

Нейрохимия, 2019, T. 36, № 4, стр. 275-291
Возможный механизм появления ночных кошмаров при посттравматическом стрессовом расстройстве и подходы к их предотвращению
И. Г. Силькис
Федеральное государственное бюджетное учреждение науки Институт высшей нервной деятельности и нейрофизиологии Российской академии наук
Москва, Россия
Поступила в редакцию 09.01.2019
После доработки 08.04.2019
Принята к публикации 11.04.2019
Аннотация
Выдвигается гипотеза, что в основе появления ночных кошмаров при посттравматическом стрессовом расстройстве (ПТСР) лежит модификация синаптической передачи в нейронных цепях, каждая из которых содержит одну из следующих структур: область зрительной коры, префронтальную кору (ПфК), базолатеральную миндалину (БЛМ), гиппокамп и связанные с ними ядра таламуса и базальных ганглиев (БГ). Появлению сновидений способствует индукция длительной потенции возбудительных входов к связанным со зрительной корой стрионигральным клеткам и длительной депрессии входов к стриопаллидарным клеткам, дающим начало соответственно прямому и непрямому пути через БГ. Это приводят к синергичному растормаживанию со стороны выходных ядер БГ определенных групп нейронов в таламусе и формированию в связанных с ними областях зрительной коры и гиппокампа паттернов активности, отображающих содержание сновидений. Появление в сновидениях негативно окрашенных эпизодов является следствием повышения при ПТСР активности БЛМ, сигналы от которой суммируются с сигналами из гиппокампа и ПфК на нейронах вентрального стриатума. Из механизма следует, что для предотвращения ночных кошмаров необходимо индуцировать длительную депрессию на возбудительных входах к нейронам лимбических структур и к стрионигральным клеткам. Нежелательно воздействовать на рецепторы на стриопаллидарных клетках, чтобы одновременно не индуцировать длительную потенциацию на возбудительных входах к нейронам ПфК, БЛМ и гиппокампа, на которых располагаются рецепторы тех же типов. С учетом известных данных о типах рецепторов на нейронах стриатума, из сформулированных нами ранее правил модуляции синаптической передачи следует, что предотвращению ночных кошмаров могут способствовать антагонисты дофаминовых Д1 рецепторов и альфа1 адренорецепторов, а также агонисты глюкокортикоидных, серотониновых 5-НТ1В и каннабиноидных СВ1 рецепторов. Использовать агонисты минералокортикоидных и серотониновых 5-НТ2А рецепторов, а также антагонисты альфа2 адренорецепторов и дофаминовых Д2 рецепторов (входящих в состав некоторых антидепрессантов и антипсихотических препаратов) нежелательно во избежание побочных эффектов. Воздействие на рецепторы должно быть кратковременным и применяться только перед сном, чтобы не ухудшать двигательную активность и сенсорное восприятие в состоянии бодрствования. Следствия предлагаемого механизма согласуются с известными результатами клинических исследований.
Принятые сокращения:
БГ – базальные ганглии;
БЛМ – базолатеральная миндалина;
ГКР – глюкокортикоидные рецепторы;
ГП – гиппокамп;
ДД – длительная депрессия эффективности возбудительных входов к нейрону;
ДП – длительная потенциация эффективности возбудительных входов к нейрону;
МКР – минералокортикоидные рецепторы;
ПС – парадоксальный сон (сон с быстрыми движениями глаз);
ПТСР – посттравматическое стрессовое расстройстве;
ПфК – префронтальная кора;
ПЯ – прилежащее ядро (вентральный стриатум);
С-Н – стрионигральные шипиковые клетки;
С-П – стриопаллидарные шипиковые клетки.
Ночные кошмары являются отличительным признаком посттравматического стрессового расстройства (ПТСР) [1]. Это заболевание диагностируют у 5–20% участников войн в Ираке и Афганистане. В зависимости от тяжести заболевания у пациентов с ПТСР ночные кошмары появляются в 19–71% случаев [2]. Кроме того, у 70% пациентов с ПТСР имеются проблемы со сном [3], в 70–91% случаев имеются трудности с засыпанием [2] и они часто страдают бессонницей [4]. Симптомы этого заболевания проявляются и в тенденции к снижению времени нахождения в состоянии парадоксального сна (ПС) [5], в котором появляются сновидения. Исследования показали, что быстрое пробуждение и бессонница коррелируют с нарушениями дыхания и движений ног во время сна, а также с учащением сердцебиений при пробуждении, которые чаще наблюдаются у пациентов с ПТСР, чем у популяции в целом [2, 6]. Хроническое ограничение ПС в молодом возрасте, в свою очередь, приводит к увеличению уровня норадреналина в миндалине и вентральном гиппокампе (ГП), нарушает физическое развитие и способствует тревожному поведению [7]. Однако имеются данные и об увеличении времени нахождения в состоянии ПС при ПТСР или об отсутствии изменений. Так, у некоторых пациентов с ПТСР увеличение времени нахождения в состоянии ПС ассоциировалось с тревожностью и самоощущением состояния депрессии [5]. Было высказано предположение, что ПТСР может быть связано с вызванным травмой первоначальным уменьшением ПС, которое сопровождается его увеличением со временем [8]. Показано, что ПС может усиливаться или ослабляться в зависимости от специфических характеристик стресса или экспериментальных условий, в которой стимул вызывает страх [9]. Обычно изменения ПС не позволяют предсказать выраженность памяти о стрессе, вызванном предъявлением только контекста, в котором раньше подавался стрессовый стимул [10]. Тот факт, что ПС усиливается после контролируемого стресса, а не после не контролируемого, согласуется с гипотезой о том, что ПС важен для формирования эмоциональной памяти [11–14]. Возможно, такие отличительные симптомы ПТСР как ночные кошмары и бессонница, связаны с желанием уменьшить переживания страха во сне. Существует мнение, что депривация ПС может снизить память о воздействии стресса [15]. В результате этого может развиваться бесконечный замкнутый круг, когда нарушения сна из-за ночных кошмаров увеличивают риск появления ПТСР [16]. В свою очередь, ПТСР ведет к увеличению фрагментации сна, увеличению частоты появления ночных кошмаров. Поскольку симптомы ПТСР могут усиливаться с течением времени, предполагают, что продолжающиеся нарушения сна могут поддерживать и усугублять проявления болезни [17]. На основании обзора литературных данных был сделан вывод, что нарушения ПС могут представлять фактор риска для появления психических заболеваний [18], а ночные кошмары при ПТСР могут являться фактором риска для суицида [19]. В последние годы увеличивается число исследований, направленных на понимание роли сна в развитии и течении ПТСР, а также на поиск способов устранения симптомов ПТСР [20]. Обращено внимание клиницистов на то, что поскольку хронические нарушения сна, связанные с ночными кошмарами, могут влиять на эффективность лечения ПТСР, только направленное лечение может ускорить восстановление после ПТСР [18]. Из вышеизложенного следует, что устранение ночных кошмаров может являться одним из эффективных способов лечения ПТСР. Однако терапевтические методы для ослабления нарушений сна при ПТСР ограничены [16], поэтому необходим поиск новых подходов.
