

Нейрохимия, 2019, T. 36, № 2, стр. 128-139
Влияние тяжелой гипоксии на HIF1- и Nrf2-опосредованные механизмы антиоксидантной защиты в неокортексе крыс
К. В. Сариева 1, 2, А. Ю. Лянгузов 3, О. В. Галкина 2, О. В. Ветровой 1, 2
1 ФГБУН Институт физиологии им. И.П. Павлова РАН
Санкт-Петербург, Россия
2 ФГБОУ ВО Санкт-Петербургский государственный университет, биологический факультет,
кафедра биохимии
Санкт-Петербург, Россия
3 ФГБОУ ВО Санкт-Петербургский государственный университет, ресурсный центр
“Обсерватория экологической безопасности”, Научный парк
Санкт-Петербург, Россия
Поступила в редакцию 25.06.2018
После доработки 20.07.2018
Принята к публикации 01.08.2018
Аннотация
Одним из важнейших факторов адаптации к хронической гипоксии мозга является транскрипционный фактор (ТФ) HIF1. Однако в условиях острой гипоксии и реоксигенации особое значение приобретает ТФ стрессорного ответа – NRF2. Взаимодействие этих белков на уровне регуляции антиоксидантной защиты и глюкозного метаболизма ранее было показано в чувствительных к гипоксии раковых опухолях. В данном исследовании изучено влияние тяжелой гипобарической гипоксии (ТГ) на HIF1- и NRF2-зависимые процессы в неокортексе крыс. Была выявлена совместная регуляция глутатион-зависимой антиоксидантной системы этими белками, что оказывало влияние на общую антирадикальную активность и клеточный окислительно-восстановительный статус. В частности, ингибирование HIF1 предотвращало вызываемый ТГ окислительный сдвиг через день после реоксигенации, что сопровождалось увеличением количества общего глутатиона и глутатион редуктазной активности. Оба эти эффекта носят NRF2-зависимый характер и свидетельствуют об активации этого транскрипционного фактора в ответ на ТГ в сочетании с ингибированием HIF1. Полученные данные подтверждают ранее высказанную гипотезу о дезадаптивном эффекте HIF1 в условиях острой гипоксии и реоксигенации, а также свидетельствуют о вкладе NRF2 в реализацию защитных механизмов в постгипоксический период. Гипотеза о взаимодействии этих транскрипционных факторов в (пост)гипоксический период требует дальнейшей проверки и может существенно повлиять на понимание молекулярных патомеханизмов гипоксии мозга.
ВВЕДЕНИЕ
Гипоксия – состояние, обусловленное пониженным содержанием кислорода в тканях. В связи с высокими энергетическими потребностями, головной мозг часто встречается с локальным недостатком кислорода и должен обладать механизмами быстрой компенсации кислородного голодания и защиты от его последствий. Физиологическая активность мозга влияет на кислородное и глюкозное снабжение с вовлечением сигнальных процессов, приводящих к расширению сосудов, гидролизу гликогена и, вероятно, усилению роли лактатного шунта [1–3]. Однако продолжительная гипоксия требует адаптации метаболизма, направленной на обеспечение функционирования в измененных условиях, что осуществляется с вовлечением транскрипционных факторов (ТФ) [4, 5]. Долгосрочная гипоксия может носить как физиологический, так и патологический характер. В обоих случаях основными регуляторами клеточного ответа на гипоксию и/или реоксигенацию являются транскрипционные факторы HIF1 (hypoxia-inducible factor 1) и NRF2 (nuclear factor (erythroid-derived 2)-like 2) [6, 7].
В условиях гипоксии начальным этапом запуска HIF1-опосредованной метаболической перестройки является ингибирование пролил-гидроксилазы [8], которое приводит к стабилизации HIF1 и его транслокации в ядро. Временны́е характеристики реализации HIF1-индуцированных процессов таковы: в первые несколько часов после начала гипоксического воздействия происходит накопление этого ТФ в ядре и активация транскрипции его генов-мишеней [9]; в период до нескольких суток происходит трансляция мРНК и реализация функций, опосредуемых синтезированными белками [10]. К транскрипционным мишеням HIF1 относятся гены, отвечающие за перестройку гематоэнцефалического барьера и ангиогенез (VEGF), гены ферментов гликолиза, лактат дегидрогеназы, цитокина эритропоэтина и многие другие [8, 11, 12]. Эффекты HIF1 можно наблюдать уже через день после гипоксического воздействия, причем характер изменений скорее предполагает адаптацию к хронической гипоксии [11], нежели к краткосрочной патологической гипоксии, как, например, в случае ишемического инсульта. Так, в моделях гипоксического прекондиционирования показана HIF1-опосредованная толерантность к тяжелой гипоксии [9, 13, 14].
Напротив, NRF2 представляет собой ТФ, участвующий в реализации механизмов защиты от окислительного стресса и вероятней всего связан с острыми гипоксическими состояниями и последующей реоксигенацией [6, 15]. Среди множества транскрипционных мишеней NRF2 находятся гены ферментов семейства глутатион-S-трансфераз [15], ферментов синтеза глутатиона [16], некоторых транспортеров суперсемейства ABC [17, 18], которые осуществляют экспорт окисленного глутатиона и глутатионилированых субстратов из клетки [19–21]. Таким образом, NRF2 выполняет функцию ключевого регулятора антиоксидантной защиты клеток.
