты в интактном и поврежденном 6‑OHDA стриатуме мышей линии CD-1. Обозначения как на рис. 1.
Однократное интрастриатное введение 6-OHDA не оказало влияния на норадренергическую и серотонинергическую систему поврежденного и интактного стриатумов (табл. 1).
ВЭЖХ исследование показало, что введение афобазола мышам в дозе 2.5 мг/кг на протяжении 14 сут приводило к двукратному увеличению медианы содержания ДА в поврежденном стриатуме опытных животных до 69.8 нмоль/мг ткани по сравнению с поврежденным стриатумом опытных животных, получавших плацебо (рис. 1). Афобазол статистически значимо восстанавливал уровень ДА до значений, зарегистрированных как в поврежденном (74.7 нмоль/мг), так и интактном стриатумах (71.3 нмоль/мг) ложно-оперированных животных, получавших плацебо, таким образом обращая эффект однократного введения 6-OHDA (рис. 1). В группе опытных животных афобазол оказывал незначительное влияние на содержание метаболитов ДА – ДОФУК и ГВК в поврежденном стриатуме, которое оставалось на уровне, определенном для опытных животных, получавших плацебо (рис. 2, 3). Сниженные значения метаболических отношений ДОФУК/ДА, ГВК/ДА и (ДОФУК + ГВК)/ДА (табл. 1) в поврежденном стриатуме опытных животных, получавших афобазол, в сравнении с группой плацебо соответствуют изменению содержания ДА и его метаболитов.
Введение афобазола опытным животным приводило к снижению содержания 5‑ОТ и его метаболита 5‑ОИУК в интактном стриатуме по сравнению с соответствующим стриатумом ложно-оперированных животных, получавших плацебо (табл. 1).
Введение афобазола ложно-оперированным и опытным животным не влияло на содержание НА как в поврежденном, так и интактном стриатуме и его уровень не отличался от зарегистрированных значений у ложно-оперированных животных, получавших плацебо (табл. 1).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Зафиксированное в эксперименте снижение уровня ДА в поврежденном стриатуме опытных мышей согласуется с результатами нейрохимических исследований при моделировании болезни Паркинсона интрастриатным унилатеральным введением 6‑OHDA [12, 46–48]. В работе Alvarez‑Fischer, D. et al. введение 4 мкг 6‑OHDA в идентичный локус стриатума мышей приводило к значительному уменьшению содержания ДА в стриатуме и черной субстанции через 14 сут, наряду с двукратным снижением количества TH+ нейронов [12]. В том же исследовании показано, что стойкий негативный эффект однократного введения 6-OHDA на уровень ДА стриатума развивается на третий день, и далее его содержание остается значимо сниженным в течение 56 сут [12]. В научной периодике отражены эффекты разных комбинаций доз 6‑OHDA и времени до нейрохимического исследования при моделировании болезни Паркинсона. Например, через 36 сут после введения 5 мкг 6‑OHDA содержание ДА стриатума уменьшалось приблизительно в два раза в сравнении с ложно-оперированными мышами [47], что соответствовало кратности снижения ДА в настоящем исследовании. Однако введение 18 мкг 6‑OHDA приводило через 14 сут более чем к шестикратному снижению ДА стриатума, сочетавшемуся с повреждением TH+ терминалей стриатума и нейронов черной субстанции [46]. Аналогичные ответы дофаминергической системы на интрастриатное унилатеральное введение 6-OHDA зафиксированы на крысах. Так введение 12 и 8 мкг 6‑OHDA крысам соответственно в течение 14 и 28 сут вызывало пятикратное снижение уровня ДА в стриатуме [49, 50]. Зафиксированное в нашей работе увеличение метаболических отношений ГВК/ДА и (ДОФУК + ГВК)/ДА свидетельствует о характерном для использованной модели компенсаторном увеличении оборота ДА в период 2–4 нед после повреждения [51].