Очевидно, что для предотвращения ночных кошмаров, которые представляют собой разновидность сновидений, важно представлять не только механизмы появления сновидений, но и механизмы включения в них ночных кошмаров. Гипотетический механизм появления сновидений был выдвинут нами ранее [21]. Предположено, что в их возникновении участвуют нейронные цепи кора–базальные ганглии–таламус–кора (К–БГ–Т–К), включающие зрительные области коры и связанные с ними ядра таламуса и базальных ганглиев (БГ), а также лимбические цепи, включающие префронтальную кору (ПфК), миндалину, гиппокамп (ГП), вентральную часть стриатума – прилежащее ядро (ПЯ), таламические ядра – медиодорзальное и реуниенс. На функционирование указанных цепей существенное влияние оказывают различные нейромодуляторы. Современные исследования указывают на то, что симптомы ПТСР связаны с патологией адренергической, дофаминергической, ееротонинергической, кортикоидной, глутаматергической, ГАМКергической, каннабиноидной, опиоидной и других нейротрансмиттерных и нейроэндокринных систем [1, 22, 23]. В течении ряда лет нами анализировались особенности влияний на функционирование указанных цепей таких нейромодуляторов, как дофамин, аденозин, ацетилхолин, серотонин, опиоиды и каннабиноиды [24–30], но не анализировались влияния норадреналина и кортикоидов на эффективность возбудительных входов в стриатум. Хотя в норме при ПС норадреналин не выделяется, но у ветеранов войн с ПТСР его концентрация при ПС увеличена [31], так что его влияние необходимо учитывать. Согласно предложенному нами механизму возникновения сновидений, влияние нейромодуляторов на их содержание может реализоваться за счет изменения условий для индукции длительной потенциации и длительной депрессии эффективности возбудительных входов (ДП и ДД) к шипиковым нейронам стриатума, а также к нейронам ПфК, ГП и БЛМ [21].
Нам не удалось найти в литературных источниках каких-либо теорий, которые позволяли бы объяснить механизмы появления сновидений и, в частности, механизмы включения в них ночных кошмаров. В настоящей работе выдвинут гипотетический нейронный механизм появления ночных кошмаров при ПТСР. На его основе предположено, что для предотвращения ночных кошмаров следует создать условия, при которых будет затруднено появление сновидений в состоянии ПС. Целью настоящей работы являлся анализ возможностей предотвращения ночных кошмаров при ПТСР за счет изменения знака модификации (ДП или ДД) эффективности входов к нейронам структур, участвующих в возникновении сновидений. Необходимой направленности изменений можно добиться определенным воздействием на рецепторы, расположенные на указанных нейронах.
ГИПОТЕТИЧЕСКИЙ МЕХАНИЗМ ПОЯВЛЕНИЯ НОЧНЫХ КОШМАРОВ
Гипотетический механизм возникновения сновидений, выдвинутый нами ранее. Нами было предложено, что для возникновения сновидений необходимо ослабить торможение со стороны БГ холинергических нейронов педункулопонтийного и латеродорзального тегментального ядер, а также нейронов нижнего коленчатого тела, чья активность способствует генерации понто-геникуло-оципитальных волн и возникновению ПС [21]. Упрощенная схема организации связей между нейронами структур, участвующих в появлении сновидений, представлена на рис. 1. С целью упрощения, на этом рисунке не представлены педункулопонтийное и латеродорзальное тегментальное ядра, а также субталамическое ядро, возбуждающее эти ядра и выходные ядра БГ. В отсутствие входов из сетчатки, зрительные образы в сновидениях являются результатом уменьшения торможения со стороны БГ (растормаживания) нейронов таламических ядер, проецирующихся в зрительные области коры, в ПфК и в ГП. Затем сигналы из коры, ГП и миндалины поступают обратно во входную структуру БГ – стриатум. Нейромодуляторы оказывают существенное влияние на функционировании БГ, поскольку воздействуют на рецепторы, расположенные на стрионигральных (С-Н) и стриопаллидарных (С-П) клетках. При разработке модели были использованы известные данные о расположении рецепторов разных типов на нейронах стриатума, а также сформулированные нами ранее правила модуляции эффективности возбудительных входов к нейронам разных структур (табл. 1) [32, 33]. При выделении в стриатуме дофамина одновременно активируются Д1 рецепторы, располагающиеся преимущественно на С-Н нейронах, дающих начало прямому растормаживающему пути через БГ, и Д2 рецепторы, располагающиеся преимущественно на С-П нейронах, дающих начало непрямому ингибирующему пути через БГ (рис. 2). В результате последующей индукции ДП на С-Н клетках и ДД на С-П клетках должно синергично усилиться растормаживание со стороны выходных ядер БГ определенной группы нейронов таламуса и их клеток-мишеней в разных областях коры и гиппокампа. Эти группы являются нейронным отображением в сновидениях зрительных образов и связанного с ними контекста. Модификации синапсов способствуют характерные для ПС понто-геникуло-оципитальныe волны, поскольку синхронизация активности в разных структурах мозга облегчает выполнение правила Хебба (совпадение во времени активности пре- и постсинаптических клеток). Упрочение модификации синапсов вследствие повторных прохождений активности через БГ может лежать в основе участия БГ в консолидации следов памяти при ПС [21].
Рис. 1.
Упрощенная схема топически организованных нейронных цепей, участвующих в появлении ночных кошмаров. Их предотвращению способствует длительная депрессия (ДД) возбудительных входов к стрионигральным нейронам под действием антагонистов дофаминовых Д1 и α1 адренорецепторов и последующее усиление ингибирования нейронов таламуса. БЛМ – базолатеральная миндалина; Гипп. – гиппокамп; мПфК – медиальная префронтальная кора; ПЯ – прилежащее ядро (вентральный стриатум); ЧВк и ЧВр – компактная и ретикулярная части черного вещества, соответственно; ВП – вентральный паллидум; ВПП – вентральное поле покрышки; ГП – голубое пятно. Тал. –таламус; 1, 2, 3, 4 – таламические ядра: медиодорзальное, реуниенс, зрительные, вентролатеральное, соответственно. Кружки со штриховкой с наклоном вправо – стрионигральные шипиковые клетки; (стриопаллидарные клетки не представлены); светлые кружки – возбудительные клетки; черные кружки – ГАМКергические клетки; кружки с сетчатой штриховкой – дофаминергические клетки; серый кружок – норадренергическая клетка. Ядра базальных ганглиев обведены толстыми линиями. Стрелки одно- и двухсторонние – однонаправленные и реципрокные возбудительные связи; линии со стрелками – возбудительные входы; линии с ромбами – тормозные входы; штрих-пунктирные линии со стрелкой – дофаминергические входы; штрих-пунктирные линии с двумя точками со стрелкой – адренергические входы. Утолщенные и тонкие линии – сильные и слабые входы, соответственно.
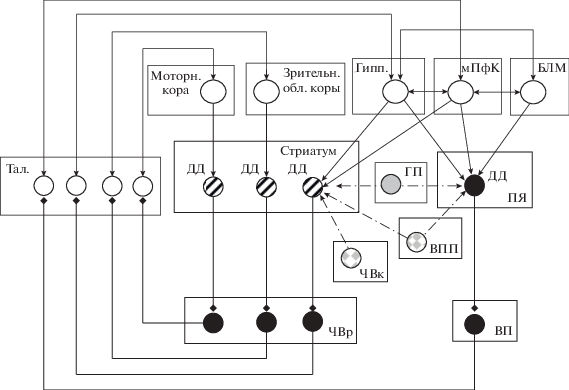
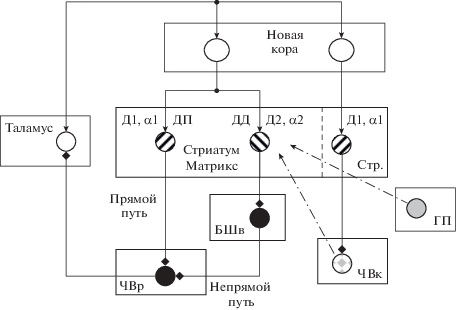
Рис. 2.