Для моделирования условий повреждающей гипоксии в Лаборатории регуляции функций нейронов мозга Института Физиологии им. И.П. Павлова РАН используется модель тяжелой гипобарической гипоксии (ТГ), приводящую к широкому спектру нарушений на различных уровнях, от биохимического до поведенческого [22–26], что сопровождается запуском апоптотических процессов [27], а также уменьшением экспрессии нейропротективных белков [28]. В частности, следует отметить выявленную ранее ап-регуляцию HIF1α через день после ТГ (Vetrovoy et al., 2018, DOI 10.1007/s11064-018-2681-x), что потенциально может играть как протективную, так и дезадаптивную роль [10]. Большинство ранних исследований проведено на гиппокампе – наиболее чувствительной к гипоксии структуре мозга. В данном эксперименте были исследованы реакции неокортекса на идентичную патологическую ситуацию. В частности, различия между гиппокампом и неокортексом касаются активности антиоксидантных систем. В связи с этим целью данной работы была оценка роли HIF1 в развитии постгипоксического состояния; а также оценка функционального состояния глутатион-зависимой антиоксидантной системы, которая находится под частичным контролем NRF2 и HIF1.
МЕТОДЫ ИССЛЕДОВАНИЯ
Животные. Работа проведена на взрослых самцах крыс линии Вистар весом 220–250 г. из биоколлекции “Коллекция лабораторных млекопитающих разной таксономической принадлежности” Института физиологии им. И.П. Павлова РАН, поддержанной программой биоресурсных коллекций ФАНО России. Животные были выращены в стандартных условиях вивария Института физиологии им. И.П. Павлова РАН и содержались в лабораторных условиях при свободном доступе к воде и пище. При проведении экспериментов соблюдались требования, сформулированные в Директивах Совета Европейского сообщества (86/609/EEC) об использовании животных для экспериментальных исследований. Протоколы опытов были утверждены Комиссией по гуманному обращению с животными Института физиологии им. И.П. Павлова РАН.
Инъекции топотекана – ингибитора HIF1. За 10 мин до начала гипоксического сеанса животным интраперитонеально вводили 200 мкл топотекана гидрохлорида в смеси ДМСО : 0.9% NaCl = = 1 : 1 в расчете 5 мг/кг либо смесь ДМСО : 0.9% NaCl в таком же объеме (в качестве инъекционного контроля) [29, 30]. Действие ингибитора HIF1 в отсутствие гипоксического воздействия проверяли путем ежедневного введения указанных доз препарата.
Тяжелая гипобарическая гипоксия. Условия ТГ создавали в барокамере проточного типа при давлении 160–180 мм рт. ст. Продолжительность воздействия 3 ч. Продув камеры воздухом осуществляли каждые 20 мин с целью поддержания нормального соотношения газов и избегания гиперкапнии [31].
Схема эксперимента представлена на рис. 1. Для анализа биохимических показателей животных декапитировали гильотиной через 1, 2 и 4 дня после ТГ. Животным из группы, не подвергавшейся гипоксическому воздействию, вводили ингибитор HIF1 раз в день на протяжении 4 дней и выводили из эксперимента через 1 день после последней инъекции (рис. 1а). Впоследствии забирали мозг, выделяли сенсомоторную кору и быстро замораживали в жидком азоте. В качестве контрольной группы использовали животных, не подвергавшихся гипоксическому воздействию.
Рис. 1.
Дизайн экспериментов, направленных на измерение цитозольной антирадикальной активности, количества тиоловых групп и общего глутатиона, глутатион редуктазной и пероксидазной активностей (а), а также иммуногистохимического анализа и определения оснований Шиффа (б). ТГ, тяжелая гипобарическая гипоксия; ОШ, основания Шиффа; ИГХ, иммуногистохимия.

Для иммуногистохимического анализа и анализа количества оснований Шиффа животных выводили из эксперимента через 1, 2 и 4 дня после ТГ. Животных, подвергшихся инъекции ингибитора HIF1 без ТГ, декапитировали через день после инъекции (рис. 1б).
Выделение цитозольной фракции. Сенсомоторную кору гомогенизировали на льду в системе стекло–стекло (гомогенизатор Поттера) в 1 мл 0.3 М раствора сахарозы на 1 мМ ЭДТА, 0.2 М Трис-HCl (рН 7.4). Центрифугирование проводили в течение 10 мин при 1000 g и 4°С. Супернатант переносили в чистые пробирки и подвергали повторному центрифугированию в течение 20 мин при 2000 g и 4°С [32], аликвотировали и замораживали при –80°С.
Измерение содержания белка. Количество белка в пробах определяли фотометрически непосредственно перед каждым экспериментом с помощью фотометра Biophotometer plus (Eppendorf, Германия) согласно стандартному трехволновому (230, 280, 340 нм) протоколу.
Измерение цитозольной антирадикальной активности методом ORAC (Oxygen Radical Absorption Capacity). Образцы цитозольной фракции инкубировали с 8.6 нМ раствором флуоресцеина (на 10 мМ фосфатном буфере, рН 7.4) на протяжении 30 мин при 37°С. Измеряли базальную интеносивность флуоресценции с использованием спектрофлуориметрического планшетного ридера FLUOstar Omega (BMG Labtech, Германия) (Ex. 485 нм, Em. 520 нм). С помощью инжекторной системы прибора в лунки планшета вводили инициатор образования алкилперекисных радикалов AAPH (2,2'-азобис(2-амидино-пропан)) до конечной концентрации 30 мМ [33], после чего продолжали измерение интенсивности флуоресценции до ее полного падения. Антирадикальную активность оценивали как отношение светосуммы (площади под кривой изменения флуоресценции с течением времени) к количеству белка в пробе. Среднее и стандартную ошибку среднего выражали в % от контроля. Контроль принят за 100%. Каждый эксперимент повторяли дважды.