Анализ научной литературы показывает, что выявленная способность афобазола восстанавливать содержание ДА в поврежденном 6-OHDA стриатуме мышей согласуется с нейропротекторным действием в данной модели болезни Паркинсона [46, 47, 49, 50, 52]. Соответствие увеличения содержания ДА в поврежденном 6-OHDA стриатуме и нейропротекторной активности установлено для различных терапевтических воздействий. Повышение уровня ДА в стриатуме мышей, зафиксированное при пероральном введении хризина (5,7‑dihydroxyflavone) в дозе 10 мг/кг в течение 28 дней, сопровождалось увеличением содержания нейротрофинов и количества TH+ нейронов [47]. Позитивное действие на уровень ДА и усиление окрашивания TH+ нейронов в поврежденном стриатуме крыс оказывало 14-суточное введение кофеина в дозе 10 мг/кг в/б [49] и 28‑сут введение производного имидазола (1,3-bisbenzylimidazolium bromide) в дозе 6 мг/кг в/б [50]. В условиях унилатерального повреждения стриатума мышей 6‑OHDA введение трансфицированных астроцитов, синтезирующих GDNF, увеличивало уровень ДА и плотность TH+ терминалей нейронов в стриатуме [46]. Инъекция в стриатум крыс трансформированных мезенхимальных стволовых клеток, продуцирующих нейротрофины, вызывала аналогичный эффект [52]. В приведенных публикациях был использован схожий с настоящей работой дизайн эксперимента, однако, содержание ДА не достигало уровня ложно-оперированной группы, в то время как афобазол, действуя в меньших дозах, приводил уровень ДА к контрольным значениям. Выраженное влияние препарта на содержание ДА в поврежденном стриатуме в сравнении с другими соединениями может быть объяснено с позиций мультитаргетного действия афобазола, в механизм которого включена регуляция как NQO2, так и SigmaR1 [35]. Такое сочетание может иметь следствием с одной стороны подавление патологических процессов, с другой – активацию компенсаторных и регенераторных механизмов [24, 53–55].
Гиперпродукция NQO2 положительно коррелирует с развитием идиопатической формы болезни Паркинсона [56] и болезни Альцгеймера [57], поэтому снижение активности фермента может оказывать нейропротекторное действие. Связываясь с мелатонинзависимым регуляторным сайтом NQO2 (МТ3 рецептор), афобазол ингибирует фермент [39], что препятствует двухэлектронному восстановлению DAQ в цитозоле, возобновлению цикла генерации супероксид анион-радикала и развитию окислительного стресса [33]. Ранее в модели менадионовой генотоксичности in vitro продемонстрирован вклад NQO2 в цитотопротекторное действие афобазола, выражающееся в снижении окислительного повреждения геномной ДНК [40, 58]. Защитный эффект афобазола согласуется с нейропротекторной активностью ингибитора NQO2 NMDPEF, введение которого в черную субстанцию ГМ мышей в условиях нейродегенерации, вызванной паракватом, снижало содержание маркера перекисного окисления липидов малонового диальдегида в тканях мозга [59]. В более позднем исследовании того же авторского коллектива на модели нейродегенерации показана определяющая роль NQO2 в негативном влиянии длительного окислительного стресса на процесс аутофагии астроцитов. Снижение активности NQO2 приводило к восстановлению аутофагии [53]. Таким образом, ингибирование NQO2 афобазолом в условиях нейродегенерации, с одной стороны, может снижать продукцию АФК, предотвращая окислительную модификацию макромолекул [53], с другой – оказывать положительное влияние на компенсаторные процессы аутофагии и активировать утилизацию белковых агрегатов и поврежденных органелл [60].