Растормаживание активности нейронов таламуса и коры со стороны базальных ганглиев под действием дофамина и норадреналина. БШв – внутренняя часть бледного шара. Кружки со штриховкой с наклоном влево – стриопаллидарные шипиковые клетки. Стр. – стриозомы. Пунктирной чертой отделена область стриозом стриатума. Д1 и Д2 – дофаминовые рецепторы; α1 и α2 – адренорецепторы; ДП – длительная потенциация корково-стриатных входов. Другие нейромодуляторные входы и чувствительные к ним рецепторы не представлены с целью упрощения. Остальные обозначения как на рис. 1.
Хотя принято считать, что во время ПС норадренергические и серотонинергические нейроны не разряжаются, в отличие от дофаминергических [34], однако остаточное количество норадреналина в мозге имеется вследствие его диффузного экстрасинаптического выделения и отсутствия быстрого обратного захвата, причем это может приводить к ментальным изменениям во время сновидений [35]. При ПС больше активированы экстрастриарные области коры и относительно меньше активность нейронов в первичной зрительной коре [36, 37] (по-видимому, вследствие отсутствия внешних зрительных сигналов). Во время ПС активированные области зрительной коры шире, чем при нормальной зрительной стимуляции [38], что указывает на усиление их возбуждения в этой фазе сна. Причудливость сновидений была объяснена нами тем, что при характерном для состояния ПС изменении состава нейромодуляторов ослабляется эффективность передачи по трисинаптическому пути через ГП, но эффективной остается связь ГП с энторинальной корой, а через нее с другими областями коры [39]. В результате в сновидениях искажена эпизодическая информация, хранящаяся, по нашему предположнению, в нейронах разных полей ГП с возрастающей степенью обобщенности по мере продвижения по трисинаптическому пути [40]. Поскольку когерентность в ЭЭГ ассоциируют со связыванием в целостное событие пространственно разделенных, но объединенных во времени стимулов и поскольку во время ПС уменьшена корреляция между активностью во фронтальной коре и в сенсорных зонах коры [41], можно прлагать, что диссоциация активности в ЭЭГ тоже лежит в основе причудливости сновидений. В работе [42] также было высказано предположение, что ослабление связей между зрительными и фронтальными областями коры во время ПС лежит в основе большей гибкости когнитивных процессов при ПС, что отражается в причудливости и гиперассоциативности сновидений.
Вклад лимбических структур в содержание сновидений (сравнение нормы и состояния стресса). В предлагаемом нами механизме возникновения сновидений такие структуры, как миндалина, ГП, ПфК, а также связанная с ними лимбическая часть БГ, вносят существенный вклад в содержание сновидений [21]. Этот вклад зависит от результирующего воздействия на активность ПЯ конвергирующих входов из трех указанных структур (рис. 1). Из БЛМ поступает эмоционально значимая информация, вход из поля СА1/вентрального субикулюма обеспечивает поступление информации о контексте, а ПфК обеспечивает интегративное влияние [43]. В современных нейробиологических моделях предполагается, что миндалина и ПфК играют основную роль в процессах, связанных со стрессом [44]. Информация об эмоциональном состоянии передается от нейронов БЛМ также в ГП и ПфК, причем БЛМ связана с этими структурами реципрокно (рис. 1), как и с областями заднего мозга, нейроны которого запускают вызванную норадреналином реакцию на стресс [45]. Возможный механизм влияния на функционирование БГ динамического взаимодействия между БЛМ, вентральной частью ГП и ПфК также анализировался нами ранее [46]. Этот механизм базируется на известных данных о том, что входы в ПЯ из ГП, миндалины и ПфК организованы топически и могут модифицироваться. Реакции шипиковых клеток ПЯ зависят от степени их возбуждения со стороны нейронов БЛМ, ГП и ПфК, а также от того, в какой последовательности возбуждение поступает в ПЯ. Гиппокампальной формации отводят особую роль во влиянии на синаптическую интеграцию возбуждения в нейронах стриатума, поскольку сигналы из ГП может перевести шипиковую клетку ПЯ в верхнее деполяризованное состояние [47]. Показано, что поступление возбуждения из ГП способствует прохождению через ПЯ следующих за ним сигналов из ПфК [48] и БЛМ [49]. Совместная тетанизация входов в ПЯ из ГП и БЛМ приводит к ДП обоих входов [50]. Активация БЛМ, предшествующая активации ГП, вызывает в медиальной части ПЯ усиление ответа на стимуляцию ГП [51] и мПфК [52]. Эти данные указывают на существенный вклад БЛМ в обработку информации в нейронных цепях, включающих ГП и ПфК.
Известно, что в норме в состоянии ПС активность миндалины такая же, как при бодрствовании [53], но в процессах, связанных с эмоциями и памятью о страхе, активность миндалины и ГП во время ПС усиливается [54]. Полагают, что гипервозбудимость основных клеток БЛМ при стрессе может лежать в основе нарушений, характерных для ПТСР [45]. Показано, что вследствие высокой активности миндалины при стрессе усиливается выделение глутамата в синапсах, образованных аксонами нейронов БЛМ на нейронах ПфК и вентрального ГП [55, 56]. За счет последующего повышения активности нейронов ПфК и ГП должны усилиться и реакция шипиковых клеток ПЯ на возбуждение, поступающее из этих структур. Поэтому, высокая активность БЛМ и ГП при ПТСР может способствовать растормаживанию нейронов таламуса со стороны лимбической части БГ и появлению в сновидениях фрагментов связанных с травмой негативно окрашенных эпизодов, память о которых сохранилась в активности нейронов ГП и ПфК. Из предлагаемого механизма следует, что если ПТСР было вызвано сильным стрессом во время боевых действий, может возрасти вероятность воспроизведения в ночных кошмарах фрагментов эпизодов, связанных с войной. Действительно показано, что только у ветеранов войны с ПТСР в ночных кошмарах присутствовали боевые действия [57]. У ветеранов вьетнамской войны элементы боевых эпизодов встречались в сновидениях в 50% случаев [58]. Содержание ночных кошмаров повторялось и часто казалось точной копией стрессового события [59]. С точки зрения предлагаемого механизма, повторение содержания сновидений является следствием усиления модификации синапсов между определенными группами нейронов в цепях К–БГ–Т–К по мере повторяющейся циркуляции активности в этих цепях.