Измерение содержания тиоловых групп и общего глутатиона. Для оценки количества тиоловых групп к 25 мкл образца цитозольной фракции приливали 40 нг/мл ДТНБ (5,5'-дитиобис-2-нитробензойная кислота) (на 0.1 М фосфатном буфере, 1 мМ ЭДТА, рН 7.4) и измеряли оптическую плотность при 412 нм на спектрофотометричеком планшетном ридере SPECTROstar Nano (BMG Labtech, Германия).
Для измерения количества общего глутатиона экстракты цитозольной фракции предварительно депротеинизировали: 80 мкл образца разводили с равным количеством 2 М HClO4 (перхлорной кислоты) и инкубировали в течение 5 мин при комнатной температуре. Далее образцы центрифугировали при 5000 g в течение 5 мин, переносили супернатант в чистые пробирки и нейтрализовали с помощью 2 М КОН. Инкубировали образцы в течение 5 мин при комнатной температуре и подвергали повторному центрифугированию при 5000 g в течение 5 мин. 25 мкл супернатанта разводили реакционной смесью, содержащей 0.19 мг/мл НАДФН (AppliChem., Германия) и 25 нг/мл ДТНБ (на 0.1 М фосфатном буфере, 1 мМ ЭДТА, рН 7.4). Добавляли глутатион редуктазу до конечной активности 6 U/мл и измеряли оптическую плотность при 412 нм на спертрофотометричеком планшетном ридере SPECTROstar Nano [34].
Концентрацию тиоловых групп и общего глутатиона выражали в количестве, нормализованном на количество общего белка (нмоль/мг белка). Эксперимент проводили дважды. Среднее и стандартная ошибка среднего представлены в % от контроля, контроль принят за 100%.
Измерение глутатион пероксидазной (ГП) активности. Метод основан на измерении снижения оптической плотности раствора НАДФН при 340 нм. К 50 мкл образца приливали реакционную смесь, содержащую в конечных концентрациях 4 мМ GSH (восстановленный глутатион), 2 мМ NaN3 (азид натрия), глутатион редуктазу 20 U/мл, 0.4 мМ НАДФН и 0.25 мМ H2O2. Оптическую плотность измеряли при 340 нм и температуре 37°C на спектрофотометрическом планшетном ридере SPECTROstar Nano [35].
ГП активность выражали в количестве потребленного НАДФН в минуту и нормализовали на количество общего белка (пмоль НАДФН/(мин мг белка). Эксперимент проводили дважды. Результаты выражали в виде среднего и стандартной ошибки среднего. Среднее и стандартная ошибка среднего представлены в % от контроля, контроль принят за 100%.
Измерение глутатион редуктазной (ГР) активности. 80 мкл образца разводили реакционной смесью, содержащей в конечной концентрации 0.4 мМ НАДФН и 4 мМ GSSG (окисленный глутатион) (на 0.2 М фосфатном буфере, 2 мМ ЭДТА, рН 7.0) [36]. Оптическую плотность измеряли при 340 нм и температуре 37°C на спектрофотометрическом планшетном ридере SPECTROstar Nano.
ГР активность выражали в количестве потребленного НАДФН в мин и нормализовали на количество общего белка (пмоль НАДФН/(мин мг белка). Эксперимент проводили дважды. Результаты выражали в виде среднего и стандартной ошибки среднего. Среднее и стандартная ошибка среднего представлены в % от контроля, контроль принят за 100%.
Измерение содержания оснований Шиффа (ОШ). Экстракцию липидов из сенсомоторной коры осуществляли методом Фолча с помощью смеси хлороформ : метанол = 2 : 1 [37]. Экстракт отмывали от нелипидных примесей охлажденным 0.09% раствором NaCl, а поверхность нижней фазы – дважды смесью хлороформ : метанол : H2O (3 : 48 : 47). Смесь выпаривали и растворяли в хлороформе. Интенсивность флуоресценции ОШ измеряли с помощью спектрофлуориметрического планшетного ридера FLUOstar Omega (Ex. 370 нм, Em. 450 нм) [38]. Результаты выражали в условных единицах (единицах флуоресценции) в расчете на 1 мг фосфолипидов. Количество фосфолипидов оценивали (спектрофотометрически при 830 нм) по содержанию неорганического фосфора методом Бартлетта [39].
Флуоресцентная иммуногистохимия. Образцы ткани мозга обрабатывали согласно стандартному гистологическому протоколу: фиксировали в фиксаторе FineFix (разведение: 28 мл фиксатора + + 72 мл 96° этанола, Milestone, Italy) в течение 24 ч при температуре 4°C. Затем образцы проводили через этанол возрастающих концентраций: 70° (1.5 ч) → 96° этанол/100% изопропиловый спирт 80/20 (3 ч) → 100% изопропиловый спирт (3 ч) при 60°C. После этого материал помещали в парафин и выдерживали в термостате при температуре 56°C в течение часа. Процедуру повторяли дважды, после чего изготавливали парафиновые блоки. На ротационном микротоме (Reichert, Austria) изготавливали серийные срезы мозга во фронтальной плоскости толщиной 7 мкм на уровне –2.80 мм от брегмы. Полученные срезы монтировали на предметные стекла, обработанные полилизином. Далее срезы депарафинизировали в ксилоле (2 смены по 5 мин), подвергали регидратации в спиртах (96° → 96° → 96° → 70° по 5 мин в каждом) и демаскировали кипячением в цитратном буфере (рН 6.0).