Увеличение содержания ДА в поврежденном стриатуме опытных животных при введении афобазола в сравнении с группой плацебо может быть обусловлено активацией шаперона SigmaR1 [61]. В экспериментах показано, что SigmaR1 включен в защитное действие афобазола, регулируя внутриклеточные процессы, имеющие важное значение для патогенеза болезни Паркинсона. В процессе нейродегенерации трансформируется специфичный паттерн кальциевых токов нейронов черной субстанции, что приводит к избыточному поступлению кальция в клетку и стимулирует развитие окислительного повреждения [62, 63]. Афобазол в экспериментах in vitro на модели бета‑амилоидной нейротоксичности (Aβ25-35) оказывал нейропротекторное действие, которое сопровождалось снижением концентрации внутриклеточного кальция в культуре кортикальных нейронов. В этих же исследованиях установлена способность афобазола уменьшать образование NO [41], которая позднее была подтверждена in vivo [64] и соответствует ингибирующему действию селективного агониста Sigma1R (+)‑пентазоцина на NOS [65]. На различных моделях болезни Паркинсона показано, что увеличенный синтез NO усиливает повреждение нейронов, а ингибирование продукции NO в условиях эксайтотоксичности и повышения концентрации внутриклеточного кальция способствует выживаемости нейронов [66]. Важным фактором патогенеза болезни Паркинсона является активация микроглии с последующим высвобождением провоспалительных цитокинов и АФК [67]. Терапевтическое введение афобазола уменьшало число активированных клеток микроглии при ишемии на модели окклюзии средней мозговой артерии, наряду с увеличением их общего количества [64]. Известно, что при болезни Паркинсона снижается экспрессия гена BDNF на уровне мРНК и белка [68, 69]. Эти данные коррелируют со снижением ретроградного аксонального транспорта BDNF и ингибированием его сигнальных путей при накоплении α‑синуклеина в нейронах [70]. Повышение уровня ДА в поврежденном стриатуме опытных животных при терапевтическом введении афобазола может быть следствием нейропротекторного действия препарата, в механизм которого включена SigmaR1 зависимая регуляция нейротрофинов и активация нейропластичности. Это предположение соответствует способности афобазола увеличивать содержание нейротрофинов в культуре нейронов и головном мозге мышей [71, 72], а также согласуется со стимулирующим действием агонистов SigmaR1 на секрецию BDNF в нейронах [73].
Позитивное влияние афобазола на содержание ДА в поврежденном стриатуме опытных животных схоже с действием селективного агониста SigmaR1 PRE‑084, внутрибрюшинное введение которого мышам на модели болезни Паркинсона, вызванной токсическим действием 6‑OHDA, приводило к повышению числа дофаминергических волокон и уровня дофамина в денервированных областях стриатума [74]. Увеличение содержания ДА наряду с отсутствием статистически значимого изменения уровня ДОФУК и ГВК (табл. 1, рис. 1) при введении афобазола согласуется с ингибирующим действием агонистов SigmaR1 на высвобождение ДА в стриатуме [75]. Показано, что агонисты SigmaR1 вызывают изменение конформации DAT в стриатуме, приводя к повышению чувствительности к ингибиторным воздействиям [76]. Авторы исследования указывают на корреляцию полученных данных с изменением липидного микроокружения переносчика и связывают действие агонистов с активацией шаперонных функций SigmaR1 в отношении DAT.
ЗАКЛЮЧЕНИЕ
В настоящем исследовании при интрастриатном унилатеральном введении 6-OHDA мышам установлено восстанавливающее влияние афобазола на содержание ДА в поврежденном стриатуме, что позволяет предположить наличие у препарата нейропротекторного действия в данной модели болезни Паркинсона. Анализ собственных данных и научной периодики показывает, что выявленный эффект может быть следствием регуляции афобазолом NQO2 и шаперона SigmaR1. Целесообразно дальнейшие изучение вклада каждой мишени препарата в нейропротекторное действие в различных моделях болезни Паркинсона.
Список литературы
Cacabelos R.// Int. J. Mol. Sci. 2017. V. 18. № 3. P. E551.
Moreno J.A., Tiffany-Castiglioni E.// Neurochem. Res. 2015. V. 40. № 2. P. 329–335.
Zeeshan H.M., Lee G.H., Kim H.R., Chae H.J.// Int. J. Mol. Sci. 2016. V. 17. № 3. P. 327.
Dias V., Junn E., Mouradian M.M.// J. Parkinsons Dis. 2013. V. 3. № 4. P. 461–491.
Ebrahimi-Fakhari D., Wahlster L., McLean P.J. // J. Parkinsons Dis. 2011. V. 1. № 4. P. 299–320.
Gorbatyuk M.S., Gorbatyuk O.S.// J. Genet. Syndr. Gene Ther. 2013. V. 4. № 2. P. 128.
Whitworth A.J., Pallanck L.J.// SEB Exp. Biol. Ser. 2008. V. 60. P. 93–113.