Некоторые эффекты, в которых участвует БЛМ, могут реализовываться через нейронные цепи, вовлеченные как в регуляцию ПС, так и в запоминание. Поэтому при поиске методов лечения ПТСР исследуют различные возможности угашения памяти о страхе. При этом в качестве мишеней воздействия рассматривают связанные с БЛМ вентромедиальную ПфК и ГП [60]. При обучении реагировать на стимул, вызывающий страх, выявлена синхронизация активности нейронов миндалины, ГП и коры [61]. Поскольку при такой синхронизации выполняется правило Хебба и улучшаются условия для модификации эффективности связей между нейронами лимбических структур, должно облегчаться и формирование следов памяти. На грызунах показано, что синхронизация в тета-ритме активности вентромедиальной ПфК и миндалины коррелирует с консолидацией памяти о страхе [62]. Эксперименты показали также, что если снизить связанную со страхом активность в таких структурах, как ГП, миндалина и вентромедиальная ПфК, а также ослабить связи между ними, базовый уровень ПС повысится [63]. Полагают, что измерение базового уровня ПС может являться неинвазивным биомаркером устойчивости к травме или развития ПТСР после травмы [63]. Поскольку при стрессе усиливается взаимодействие нейронов в цепи, включающей миндалину, ГП и мПфК, было высказано предположение, что должна существовать цепь, которая регулирует затухание страха при ПС, а прерывание последней приводит к появлению ночных кошмаров [64].
ПРЕДЛАГАЕМЫЕ СПОСОБЫ ПРЕДОТВРАЩЕНИЯ НОЧНЫХ КОШМАРОВ
Предлагаемый нами механизм возникновения ночных кошмаров при ПТСР позволяет выдвинуть гипотезу, что для их предотвращения необходимо таким образом изменить характер функционирования цепей К–БГ–Т–К, БЛМ–БГ–Т–БЛМ, ГП–БГ–Т–ГП, чтобы усилить ингибирование различных таламических ядер со стороны БГ. Это приведет к подавлению или исчезновению сновидений при ПС, а также к уменьшению возбуждения БЛМ, ГП и мПфК. Снижение активности нейронов в этих лимбических структурах может способствовать ослаблению и других симптомов ПТСР. То, что БГ влияют на активность нейронов в миндалине, вентромедиальной ПфК и ГП, показано в моделях ПТСР на животных [65]. Из предлагаемого механизма функционирования БГ следует, что для достижения поставленной цели необходимо таким образом воздействовать на рецепторы, расположенные на С-Н и С-П нейронах, чтобы индуцировать ДД и ДП на их возбудительных входах, соответственно. Поскольку однотипные рецепторы располагаются не только на С-П клетках, но и на нейронах ПфК, миндалины и ГП, одновременно с ДП на С-П клетках будет индуцироваться ДП на нейронах лимбических структур и вследствие этого повышаться их активность. Поэтому, чтобы не усилить симптомы ПТСР, нежелательно воздействовать на рецепторы тех типов, которые располагаются на С-П клетках. Из этого следует, что для предотвращения ночных кошмаров следует избирательно воздействовать только на рецепторы, расположенные на С-Н клетках (рис. 1). Кроме того, необходимо учитывать характер влияния подбираемых веществ на параметры ПС, поскольку прерывание сновидений не должно препятствовать нахождению в парадоксальной фазе сна.
Поиск веществ для воздействия на рецепторы на С-Н клетках базируется на сформулированных нами ранее унифицированных правилах модуляции сильных возбудительных входов к шипиковым клеткам [32], а также к нейронам ГП и новой коры [33]. Эти правила позволяют определить знак модификации (ДП или ДД) при активации рецептора, связанного с одним из трех типов G белков (табл. 1). Из правил модуляции следует, что для индукции ДД на С-Н клетках можно использовать агонисты рецепторов, связанных с Gi/0 белками, и/или антагонисты рецепторов, связанных с Gs и Gq/11 белками. Нежелательно использовать агонисты рецепторов, связанных с Gs и Gq/11 белками, и антагонисты рецепторов, связанных с Gi/0 белками, поскольку они могут способствовать индукции ДП не только на С-П клетках, но и на нейронах лимбических структур, проецирующихся в ПЯ. Следует также иметь в виду, что часть С-Н нейронов, расположенных в стриозомах дорзального стриатума и в ПЯ, проецируется в дофаминергические структуры – вентральное поле покрышки и компактную часть черного вещества. Поскольку шипиковые клетки являются ГАМКергическими, индукция на них ДД может способствовать ослаблению ингибирования дофаминергических клеток, увеличению их активности и повышению концентрации дофамина. Однако дофамин, воздействуя на Д1 рецепторы, может препятствовать индукции ДД на С-Н клетках и одновременно способствовать увеличению активности нейронов лимбических структур, что ухудшит условия для предотвращения ночных кошмаров.
Таблица 1.
Влияние нейромодуляторов на модификацию эффективности возбудительных входов к нейронам разных структур, участвующих в формировании сновидений
| Нейромоду-лятор | Типы рецепторов | Тип G-белка | *Влияние активации рецепторов на ДП возбудительных входов к нейронам | ||||
|---|---|---|---|---|---|---|---|
| **стриатума | ПфК | БЛМ | гиппокампа | ||||
| С-Н | С-П | ||||||
| Дофамин | Д1, Д5 | Gs | ↑ | ↑ | ↑ | ↑ | |
| Д2, Д4 | Gi/0 | ↓ | ↓ | ↓ | ↓ | ||
| Ацетилхолин (мускарин) | М1, М3 | Gq/11 | ↑ | ↑ | ↑ | ↑ | |
| М2; М4 | Gi/0 | ↓ | ↓ | ↓ | ↓ | ||
| Норадреналин | α1 | Gq/11 | ↑ | ↑ | ↑ | ↑ | |
| α2 | Gi/0 | ↓ | ↓ | ↓ | ↓ | ||
| β | Gs | ↑ | ↑ | ↑ | |||
| Аденозинин | А1 | Gi/0 | ↓ | ↓ | ↓ | ↓ | |
| А2А | Gq/11 | ↑ | ↑ | ↑ | ↑ | ||
| Опиоиды | κ | Gs | ↓ | ||||
| δ | Gs | ↓ | |||||
| μ | Gs | ↓ | |||||
| Кортикоиды | ГКР | Gi/0 | ↓ | ↓ | ↓ | ↓ | |
| МКР | Gs | ↑ | ↑ | ↑ | ↑ | ||
| Каннабиноиды | СВ1 | Gi/0 | ↓ | ↓ | |||
| Серотонин | 5-НТ1В | Gi/0 | ↓ | ↓ | ↓ | ↓ | |
| 5-НТ2А | Gq/11 | ↑ | ↑ | ↑ | ↑ | ||
* Инактивация рецепторов приводит к противоположному изменению. ** Эффект развивается в случае “сильного” возбудительного входа, позволяющего открыть НМДА каналы на шипиковом нейроне. С-Н – стрионигральные нейроны; С-П – стриопаллидарные нейроны. ДП – длительная потенциация; ДД – длительная депрессия; Gs – цАМФ↑; Gq/11 – ИнФ3/диацилглицерол↑; Gi/0 – цАМФ↓. Стрелки вверх и вниз – увеличение и уменьшение, соответственно.
Нами было предположено, что одни те же принципы характерны для функционирования сенсорных (например, зрительных) и моторных цепей К–БГ–Т–К [21]. Поэтому не исключено, что если при ПТСР ночные кошмары являются следствием увеличения растормаживания нейронов таламуса со стороны БГ, то и в моторных цепях растормаживание будет увеличено, что отразится в повышенной двигательной активности при ПС. Действительно при ПТСР чаще наблюдался синдром беспокойных ног [2, 6, 66]. При исследовании 214 ветеранов вьетнамской войны было обнаружено, что при ПТСР (108 человек) ночные кошмары наблюдались в 91% случаев, а синдром беспокойных ног – в 45%; в отсутствие ПТСР ночные кошмары наблюдались в 29% случаев, а синдром беспокойных ног – в 25% [67]. После травматического воздействия поведенческие нарушения при ПС могут проявляться также в виде сомнамбулизма или движений, характерных для драки [68]. Мы полагаем, что при системном введении веществ, препятствующих появлению ночных кошмаров, двигательная активность в состоянии ПС также будет подавляться.