Для оценки содержания Nrf2 и его колокализации с нейрональным маркером NeuN (neuronal nuclear antigen) использовали иммуногистохимический метод. Основные этапы метода: 1) инкубация с поликлональными антителами к Nrf2 1 : : 200 (ABE413, Merck KGaA, Германия) и моноклональными антителами к NeuN 1 : 100 (MAB377A5, Merck KGaA, Германия) при 4°C; 2) инкубация с вторичными конъюгированными антителами. Анализ препаратов проводили с помощью инвертированного флуоресцентного микроскопа Leica DMI6000 (Leica, Германия) на базе Научного парка СПбГУ, Ресурсный центр “Развитие молекулярных и клеточных технологий”. Оценку площади колокализации Nrf2 и NeuN проводили с помощью программного обеспечения ImageJ.
Статистическая обработка. Статистическую обработку проводили с использованием скриптов на свободном статистическом языке R (https:// cran.r-project.org/) в Ресурсном центре “Обсерватория экологической безопасности” Научного парка СПбГУ. Для построения графиков использовали метод бутстрепа. Результаты выражены в виде графиков типа “ящик-с-усами”, где представлены среднее, “ящик” – 68% доверительный интервал для среднего (приблизительно ± стандартная ошибка среднего), “усы” – 95% доверительный интервал (приблизительно ± двукратная стандартная ошибка среднего). Оценку статистической значимости проводили с использованием непараметрического теста Крускала–Уоллиса (The Kruskal–Wallis test). Результаты считали достоверными при р < 0.05. В качестве post-hoc процедуры использовали Данн тест (Dunn’s test) для множественных выборок при уровне значимости α = 0.05 [40].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Влияние ТГ и ТГ в сочетании с введением ингибитора HIF1 на окислительно-восстановительный статус клеток и интенсивность перекисного окисления липидов. Важнейшим фактором патогенеза постгипоксических состояний является окислительный стресс. Характерными маркерами последнего может служить интенсификация свободнорадикального повреждения макромолекул (в том числе перекисное окисление липидов ПОЛ) и изменение окислительно-восстановительного статуса клетки (редокс статус) в сторону более окисленного [41]. Количество тиоловых групп, характеризующее редокс статус, снижалось в неокортексе крыс через день после ТГ, после чего возвращалось к контрольным значениям. В то же время введение ингибитора HIF1 приводило к увеличению данного показателя на протяжении всего времени эксперимента в неокортексе животных, не подвергавшихся гипоксии. Введение ингибитора HIF1 в сочетании с ТГ стабилизировало количество тиоловых групп на уровне контрольных значений (рис. 2а).
Рис. 2.
Влияние ТГ и ТГ в сочетании с введением ингибитора HIF1 (ТГ + i) на количество тиоловых групп (а) и оснований Шиффа (б) в неокортексе через 1, 2 и 4 дня после сеанса ТГ. * Различия достоверны по отношению к контролю, ** Различия достоверны по отношению к группе Контроль + i. *** Различия достоверны между группами ТГ и ТГ + i, p < 0.05.
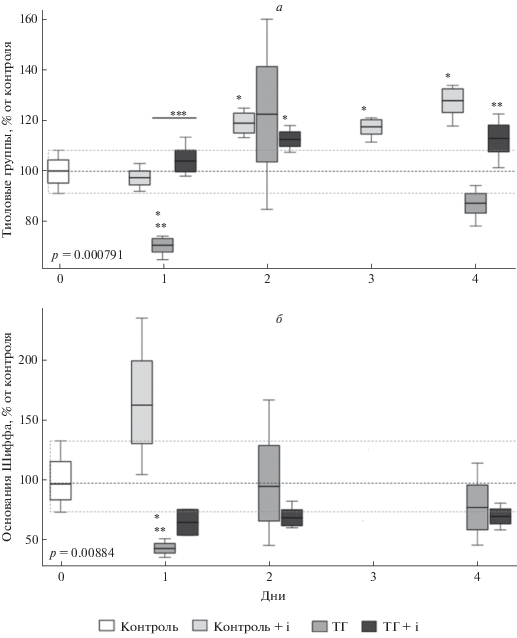
При определении количества ОШ, свидетельствующего об интенсивности процессов свободно-радикального окисления липидов, нами не было обнаружено достоверных изменений этого показателя ни в одной из экспериментальных групп, за исключением ТГ, в которой через день после реоксигенации количество ОШ было ниже контрольных значений, в дальнейшем возвращаясь к контрольным значениям (рис. 2б).
Таким образом, наши данные указывают на то, что ингибирование HIF1 не приводит к развитию окислительного стресса в неокортексе крыс и даже несколько смещает редокс статус в сторону восстановленного состояния, что смягчает реакцию организма на ТГ. Нами показано, что в присутствии ингибитора HIF1, в первый день после ТГ не наблюдается развития окислительного стресса. Однако важно отметить показанную нами ранее относительную устойчивость неокортекса к патологической гипоксии по сравнению гиппокампом (Сариева и др., 2018, DOI 10.1134/S1027813318030123).
Влияние ТГ и ингибитора HIF1 на некоторые показатели антиоксидантной защиты. Важнейшим фактором защиты от окислительного стресса в постгипоксических условиях становится мобилизация систем антиоксидантной защиты [42]. Глутатион-зависимая система является ключевой антиоксидантной системой мозга [43], причем на многих этапах метаболизма глутатиона задействованы белки, кодируемые генами-мишенями как HIF1 [44], так и NRF2 [7]. В связи с этим мы определили активность ферментов, вовлеченных в обеспечение антиоксидантной функции глутатиона, а также измерили общую антиоксидантную активность в цитоплазме.