Hong J.S.// Ann. N. Y. Acad. Sci. 2005. V. 1053. P. 151–152.
Gluck M.R., Zeevalk G.D.// J. Neurochem. 2004. V. 91. № 4. P. 788–795.
Dimant H., Ebrahimi-Fakhari D., McLean P.J. // Neuroscientist. 2012. V. 18. № 6. P. 589–601.
Blum D., Torch S., Lambeng N., Nissou M., Benabid A.L., Sadoul R., Verna J.M. // Prog. Neurobiol. 2001. V. 65. № 2. P. 135–172.
Alvarez-Fischer D., Henze C., Strenzke C., Westrich J., Ferger B., Hoglinger G.U., Oertel W.H., Hartmann A. // Exp. Neurol. 2008. V. 210. № 1. P. 182–193.
Thiele S.L., Warre R., Nash J.E. // J. Vis. Exp. 2012. № 60.
Holtz W.A., Turetzky J.M., Jong Y.J., O’Malley K.L. // J. Neurochem. 2006. V. 99. № 1. P. 54–69.
Ryu E.J., Harding H.P., Angelastro J.M., Vitolo O.V., Ron D., Greene L.A. // J. Neurosci. 2002. V. 22. № 24. P. 10690–10698.
Braakman I., Hebert D.N. // Cold Spring Harb. Perspect. Biol. 2013. V. 5. № 5. P. a013201.
Peth A., Nathan J.A., Goldberg A.L.// J. Biol. Chem. 2013. V. 288. № 40. P. 29215–29222.
Mercado G., Castillo V., Vidal R., Hetz C.// Front. Aging Neurosci. 2015. V. 7. P. 39.
Mercado G., Valdes P., Hetz C. // Trends Mol. Med. 2013. V. 19. № 3. P. 165–175.
Mercado G., Castillo V., Soto P., Sidhu A. // Brain Res. 2016. V. 1648. Pt B. P. 626–632.
Hayashi T., Su T.P. // Cell. 2007. V. 131. № 3. P. 596–610.
Mori T., Hayashi T., Hayashi E., Su T.P. // PLoS One. 2013. V. 8. № 10. P. e76941.
Su T.P., Su T.C., Nakamura Y., Tsai S.Y. // Trends Pharmacol. Sci. 2016. V. 37. № 4. P. 262–278.
Ruscher K., Wieloch T. // J. Pharmacol. Sci. 2015. V. 127. № 1. P. 30–35.
LaVoie M.J., Hastings T.G. // J. Neurosci. 1999. V. 19. № 4. P. 1484–1491.
Glinka Y.Y., Youdim M.B. // Eur. J. Pharmacol. 1995. V. 292. № 3–4. P. 329–332.
Berman S.B., Hastings T.G. // J. Neurochem. 1999. V. 73. № 3. P. 1127–1137.
Kuhn D.M., Arthur R.E., Jr., Thomas D.M., Elferink L.A. // J. Neurochem. 1999. V. 73. № 3. P. 1309–1317.
Whitehead R.E., Ferrer J.V., Javitch J.A., Justice J.B. // J. Neurochem. 2001. V. 76. № 4. P. 1242–1251.
Terland O., Flatmark T., Tangeras A., Gronberg M. // J. Mol. Cell. Cardiol. 1997. V. 29. № 6. P. 1731–1738.
Vella F., Ferry G., Delagrange P., Boutin J.A.// Biochem. Pharmacol. 2005. V. 71. № 1 –2. P. 1–12.
Reybier K., Perio P., Ferry G., Bouajila J., Delagrange P., Boutin J.A., Nepveu F. // Free Radic. Res. 2011. V. 45. № 10. P. 1184–1195.
Segura-Aguilar J., Paris I., Munoz P., Ferrari E., Zecca L., Zucca F.A. // J. Neurochem. 2014. V. 129. № 6. P. 898–915.
Середенин С.Б., Воронина Т.А., Незнамов Г.Г., Бледнов Ю.А., Бадыштов Б.А., Виглинская И.В., Козловская М.М., Колотилинская Н.В., Яркова М.А., Савельев В.Л., Гарибова Т.Л., Вальдман Е.А. // Вестник РАМН. 1998. Т. 11. С. 3–9.