АНАЛИЗ ВЛИЯНИЙ НЕЙРОМОДУЛЯТОРОВ НА АКТИВНОСТЬ НЕЙРОННЫХ ЦЕПЕЙ, УЧАСТВУЮЩИХ В ПОЯВЛЕНИИ НОЧНЫХ КОШМАРОВ С ЦЕЛЬЮ ПОИСКА ПРЕПАРАТОВ ДЛЯ ИХ ПРЕДОТВРАЩЕНИЯ
Влияние дофамина. В нашей предшествующей работе [32] приведены известные из литературы данные о расположении чувствительных к дофамину и ацетилхолину рецепторов на С-Н и С-П клетках (табл. 1). Предлагаемый гипотетический механизм позволяет предположить, что для предотвращения ночных кошмаров могут быть полезны антагонисты Д1 рецепторов, поскольку они должны способствовать индукции ДД на С-Н клетках (табл. 1). В согласии с предлагаемым механизмом показано, что подавление цАМФ в экспрессирующих динорфин нейронах ПЯ, т.е. С-Н клетках, оказывает такой же эффект, как антидепрессанты [69]. Поскольку показано, что воздействие на Д1 рецепторы способствует увеличению активности нейронов ПфК и поля СА1 ГП [70], можно полагать, что антагонисты Д1 рецепторов будут препятствовать ДП возбуждения этих нейронов. С учетом данных о том, что увеличенная активация Д1 рецепторов в ПфК может вносить вклад в уязвимость к различным психиатрическим расстройствам [71], есть основание полагать, что антагонисты Д1 рецепторов позволят ослабить такие расстройства или их предотвратить.
Влияние каннабиноидов. Известно, что стресс стимулирует выделение эндоканнабиноидов, в частности, в БЛМ [72]. Из нашей предшествующей работы следует, что воздействие каннабиноидов на СВ1 рецепторы на нейронах стриатума может привести к усилению ингибирования таламических ядер со стороны БГ [25]. Поэтому каннабиноиды могут препятствовать появлению ночных кошмаров. Кроме того показано, что каннабиноиды ослабляют тревожность [73], а их введение в БЛМ или ГП способствует затуханию условно-рефлекторной реакции на стимул, вызывающий страх [74]. По-видимому, эффект связан с тем, что активация СВ1 рецепторов облегчает индукцию ДД на нейронах БЛМ и ГП (табл. 1). Следует отметить, что хотя введение агонистов каннабиноидных рецепторов уменьшает как глутаматергическую, так и ГАМКергическую передачу в латеральной миндалине, эффект от воздействия каннабиноидов на эффективность возбуждения превалирует над воздействием на эффективность торможения, в результате чего уменьшается активность основных клеток [75]. Аналогичный результат продемонстрирован и для поля СА1 гиппокампа [76]. Отмечено, что агонист СВ1 рецепторов способствовал индукции ДД на пирамидных клетках поля СА1, вызванных стимуляцией не только коллатералей Шаффера, но иперфорантных волокон [77]. Эти данные указывают на ослабление под действием каннабиноидов связей ГП через энторинальную кору с другими областями коры, что должно уменьшать вероятность появления в сновидениях фрагментов эпизодов, связанных с травмой.
В согласии с предложенным нами гипотетическим механизмом показано, что при использовании агониста СВ1 рецепторов увеличивается концентрация дофамина [78]. Но с другой стороны, имеются данные о том, что после локального введения агониста Д2 рецепторов выброс эндоканнабиноида анандамида возрастал в 8 раз [79]. Поэтому, если в результате воздействия увеличенной концентрации дофамина на Д2 рецепторы повысится концентрация эндоканнобиноида, то последующее увеличение воздействия на СВ1 рецепторы может компенсировать потенциирующее действие активации Д1 рецепторов на возбуждение С-Н клеток.
Влияние норадреналина. В норме при ПС норадреналин не выделяется, однако при ПТСР концентрация норадреналина повышена, особенно при навязчивых ночных кошмарах и увеличенной вероятности пробуждения [80]. Полагают, что норадреналин критически вовлечен в реакцию на стресс и патофизиологию ПТСР [81]. Для определения знака модулирующего действия норадреналина на эффективность возбудительных входов к шипиковым клеткам (ДП или ДД), необходимо знать типы располагающихся на них адренорецепторов. Показано, что на постсинаптической мембране шипиковой клетки раковины ПЯ ко-локализованы Д1 рецепторы и альфа1 адренорецепторы [82]. Поскольку Д1 рецепторы преимущественно располагаются на С-Н клетках, можно полагать, что на них располагаются и альфа1 адренорецепторы (рис. 2). Кроме того, Д1 рецепторы и альфа1 адренорецепторы ко-локализованы на дендритах нейронов ПфК [83]. В согласии с правилами модуляции (табл. 1) показано, что агонист связанных с Gq/11 белками альфа1 адренорецепторов увеличивает частоту срабатывания шипиковых клеток [84], а также нейронов зрительной коры за счет увеличения активности протеинкиназы С [85]. Показано также, что при использовании агониста альфа1-адренорецепторов облегчается индукция посттетанической ДП на нейронах поля СА1, тогда как антагонист альфа1 адренорецепторов блокирует индукцию ДП [86]. Эти данные дают основание полагать, что использование антагонистов альфа1-адренорецепторов может способствовать индукции ДД на С-Н клетках, последующему ингибированию со стороны БГ активности нейронов таламуса и связанных с ними клеток зрительной коры, а также ослаблять возбуждение нейронов ПЯ со стороны лимбических структур (рис. 1), снижая вероятность появления ночных кошмаров. Кроме того, известно, что повышение уровня норадреналина ухудшает условия для ПС [87], тогда как подавление активности норадренергических клеток голубого пятна или подавление синтеза норадреналина в педункулопонтийном ядре способствует появлению эпизодов ПС [87]. Поэтому можно ожидать, что использование антагонистов альфа1 адренорецепторов будет способствовать нормализации ПС.
Что касается связанных с Gi/0 белками альфа2 адренорецепторов, то показано [88, 89], что системное введение их антагонистов усиливает действие антагонистов также связанных с Gi/0 белками Д2 рецепторов, которые преимущественно располагаются на С-П клетках. Эти данные косвенно указывают на присутствие альфа2 адренорецепторов на С-П клетках. Использование агонистов этих рецепторов нежелательно, поскольку будет способствовать индукции ДД на С-П клетках. Блокада альфа2 адренорецепторов не только ослабляет депрессирующее действие дофамина на С-П клетки через Д2 рецепторы, но и усиливает потенциирующий эффект дофамина на С-Н клетки через Д1 рецепторы [90]. По этой причине системное использование антагонистов альфа2 адренорецепторов нежелательно, так как оно может способствовать индукции ДП на С-Н клетках.