ГП активность повышалась в ответ на ТГ в первый день, а затем возвращалась к контрольным значениям. Данные изменения хорошо согласуются с динамикой HIF1α после гипоксического воздействия (статья на рецензии). На модели колоректального рака было показано, что регуляция гена Gpx1 (одна из изоформ ГП) на уровне транскрипции осуществляется с участием HIF1 [44]. Известно, что в головном мозге крыс экспрессируются две изоформы этого фермента – ГП1 (цитозольная форма или “классическая”) и 4 (фосфолипид-гидропероксидная форма) [45]. Можно предположить, что больший вклад в общую ГП активность вносила именно ГП1. Данное наблюдение подтверждается динамикой активности фермента в присутствии ингибитора HIF1: ингибирование HIF1 как самостоятельно, так и в сочетании с ТГ, приводило к снижению ГП активности как минимум на протяжении четырех дней после воздействия. Необходимо также отметить, что ТГ в сочетании с ингибитором HIF1 существенно снижало ГП активность в неокортексе крыс на первый и второй день после воздействия по сравнению с животными, подвергшимися только воздействию ТГ (рис. 3а).
Рис. 3.
Влияние ТГ и ТГ в сочетании с введением ингибитора HIF1 (ТГ + i) на глутатион пероксидазную (ГП) активность (а) и цитозольную антирадикальную активность (ЦАА) (б) в неокортексе через 1, 2 и 4 дня после сеанса ТГ и ТГ в сочетании с ингибированием HIF1. * Различия достоверны по отношению к контролю, ** Различия достоверны по отношению к группе Контроль + i. *** Различия достоверны между группами ТГ и ТГ + i, p < 0.05.
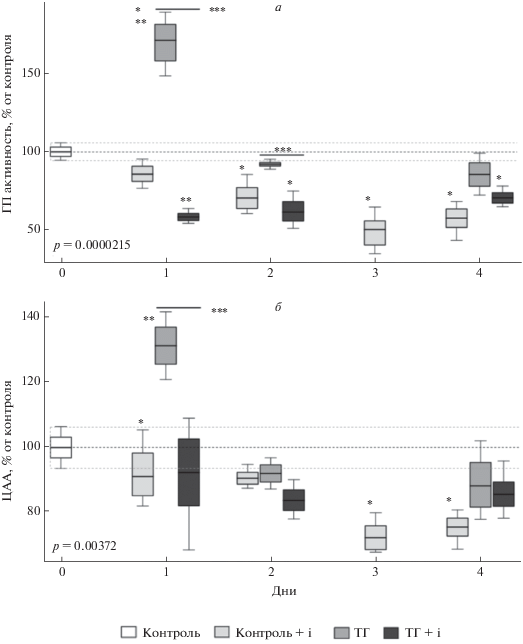
Общая антирадикальная активность цитозольной фракции, исследованная с помощью метода ORAC, повышалась на первый день после ТГ, а затем возвращалась к контрольным значениям. Ингибирование HIF1 приводило к снижению данного показателя на протяжении всего времени эксперимента, тогда как при ингибировании HIF1 в сочетании с ТГ общая антирадикальная активность цитозоля оставалась у контрольных значений (рис. 3б). Снижение антирадикальной активности в ответ на ингибирование HIF1 на фоне восстановленного состояния клетки, вызванных данным воздействием, может служить адаптацией к сниженной генерации АФК и поддержанию АФК-опосредованных сигнальных процессов [41]. Несмотря на то, что метод ORAC является широко используемым в настоящее время, общая антирадикальная активность, определяемая данным методом, является интегральным показателем, поэтому предположить механизм ее регуляции достаточно сложно.
Влияние ТГ и ингибитора HIF1 на транскрипционный фактор NRF2 и NRF2-зависимые процессы. Если экспрессия гена ГП1 регулируется транскрипционным фактором HIF1, то гены подавляющего большинства глутатион-ассоциированных белков, в частности глутамилцистеин лигазы, первого фермента в процессе синтеза глутатиона de novo, и ГР находятся под контролем NRF2 [46]. Поэтому мы исследовали активность NRF2 в ответ на ТГ в сочетании с ингибированием HIF1 либо без него. Исследование проводилось по нескольким направлениям: определяли ядерную локализацию NRF2 (рис. 4), количество общего глутатиона (рис. 5а) и ГР активность (рис. 5б).
Рис. 4.
в, г – Влияние ТГ и ТГ в сочетании с введением ингибитора HIF1 (ТГ + i) на площадь колокализации во 2 (в) и 5 (г) слое неокортекса через 1, 2 и 4 дня после сеанса ТГ и ТГ в сочетании с ингибированием HIF1. По оси абсцисс – время; по оси ординат – площадь колокализации (S), выраженная в пикселях. Данные представлены в виде среднего ± стандартная ошибка среднего. Белый столбик – контроль + ДМСО (инъекционный контроль), (n = 6); светло-серый столбик – контроль + ингибитор HIF1; серые столбики – крысы, подвергшиеся ТГ, темно-серые столбики – крысы, подвергшиеся ТГ в сочетании с ингибированием HIF1. Р-значение оценено с помощью непараметрического теста Крускала-Уоллиса с применением Данн теста в качестве post-hoc процедуры, где * – достоверные различия с контрольными значениями, ** – достоверные различия с группой Контроль + ингибитор HIF1 на соответствующей временной точке, *** – достоверные различия между группами ТГ и ТГ + ингибитор HIF1 на соответствующей временной точке при p < 0.05. а, б – репрезентативные фотографии для 2 (а) и 5 (б) слоя неокортекса. Маркер – 50 мкм.