Середенин С.Б., Воронин М.В. // Эксп. клин. фарм. 2009. Т. 72. № 1. С. 3–11.
Seredenin S.B., Antipova T.A., Voronin M.V., Kurcha-shova S.Y., Kuimov A.N. // Bull. Exp. Biol. Med. 2009. V. 148. № 1. P. 42–44.
Абрамова Е.В., Воронин М.В., Середенин С.Б. // Эксп. клин. фарм. 2017. Т. 80. № 2. С. 3–7.
Hayashi T., Su T.P. // J. Pharmacol. Exp. Ther. 2003. V. 306. № 2. P. 726–733.
Kadnikov I.A., Voronin M.V., Seredenin S.B. // Pharm. Chem. J. 2014. V. 47. № 10. P. 514–516.
Voronin M.V., Kadnikov I.A. // Pharmacol. Res. Perspect. 2016. V. 4. № 6. P. e00273.
Behensky A.A., Yasny I.E., Shuster A.M., Seredenin S.B., Petrov A.V., Cuevas J. // J. Pharmacol. Exp. Ther. 2013. V. 347. № 2. P. 468–477.
Behensky A.A., Yasny I.E., Shuster A.M., Seredenin S.B., Petrov A.V., Cuevas J. // J. Pharmacol. Exp. Ther. 2013. V. 347. № 2. P. 458–467.
Середенин С.Б., Воронин М.В., Абрамова Е.В. // Эксп. клин. фарм. 2017. Т. 80. № 9. С. 9–19.
Poewe W., Seppi K., Tanner C.M., Halliday G.M., Brundin P., Volkmann J., Schrag A.E., Lang A.E. // Nat. Rev. Dis. Primers. 2017. V. 3. P. 17013.
Roffler-Tarlov S., Sharman D.F., Tegerdine P. // Br. J. Pharmacol. 1971. V. 42. № 3. P. 343–351.
Cunningham L.A., Su C. // Exp. Neurol. 2002. V. 174. № 2. P. 230–242.
Goes A.T.R., Jesse C.R., Antunes M.S., Lobo Ladd F.V., Lobo Ladd A.A.B., Luchese C., Paroul N., Boeira S.P. // Chem. Biol. Interact. 2018. V. 279. P. 111–120.
Blandini F., Armentero M.T., Martignoni E. // Parkinsonism Relat. Disord. 2008. V. 14 Suppl 2. P. S124–129.
Aguiar L.M., Nobre H.V., Jr., Macedo D.S., Oliveira A.A., Freitas R.M., Vasconcelos S.M., Cunha G.M., Sousa F.C., Viana G.S. // Pharmacol. Biochem. Behav. 2006. V. 84. № 3. P. 415–419.
Chan H.H., Kumar S., Zhuo L. // Eur. J. Pharmacol. 2013. V. 715. № 1–3. P. 405–413.
Yuan H., Sarre S., Ebinger G., Michotte Y. // Brain Res. 2004. V. 1026. № 1. P. 95–107.
Sadan O., Bahat-Stromza M., Barhum Y., Levy Y.S., Pisnevsky A., Peretz H., Ilan A.B., Bulvik S., Shemesh N., Krepel D., Cohen Y., Melamed E., Offen D. // Stem Cells Dev. 2009. V. 18. № 8. P. 1179–1190.
Janda E., Lascala A., Carresi C., Parafati M., Aprigliano S., Russo V., Savoia C., Ziviani E., Musolino V., Morani F., Isidoro C., Mollace V. // Autophagy. 2015. V. 11. № 7. P. 1063–1080.
Nguyen L., Lucke-Wold B.P., Mookerjee S.A., Cavendish J.Z., Robson M.J., Scandinaro A.L., Matsumoto R.R. // J. Pharmacol. Sci. 2015. V. 127. № 1. P. 17–29.
Francardo V., Schmitz Y., Sulzer D., Cenci M.A. // Exp. Neurol. 2017. V. 298. Pt B. P. 137–147.