Имеются данные, что норадреналин потенциирует возбуждение дофаминергических клеток в компактной части черного вещества [91], а также увеличивает активность дофаминергических клеток вентрального поля покрышки и выделение дофамина в ПЯ. Возможность влияния на выделение дофамина в ПЯ через альфа1 адренорецепторы продемонстрирована в работе [92]. Если выделение дофамина в ПЯ увеличивается при активации альфа1 адренорецепторов, можно ожидать, что использование их антагонистов будет способствовать снижению концентрации дофамина, что должно препятствовать появлению ночных кошмаров.
С активацией бета адренорецепторов связывают изменение активности нейронов БЛМ и увеличение тревожности [93]. В согласии с правилами модуляции (табл. 1) показано, что связанные с Gs белками бета адренорецепторы вовлечены в индукцию ДП на нейронах БЛМ [94] и что агонист бета адренорецепторов изопротеренол усиливает спайковую активность основных клеток латеральной миндалины [95]. При воздействии на бета адренорецепторы увеличивалась и респонсивность нейронов поля СА1 [96], а ДП в пирамидах поля СА1 можно было индуцировать даже с помощью низкочастотной стимуляции [97]. Из этих данных следует, что для ослабления симптомов ПТСР полезно использовать антагонисты бета-адренорецепторов, поскольку они могут способствовать снижению воздействия на ПЯ со стороны БЛМ и ГП. Известно, что норадреналин может увеличить соотношение сигнал/шум при обработке сенсорной информации и усилить консолидацию памяти в миндалине и ГП за счет воздействия на альфа1 и бета адренорецепторы [98]. Поэтому можно ожидать, что использование антагонистов указанных рецепторов позволит ослабить эти эффекты.
Влияние серотонина. Проведенный нами ранее [29] анализ известных литературных данных показал, что чувствительные к серотонину связанные с Gi/0-белками 5-НТ1В рецепторы располагаются преимущественно на С-Н клетках, а связанные с Gq/11-белками 5-НТ2А рецепторы – на С-П клетках. С учетом правил модуляции (табл. 1) можно полагать, что характерное для ПС отсутствие серотонина или низкий уровень этого нейромодулятора, т.е. слабое воздействие на 5-НТ1В и 5-НТ2А рецепторы должны способствовать индукции ДП и ДД на С-Н и С-П клетках, соответственно. В результате увеличится растормаживание нейронов таламуса со стороны БГ и улучшатся условия для появления сновидений и ночных кошмаров. Наоборот, искусственное увеличение концентрации серотонина или активация с помощью агонистов постсинаптических 5-НТ1В и 5-НТ2А рецепторов на шипиковых нейронах стриатума может уменьшить вероятность появления ночных кошмаров.
Однако серотонин действует также на активность нейронов гиппокампа и ПфК. Так, показано, что при воздействии на 5-НТ2А рецепторы усиливалась ДП на возбудительном входе из поля СА3 к пирамидам поля СА1 [99]. Кроме того, активация 5-НТ2А-рецепторов увеличивала частоту возбудительных токов в апикальных дендритах пирамидных клеток слоя 5 медиальной ПфК [100]. Поэтому, в присутствии серотонина может увеличиться возбуждение нейронов ПЯ со стороны ГП что нежелательно, так как будет способствовать появлению сновидений. Из этого следует, что во избежание появления ночных кошмаров использовать вещества, активирующие 5-НТ2А рецепторы, нежелательно. На то, что активация серотониновых 5-HT2А рецепторов может способствовать появлению зрительных образов, указывают результаты работы [101]. В ней показано, что у пациентов с болезнью Паркинсона, у которых при жизни возникали зрительные галлюцинации, было увеличено связывание 5-HT2А рецепторов в корковых областях вентрального зрительного пути.
Влияние кортикоидов. Известно, что при ПТСР понижен уровень кортикостерона, хотя он и присутствует в определенной концентрации [102]. Влияние кортикоидов осуществляется через чувствительные к нему рецепторы – минералокортикоидные (МКР) и глюкокортикоидные (ГКР), которые располагаются как на пост-, так и на пресинаптических мембранах [103]. У МКР значительно больше чем у ГКР связь с кортизолом (у человека) и с кортикостероном (у грызунов). Поэтому МКР хорошо активируются уже при фоновом уровне кортикоидов, тогда как ГКР активируются только при большом уровне (при стрессе или на пике циркадного ритма) [104–106]. На основании данных о том, что воздействие на МКР усиливает активность цАМФ [107], можно полагать, что эти рецепторы связаны с Gs белками. Про ГКР известно, что они связаны с Gi/0 белками [108, 109]. Из правил модуляции (табл. 1) следует, что воздействие на МКР и ГКР должно способствовать индукции ДП и ДД (или депотенциации), соответственно. В согласии с этими правилами показано, что через ГКР кортикостерон подавлял НМДА-зависимую ДП в синапсах, образованных коллатералями Шаффера на пирамидных клетках поля СА1 [110], тогда как агонист МКР способствовал индукции ДП на этом входе [111]. Поскольку активация МКР и ГКР зависит от концентрации кортикоидов, характер изменений эффективности синаптической передачи (ДП или ДД) должен зависеть от силы стрессового воздействия.
Для понимания механизмов влияния кортикостерона на эффективность возбуждения С-Н и С-П клеток, необходимо знать расположение МКР и ГКР на клетках каждого типа. В этой связи представляют интерес данные о том, что введение в дорзальный стриатум антагониста мускариновых рецепторов одновременно с кортикостероном непосредственно после обучения реакции замирания блокировало усиливающий эффект кортикостерона на удержание эффекта [112]. Этот результат указывает на сходство влияний кортикостерона (через кортикоидные рецепторы) и ацетилхолина (через мускариновые рецепторы) на функционирование шипиковых клеток стриатума. Результаты работы [112] косвенно указывают на то, что ГКР располагаются на тех же клетках, что и связанные с Gi/0 белками М2/М4 рецепторы, т.е. на С-Н, а МКР – на тех же клетках, что и связанные с Gs белками М1 рецепторы, т.е. на С-П клетках. Нами также использованы данные о том, что аналог глюкокортикоида дексаметазон уменьшал экспрессию Д1 рецепторов, но не Д2 рецепторов или уровень энкефалина в стриатуме [113]. Поскольку известно, что Д1 рецепторы располагаются преимущественно на С-Н клетках, выделяющих динорфин, тогда как Д2 рецепторы – на С-П, выделяющих энкефалин, результаты работы [113] также косвенно указывают на присутствие ГКР на С-Н клетках. Из предложенного нами механизма возникновения сновидений следует, что для их устранения необходимо использовать агонисты ГКР. Хотя к такому же эффекту привела бы и активация МКР, но использование агонистов этих рецепторов нецелесообразно, поскольку показано, что активация МКР в вентральное поле покрышки приводит к увеличению выделения дофамина [114].
Судя по известным из литературы данным, кортикостероиды сложным образом влияют на активность основных нейронов миндалины. Этот факт можно объяснить наличием рецепторов обоих типов (ГКР и МКР) как на основных глутаматергических клетках, так и на интернейронах латеральной миндалины [115, 116]. Как уже указывалось, вход из БЛМ в ПЯ влияет на вход в ПЯ из ГП. Показано, что ведение антагониста ГКР в БЛМ препятствует вызванному стрессом нарушению ДП в пути из вентрального субикулюма в ПЯ [117]. По-видимому, в этом эффекте преобладающим являлся эффект воздействия на ГКР, располагающиеся на основных клетках БЛМ. Что касается ГП, то при низких концентрациях кортизола усиливалась ДП в поле СА1 и в зубчатой извилине, а при высоких – наблюдалось снижение выраженности ДП [106, 118]. Аналогичный эффект наблюдался и в ассоциативных путях в поле СА3 [111]. В работе [119] также показано, что активация МКР усиливает ДП, тогда как активация ГКР ее подавляет. Вещество CORT 118335, которое активирует ГКР и одновременно является антагонистом МКР, уменьшало экспрессию c-Fos в поле СА1 [120], что указывает на уменьшение активности нейронов в этом поле ГП. На основании приведенных данных можно полагать, что использование агонистов ГКР будет препятствовать растормаживанию нейронов таламуса не только за счет индукции ДД на С-Н клетках, но и за счет снижения возбуждения нейронов ПЯ со стороны БЛМ и ГП.