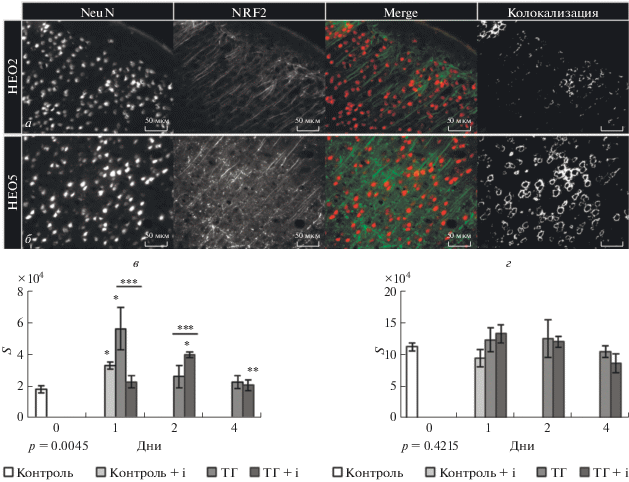
Рис. 5.
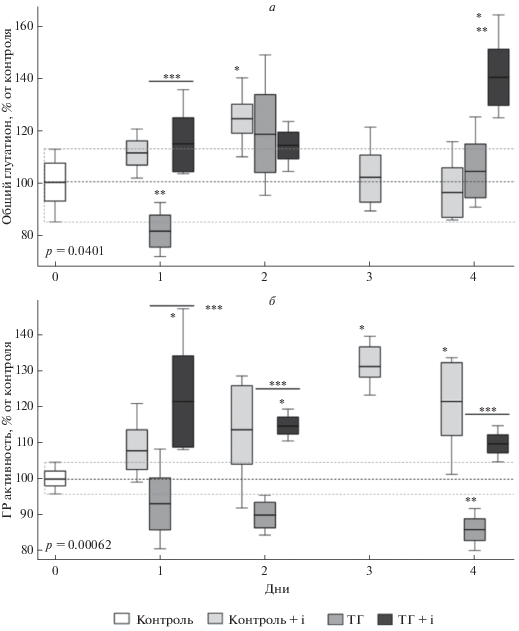
Влияние ТГ и ТГ в сочетании с введением ингибитора HIF1 (ТГ + i) на количество общего глутатиона (а) и глутатион редуктазную активность (б) в неокортексе через 1, 2 и 4 дня после сеанса ТГ и ТГ в сочетании с ингибированием HIF1. * Различия достоверны по отношению к контролю. ** Различия достоверны по отношению к группе Контроль + i. *** Различия достоверны между группами ТГ и ТГ + i, p < 0.05.
Через день после ввода ингибитора контрольным животным наблюдалось увеличение ядерной нейрональной локализации NRF2 во втором слое сенсомоторной коры (рис. 4в). Количество NRF2 повышалось в ответ на ТГ через день после воздействия, в то время как предъявление ТГ в сочетании с ингибированием HIF1 вызывало увеличение количества ядерного NRF2 через двое суток после реоксигенации. Как после ТГ, так и после ТГ на фоне ингибирования HIF1 количество ядерного NRF2 возвращалось к контрольным значениям на четвертый день реоксигенации. Для пятого слоя неокортекса не обнаружено достоверных отличий количества ядерного NRF2 от контроля (рис. 4г). Помимо ядерной локализации NRF2 была выявлена также иммунореактивность к NRF2 в нейрональных отростках (рис. 4а, 4б). Этот феномен вероятно связан с цитоплазматическим заякориванием комплекса NRF2-Keap1 на актиновый цитоскелет [47]. Keap1 является сенсором клеточного окислительно-восстановительного статуса и в физиологических условиях секвестрирует NRF2 и направляет его на протеасомную деградацию [48]. В условиях активации NRF2-зависимого пути блокируется не связь NRF2-Keap1, а создание убиквитин-лигазного комплекса, а также происходит синтез новых молекул NRF2. Существующая на данный момент модель подразумевает насыщение цитоплазматического Keap1 молекулами NRF2 в условиях окислительного или электрофильного стресса. Свободный же NRF2 транслоцируется в ядро и активирует транскрипцию генов-мишеней [49]. Нами также была отмечена преимущественно нейрональная локализация данного ТФ (неопубликованные данные об отсутствии колокализации с астроглиальным маркером GFAP), что находит как поддержку [50], так и опровержение [51] в литературе.
Далее мы определяли содержание общего глутатиона, которое также может косвенно отражать активность NRF2 поскольку, как говорилось выше, этот ТФ участвует в регуляции содержания ферментов синтеза глутатиона. Согласно полученным нами данным, уровень общего глутатиона достоверно не менялся под воздействием ТГ на всем протяжении эксперимента. Введение ингибитора HIF1 приводило к росту содержания трипептида ко второму дню эксперимента, затем его количество снижалось до исходных значений. Сочетание ингибирования HIF1 и ТГ также приводило к росту количества глутатиона, которое достоверно возрастало на 1 и 4 день после воздействия (рис. 5а). Эти данные хорошо согласуются с изменением содержания тиоловых групп и указывают на изменение редокс статуса клетки в сторону более восстановленного.
Эффективность глутатион-зависимой антиоксидантной системы зависит, в том числе, и от активности фермента ГР, ген которой, в свою очередь, также является хорошо известной транскрипционной мишенью NRF2. Воздействие ТГ, как и в случае с количеством общего глутатиона, не влияло на ГР активность в течение всего времени эксперимента. В то же время ГР активность повышалась в группе, подвергшейся ингибированию HIF1 на третий день от начала эксперимента, тогда как ТГ в сочетании с ингибированием HIF1 приводило к увеличению активности фермента уже на первый день после воздействия (рис. 5б).