Wang W., Le W.D., Pan T., Stringer J.L., Jaiswal A.K. // J. Gerontol. A. Biol. Sci. Med. Sci. 2008. V. 63. № 2. P. 127–134.
Hashimoto T., Nakai M. // Neurosci. Lett. 2011. V. 502. № 1. P. 10–12.
Kadnikov I.A., Voronin M.V., Seredenin S.B. // Bull. Exp. Biol. Med. 2015. V. 159. № 1. P. 44–47.
Janda E., Parafati M., Aprigliano S., Carresi C., Visalli V., Sacco I., Ventrice D., Mega T., Vadala N., Rinaldi S., Musolino V., Palma E., Gratteri S., Rotiroti D., Mollace V. // Br. J. Pharmacol. 2013. V. 168. № 1. P. 46–59.
Ji C.H., Kwon Y.T. // Mol. Cells. 2017. V. 40. № 7. P. 441–449.
Francardo V. // Neural Regen. Res. 2014. V. 9. № 21. P. 1882–1883.
Surmeier D.J., Schumacker P.T., Guzman J.D., Ilijic E., Yang B., Zampese E. // Biochem. Biophys. Res. Commun. 2017. V. 483. № 4. P. 1013–1019.
Lautenschlager J., Stephens A.D., Fusco G., Strohl F., Curry N., Zacharopoulou M., Michel C.H., Laine R., Nespovitaya N., Fantham M., Pinotsi D., Zago W., Fraser P., Tandon A., St George-Hyslop P., Rees E., Phillips J.J., De Simone A., Kaminski C.F., Schierle G.S.K. // Nat. Commun. 2018. V. 9. № 1. P. 712.
Katnik C., Garcia A., Behensky A.A., Yasny I.E., Shuster A.M., Seredenin S.B., Petrov A.V., Cuevas J. // J. Neurochem. 2016. V. 139. № 3. P. 497–509.
Vagnerova K., Hurn P.D., Bhardwaj A., Kirsch J.R. // Anesth. Analg. 2006. V. 103. № 2. P. 430–434.
Zhang L., Dawson V.L., Dawson T.M. // Pharmacol. Ther. 2006. V. 109. № 1–2. P. 33–41.
Subramaniam S.R., Federoff H.J.// Front. Aging Neurosci. 2017. V. 9. P. 176.
Parain K., Murer M.G., Yan Q., Faucheux B., Agid Y., Hirsch E., Raisman-Vozari R. // Neuroreport. 1999. V. 10. № 3. P. 557–561.
Howells D.W., Porritt M.J., Wong J.Y., Batchelor P.E., Kalnins R., Hughes A.J., Donnan G.A. // Exp. Neurol. 2000. V. 166. № 1. P. 127–135.
Fang F., Yang W., Florio J.B., Rockenstein E., Spencer B., Orain X.M., Dong S.X., Li H., Chen X., Sung K., Rissman R.A., Masliah E., Ding J., Wu C. // Sci. Rep. 2017. V. 7. № 1. P. 3868.
Середенин С.Б., Мелкумян Д.С., Вальдман Е.А., Яркова М.А., Середенина Т.С., Воронин М.В., Лапицкая А.С. // Эксп. клин. фарм. 2006. V. 69. № 3. P. 3–6.
Антипова Т.А., Сапожникова Д.С., Бахтина Л.Ю., Середенин С.Б. // Эксп. клин. фарм. 2009. V. 72. № 1. P. 12–14.
Fujimoto M., Hayashi T., Urfer R., Mita S., Su T.P. // Synapse. 2012. V. 66. № 7. P. 630–639.
Francardo V., Bez F., Wieloch T., Nissbrandt H., Ruscher K., Cenci M.A. // Brain. 2014. V. 137. Pt 7. P. 1998–2014.
Gonzalez-Alvear G.M., Werling L.L. // J. Pharmacol. Exp. Ther. 1994. V. 271. № 1. P. 212–219.
Hong W.C., Yano H., Hiranita T., Chin F.T., McCurdy C.R., Su T.P., Amara S.G., Katz J.L. // J. Biol. Chem. 2017. V. 292. № 27. P. 11250–11261.
Дополнительные материалы отсутствуют.