То, что воздействие на ГКР может являться мишенью для терапевтического лечения ПТСР, отмечено в работе [121]. Обнаружено, что блокада ГКР в БЛМ или ГП препятствует затуханию условно-рефлекторного страха [122] и улучшает извлечение информации из памяти [123]. Эти данные позволяют предположить, что использование агонистов ГКР ухудшит извлечение из памяти информации о стрессе, и поэтому они могут быть полезны для предотвращения ночных кошмаров и ослабления других симптомов ПСТР.
Имеются данные об увеличении уровня дофамина в стриатуме и ПфК при использовании агониста ГКР [124]. Однако показано, что синтетический глюкокортикоид дексаметазон снижает экспрессию Д1 рецепторов в коре [113]. Поэтому не исключено, что влияние дофамина через Д1 рецепторы не будет превалирующим, а его активация не приведет к индукции ДП на нейронах ПфК.
Ранее нами анализировались возможные механизмы формирования нейронных отображений ассоциаций “объект–место” в разных частях сети, включающей ГП [125]. Было указано на то, что основную роль в формировании таких отображений может играть трисинаптический путь через ГП. Проведенный нами анализ, следствия которого согласуются с известными экспериментальными данными, указал на то, что в норме при ПС увеличение концентрации ацетилхолина, дофамина и кортизола и отсутствие серотонина и норадреналина синергично способствуют усилению ДД в каждом из звеньев полисинаптического пути через гиппокамп [125]. То, что активация ГКР подавляет НМДА-зависимую ДП в синапсах, образованных коллатералями Шаффера на пирамидных клеткам поля СА1, и способствует индукции ДД в этом пути, продемонстрировано в работах [110, 126]. К такому же эффекту приводит антагонист альфа1 адренорецепторов. Из изложенного следует, что использование антагонистов альфа1 адренорецепторов и агонистов ГКР может препятствовать формированию нейронных отображений эпизодов, связанных с травмой. В свою очередь, это уменьшит вероятность внедрения такой информации в сновидения.
С учетом данных о типах рецепторов, располагающихся на С-Н и С-П клетках, и правил модуляции (табл. 1), из предлагаемого механизма возникновения ночных кошмаров следует, что для их предотвращения желательно использовать препараты, представляющие собой избирательные антагонисты дофаминовых Д1 рецепторов и альфа1 адренорецепторов, а также агонисты глюкокортикоидных, серотониновых 5-НТ1В и каннабиноидных СВ1 рецепторов, т.е. рецепторов, располагающихся на С-Н клетках. Использование агонистов МКР и серотониновых 5-HT2А рецепторов, а также антагонистов дофаминовых Д2 рецепторов, альфа2с адренорецепторов или дельта опиоидных рецепторов, т.е. рецепторов, располагающихся на С-П клетках, нежелательно во избежание побочных эффектов.
СОПОСТАВЛЕНИЕ СЛЕДСТВИЙ ПРЕДЛОЖЕННОГО МЕХАНИЗМА С ИЗВЕСТНЫМИ ИЗ ЛИТЕРАТУРЫ РЕЗУЛЬТАТАМИ ТЕРАПЕВТИЧЕСКОГО ЛЕЧЕНИЯ НОЧНЫХ КОШМАРОВ ПРИ ПТСР
При поиске препаратов, угашающих память о страхе, в качестве мишеней обычно рассматривают нейроны вентромедиальной ПфК, БЛМ и ГП [60]. Отмечено, что активация каннабиноидных СВ1 рецепторов способствует снижению активности нейронов в этих структурах [60]. Высокую нораденергическую активность и активацию альфа1 адренорецепторов в ПфК ассоциируют с вызванными травмой ночными кошмарами, нарушениями сна и повышенным бодрствованием при ПТСР [80]. Хотя в известных нам работах предлагается воздействовать на рецепторы тех же типов, что и в нашей модели, однако в них не обсуждаются механизмы возникновения сновидений и механизмов влияний лимбических структур на их содержание.
В пользу рекомендаций, вытекающих из предлагаемого нами механизма, свидетельствует ряд экспериментальных данных. Так, показано, что в тех случаях, когда симптомами ПТСР являлись ночные кошмары и бессонница, антагонист альфа1 адренорецепторов празозин позволял снизить вероятность их появления [2, 127, 128] и устранял некоторые нарушения сна [129]. Празозин приводил к ослаблению ночных кошмаров при ПТСР как у бывших военных, так и у гражданских лиц после травм [3, 80]. О пользе празозина для подавления ночных кошмаров и бессонницы свидетельствует современный обзор [130]. В нем приведены данные 12 исследований 276 пациентов, среди которых последствия травмы были у 81% участников войн и у 19% гражданских лиц. Важно подчеркнуть, что празозин характеризуется слабыми побочными эффектами [131]. Также празозин ослаблял двигательные нарушения во сне у всех исследованных пациентов [68]. Имеются данные о том, что неселективный бета адреноблокатор пропранолол был полезен для лечения ПТСР у военных, поскольку снижал память о стрессе и оказывал положительный эффект в 70% случаев [132, 133]. Эти данные свидетельствуют в пользу предлагаемого нами механизма возникновения ночных кошмаров и двигательных нарушений при ПТСР из которого следует, что для их предотвращения полезно использовать антагонисты как альфа1-, так и бета адренорецепторов.