Данные о колокализации NRF2 и NeuN должны характеризовать его транслокацию в ядро и, следовательно, свидетельствовать об его активации. Однако результаты, полученные в ходе эксперимента, неоднозначны, и должны быть оценены в комплексе с косвенными данными о функциональной активности NRF2, в нашем случае – об уровне общего глутатиона и ГР активности. В условиях ингибирования HIF1 оба эти показателя демонстрировали положительную динамику, соответствующую активации NRF2: количество общего глутатиона возрастало ко вторым суткам после воздействия, а ГР активность – к третьим. Показано, что для NRF2-зависимых мишеней характерно увеличение количества белкового продукта через день после активации ТФ [50]. Таким образом, ингибирование HIF1 имеет отложенный активирующий эффект на NRF2. В свою очередь ТГ в сочетании с ингибированием HIF1 приводило к быстрому повышению ГР активности, что заметно уже через 1 день после воздействия. В данном случае активация NRF2 скорее всего вызвана сенсором проокислительной среды Keap1 [48, 49], характерной для реоксигенации.
Ранее нами был показан HIF1-зависимый эффект гипоксического прекондиционирования, активация HIF1 приводила к более высокой устойчивости животных к последующей повреждающей гипоксии [14]. Одним из возможных объяснений этого феномена служило предотвращение резкого подъема уровня HIF1α у непрекондиционированных животных на первый день после ТГ [52], что предполагает участие HIF1 в формировании постгипоксической патологии. В нашем исследовании ингибирование HIF1 также приводит к прекондиционирующему эффекту через непрямую активацию NRF2-зависимой транскрипции.
ЗАКЛЮЧЕНИЕ
Результаты исследования свидетельствуют о взаимосвязи HIF1- и NRF2-зависимых процессов в постгипоксический период. Данный эффект реализуется на уровне совместной регуляции глутатион-зависимой антиоксидантной защиты, где ГП активность зависит от активности обоих ТФ, а синтез глутатиона и ГР реакция – от активности NRF2. Таким образом, исследуемые ТФ выполняют взаимодополняемые функции: HIF1 обеспечивает адаптацию к долгосрочной гипоксии, в то время как NRF2 ответственен за срочную защитную реакцию на острую гипоксию и последующую реоксигенацию. Детальное изучение взаимодействия между этими ТФ необходимо для дальнейшей расшифровки механизмов патологических и адаптивных реакций мозга на гипоксию и реоксигенацию.
БЛАГОДАРНОСТИ
Работа выполнена при финансовой поддержке Программы фундаментальных научных исследований государственных академий на 2014–2020 гг. (ГП-14, раздел 65), а также поддержана грантами РФФИ № 16-34-00027; 16-04-00987 и Программой поддержки биоресурсных коллекций ФАНО. Часть работ выполнена на базе ресурсных центров “Обсерватория экологической безопасности” и “Развитие молекулярных и клеточных технологий” Научного парка СПбГУ.
Вклад авторов. КС, ОВ, ОГ ответственны за разработку гипотезы, планирование экспериментов и написание статьи. КС и ОВ осуществляли эксперименты. КС и АЛ осуществляли статистический анализ.
Список литературы
Bolaños J.P., Almeida A., Moncada S. // Trends Biochem. Sci. 2010. V. 35. № 3. P. 145–149.
Mächler P., Wyss M.T., Elsayed M., Stobart J., Gutierrez R., Von Faber-Castell A., Kaelin V., Zuend M., San Martín A., Romero-Gómez I. et al. // Cell Metab. 2016. V. 23. № 1. P. 94–102.
Duran J., Saez I., Gruart A., Guinovart J.J., Delgado-García J.M. // J. Cereb. Blood Flow Metab. 2013. V. 33. № 4. P. 550–556.
Atwell D., Buchan A.M., Charpak S., Laurirzen M., MacVicar B.A., Newman E.A. // Nature. 2010. V. 468. P. 232–243.
Vetrovoy O. V, Rybnikova E.A., Glushchenko T.S., Baranova K.A., Samoilov M.O. // Neurochem. J. 2014. V. 8. № 2. P. 103–108.
Shih A.Y. // J. Neurosci. 2005. V. 25. № 44. P. 10321–10335.
Fernandez‑Fernandez S., Almeida A., Bolaños J.P. // Biochem. J. 2012. V. 443. P. 3–11.
Liu W., Shen S., Zhao X., Chen G. // Int. J. Biochem. Mol. Biol. 2012. V. 3. № 2. P. 165–178.
Bernaudin M., Nedelec A.-S., Divoux D., MacKenzie E.T., Petit E., Schumann-Bard P. // J. Cereb. Blood Flow Metab. 2002. V. 22. № 4. P. 393–403.
Sun Y., Chen X., Zhang X., Shen X., Wang M., Wang X., Liu W.-C., Liu C.-F., Liu J., Liu W. et al. // Front. Mol. Neurosci. 2017. V. 10. № August. P. 1–15.
Chavez J.C., Agani F., Pichiule P., LaManna J.C. // J. Appl. Physiol. 2000. V. 89. № 5. P. 1937–1942.
Dengler V.L., Galbraith M.D., Espinosa J.M. // Crit. Rev. Biochem. Mol. Biol. 2014. V. 49. № 1. P. 1–15.
Ann Sheldon R., Lee C.L., Jiang X., Knox R.N., Ferriero D.M. // Pediatr. Res. 2014. V. 76. № 1. P. 46–53.
Rybnikova E., Samoilov M. // Front. Neurosci. 2015. V. 9. P. 1–11.
Huang Y., Li W., Su Z.-yuan, Kong A.N.T. // J. Nutr. Biochem. 2015. V. 26. № 12. P. 1401–1413.
Wild A.C., Moinova H.R., Mulcahy R.T. // J. Biol. Chem. 1999. V. 274. № 47. P. 33627–33366.
Ji L., Li H., Gao P., Shang G., Zhang D.D., Zhang N., Jiang T. // PLoS One. 2013. V. 8. № 5. P. 1–12.
Singh A., Wu H., Zhang P., Happel C., Ma J., Biswal S. // Mol. Cancer Ther. 2010. V. 9. № 8. P. 2365–2376.
Hayes J.D., McMahon M. // Trends Biochem. Sci. 2009. V. 34. № 4. P. 176–188.
Thimmulappa R.K., Mai K.H., Srisuma S. // Cancer Res. 2002. V. 62. P. 5196–5203.
Shih A.Y., Johnson D. A., Wong G., Kraft A.D., Jiang L., Erb H., Johnson J. A., Murphy T.H. // J. Neurosci. 2003. V. 23. № 8. P. 3394–3406.
Кислин М.С., Тюлькова Е.И., Самойлов М.О. // Нейрохимия. 2010. V. 27. № 2. P. 144–149.
Rybnikova E., Vataeva L., Tyulkova E., Gluschenko T., Otellin V., Pelto-Huikko M., Samoilov M.O. // Behav. Brain Res. 2005. V. 160. № 1. P. 107–114.
Vataeva L.A., Tyul’kova E.I., Samoilov M.O. // Dokl. Biol. Sci. 2004. V. 395. № 3. P. 109–111. http://www.ncbi.nlm.nih.gov/pubmed/15255136.
Kislin M.S., Stroev S.A., Glushchenko T.S., Tiul’kova E.I., Pelto-Huikko M., Samoilov M.O. // Biomed. Khim. 2013. V. 59. № 6. P. 673–681.
Rybnikova E., Vorobyev M., Pivina S., Samoilov M. // Neurosci. Lett. 2012. V. 513. № 1. P. 100–105.
Vetrovoy O., Tulkova E., Sarieva K., Kotryahova E., Zenko M., Rybnikova E. // Neurosci. Lett. 2017. V. 639. P. 49–52.
Vetrovoi O.V., Rybnikova E.A., Glushchenko T.S., Samoilov M.O. // Neuroscience and Behavioral Physiology. 2015. V. 45. № 4. P. 367–370.
Segura C., Bandres E., Troconiz I.F., Saya O., Renedo M.J., Garrido M.J. // Pharm. Res. 2004. V. 21. № 4. P. 567–573.
Rapisarda A., Zalek J., Hollingshead M., Braunschweig T., Uranchimeg B., Bonomi C.A., Borgel S.D., Carter J.P., Hewitt S.M., Shoemaker R.H. et al. // Cancer Res. 2004. V. 64. № 19. P. 6845–6848.
Rybnikova E., Sitnik N., Gluschenko T., Tjulkova E., Samoilov M.O. // Brain Res. 2006. V. 1089. № 1. P. 195–202.
Rodrigues G. de L.A., Pellegrino A. de I. // In: General Neurochemical Techniques, 1986. P. 568.
Prior R.L., Wu X., Schaich K. // J. Agric. Food Chem. 2005. V. 53. № 10. P. 4290–4302.
Akerboom T.P.M., Sies H. // In: Methods in Enzymology. V. 77. Academic Press, 1981. P. 373–381.
Levander O.A., Smith A.D. // In: Methods in Enzymology. Academic Press, 2002. P. 113–121.
Costa L.G., Hogston E., Lawrence D.A., Reed D.J. // In: Current Protocols in Toxicology. John Wiley & Sons, 2005. P. 1141–1227.
Folch J., Lees M., Sloane G.H. // J. Biol. Chem. 1957. V. 226. № 1. P. 497–509.
Bidlack W.R., Tappel A.L. // Lipids. 1973. V. 8. № 4. P. 203–207.
Bartlett R. // J. Biol. Chem. 1959. V. 234. № 3. P. 449–458.
Dunn O.J. // J. Am. Stat. Assoc. 1961. V. 56. № 293. P. 52–64.
Ray P.D., Huang B.W., Tsuji Y. // Cell. Signal. 2012. V. 24. № 5. P. 981–990.
Galkina O.V. // Neurochem. J. 2013. V. 7. № 2. P. 89–97.
Deponte M. // Biochim. Biophys. Acta. 2013. V. 1830. P. 3217–3266.
Huang Y., Fang W., Wang Y., Yang W., Xiong B. // Int. J. Mol. Med. 2012. V. 29. № 5. P. 906–912.
Brigelius-Flohé R., Maiorino M. // Biochim. Biophys. Acta. 2013. V. 1830. № 5. P. 3289–3303.
Harvey C.J., Thimmulappa R.K., Singh A., Blake D.J., Ling G., Wakabayashi N., Fujii J., Myers A., Biswal S. // Free Radic. Biol. Med. 2009. V. 46. № 4. P. 443–453.
Liu H., Colavitti R., Rovira I.I., Finkel T. // Circ. Res. 2005. V. 97. № 10. P. 967–974.
Fukutomi T., Takagi K., Mizushima T., Ohuchi N., Yamamoto M. // Mol. Cell. Biol. 2014. V. 34. № 5. P. 832–846.
Zhang D.D. // Drug Metab. Rev. 2006. V. 38. № 4. P. 769–789.
Tanaka N., Ikeda Y., Ohta Y., Deguchi K., Tian F., Shang J., Matsuura T., Abe K. // Brain Res. 2011. V. 1370. P. 246–253.
Jimenez-Blasco D., Santofimia-Castaño P., Gonzalez A., Almeida A., Bolaños J. // Cell Death Differ. 2015. V. 22. P. 1877–1889.
Rybnikova E.A., Baranova K.A., Gluschenko T.S., Vetrovoy O., Sidorova M., Portnichenko V.I. // Int. J. Physiol. Pathophysiol. 2015. V. 6. № 1. P. 1–11.
Дополнительные материалы отсутствуют.