Результаты современных исследований указывают на то, что лечение кортикостероидами может способствовать ослаблению симптомов ПТСР [134]. В клинических условиях кортикостерон позволял смягчить такие симптомы ПТСР, как повышенная вероятность пробуждения и увеличенный ответ на стимул, вызывающий страх [135]. В согласии со следствием предлагаемого нами механизма показано, что активация ГКР способствует предотвращению возникновения ПТСР после травмы, а также восстановлению после ПТСР [136]. Из нашей гипотезы следует, что при системном введении агонистов ГКР могут подавляться и двигательные нарушения во сне. Описан случай, когда оральный прием глюкокортикоидов оказал положительный эффект при лечении синдрома беспокойных ног, тогда как другие препараты были не эффективны [137]. Полагают, что для лечения ПТСР могут быть полезны не только агонисты ГКР, но и антагонисты дофаминовых рецепторов и НМДА рецепторов [138]. Согласно нашей гипотезе, желательно использовать только агонисты ГКР и антагонисты Д1 рецепторов, поскольку антагонисты Д2 рецепторов (обычно используемые в качестве антипсихотических препаратов) могу привести к побочным эффектам. Что касается антагонистов НМДА рецепторов, мы полагаем их использование нежелательным, поскольку они широко представлены во всех областях мозга, и хотя их инактивация и позволит ингибировать активность нейронов таламуса и коры, но может вызвать различные побочные эффекты. Авторы одного из обзоров, в котором анализируются данные о лечении ПТСР 139], указывают на то, что антагонист альфа1 алренорецепторов празозин оказывает такое же действие на нарушения сна при ПТСР, как и атипичный антипсихотический препарат кветиапин. Однако этот препарат является антагонистом различных рецепторов: дофаминовых, гистаминовых Н1, серотониновых 5-НТ2А/С, альфа1- и альфа2 адренорецепторов. С точки зрения нашей гипотезы, такой неизбирательный препарат должен приводить к появлению различных побочных эффектов, поскольку воздействует на рецепторы, расположенные не только на С-Н, но и С-П клетках. Действительно, показано, что атипичный антипсихотический препарат оланзапин, по действию схожий с клозапином, хотя и способствовал устранению устойчивых ночных кошмаров и бессонницы, но давал нежелательные побочные эффекты [2]. Оланзапин имеет сродство не только с альфа1 адренорецепторами, но и с разными подтипами серотониновых 5-НТ рецепторов, дофаминовыми Д1 и Д2, а также мускариновыми М1/5 рецепторами. Некоторые из этих рецепторов располагаются на С-П клетках (таблица), что, по нашему мнению и приводило к побочным эффектам. Таким же недостатком должен обладать и используемый для лечения ПТСР антипсихотический препарат рисперидон [129], поскольку он не только блокирует 5-НТ2 рецепторы и Д2 рецепторы, но и связывается с альфа1- и альфа2 адренорецепторами. Имеются данные о том, что бессонницу и появление ночных кошмаров уменьшает антидепрессант нефазодон [2], который препятствует обратному захвату норадреналина и серотонина, в результате чего увеличивается их концентрация в головном мозге. Однако такой способ лечения вряд ли полезен, поскольку известно, что увеличение концентрации норадреналина препятствует появлению ПС [87]. Следствие предлагаемого механизма согласуется с данными о том, что антидепрессант миртазапин, усиливающий выделение серотонина и норадреналина за счет блокады альфа2 адренорецепторов, мог вызывать ночные кошмары [140]. Хотя использование трициклических антидепрессантов способствует позитивному характеру сновидений, однако имеются свидетельства того, что избирательные антагонисты обратного захвата серотонина или прерывание приема антидепрессантов приводят к появлению ночных кошмаров [141]. Описан случай, когда мощный ингибитор обратного захвата серотонина пароксети́н приводил к многочисленным побочным эффектам и вызывал сновидения, в которых пациент воевал с Саддамом Хуссейном [142].
В связи с появлением данных о вовлеченности каннабиноидной системы в эмоциональные процессы, в настоящее время проводятся исследования фармакологических возможностей влияния на эту систему. Выявлена потенциальная польза каннабиноидов для лечения разных расстройств, включая ПТСР [143–148]. В работе [149] приводится современный обзор данных о функционировании эндоканнабиноидной системы при травматических воздействиях и об изменениях связей между ПфК и миндалиной при ПТСР. С учетом этих данных была предложена модель для поиска препаратов, воздействующих на эту систему, позволившая выдвинуть предположение, что воздействие на эндоканнабиноидную систему может ослабить симптомы, связанные с чрезмерным эмоциональным воздействием [149]. Отмечено, что важно воздействовать на каннабиноидные рецепторы уже в первые часы после травмы, поскольку это влияет на последующее течение ПТСР. Так, показано на животных, что использование каннабиноидов сразу после травмы может предотвратить влияние стресса на эмоциональное состояние и память [150]. Внутрибрюшинное введение агониста СВ1/2 рецепторов через 2 ч после болевого воздействия на лапу крыс препятствовало вызванным болевым шоком изменениям в БЛМ, ПфК и поле СА1 [151]. Лечение каннабиноидами пациентов с ПТСР ослабляло такие симптомы, как частые ночные кошмары и быстрое пробуждение [150]. В литературе имеются и данные о том, что симптомы ПТСР ослабляет марихуана [152]. Пациенты с ярко выраженными симптомами ПТСР, по собственной инициативе принимавшие каннабис, сообщили, что симптомы становятся на 75% слабее [153]. Некоторым пациентам каннабис помогал лишь частично [154]. Существует предположение, что именно ослабление симптомов ПТСР вследствие активации каннабиноидной системы является причиной частого использования каннабиса [150, 155]. Но имеются и сведения об ухудшении симптомов ПТСР при использовании каннабиса [156]. Поскольку известные данные об использовании марихуаны для лечения ПТСР неоднозначны и их число ограничено, полагают, что трудно дать окончательные рекомендации для ее использования в клинике [152]. В настоящее время особое внимание для использования в клинике было уделено каннабидиолу, являющемуся фитоканнабиноидным компонентом Cannabis sativa, но без психоактивных эффектов Δ9-тетрагидроканнабинола [157]. Исследования на грызунах показали его потенциальную пригодность для ослабления симптомов ПТСР [157]. Недавно впервые показано в клиническом исследовании, что оральный прием каннабидиола в дополнение к обычному психиатрическому лечению приводит к ослаблению симптомов ПТСР у взрослых пациентов [158]. Особо отмечено, что каннабидиол давал хороший эффект у подгруппы пациентов, которые сообщали о частых ночных кошмарах [158]. Описан случай, когда каннабидиоловое масло уменьшало тревожность и улучшало сон при бессоннице, которая являлась симптомом посттравматического стресса [159].
Следует иметь в виду, что предложенный механизм влияния нейромодуляторов на функционирование нейронных цепей относится к ПС, поэтому методы лечения ночных кошмаров и бессонницы у пациентов с ПТСР могут быть не эффективны при лечении других симптомов ПТСР, характерных для состояния бодрствования [160].
Ограничения модели. При разработке механизма появления ночных кошмаров нами не учитывалось то обстоятельство, что нейромодуляторы приводят к модификации возбудительного входа не только к проекционным клеткам, но и к ГАМКергическим интернейронам стриатума, ГП и ПфК [161]. Однако, как указывалось нами ранее, при наличии однотипных рецепторов на интернейроне и его клетке-мишени и сильном тормозном действии, нейромодулятор может изменить знак модификации возбудительного входа к основной клетке на противоположный [162]. Нами не учитывался и тот факт, что результирующий эффект действия нейромодулятора зависит от его концентрации, поскольку у рецепторов с ним разное сродство [162].
С целью упрощения не учитывалось модулирующее воздействие нейромодуляторов на активность нейронов в других ядрах БГ и в субталамическом ядре. Однако каждое из этих ядер вносит свой вклад в функционирование цепей К–БГ–Т–К. Различные клетки субталамического ядра возбуждают нейроны цепи К–БГ–Т–К и цепи БГ–ППЯ–БГ. Ранее нами проведен анализ возможных механизмов влияния дофамина на вклад субталамического ядра в функционирование БГ [163]. Сложность анализа анализируемых цепей определяется функциональной неоднородностью нейронов, располагающихся в ядрах, реципрокно связанных с субталамическим ядром, а также разнонаправленным знаком модуляции эффективности входов к нейронов при воздействии на пре- и постсинаптические рецепторы различных типов. Как отмечено в нашей предшествующей работе [163], благодаря наличию чувствительных к нейромодуляторам рецепторов на терминалях С-Н и С-П клеток, может изменяться выделение ими ГАМК в черное вещество и бледный шар, и вследствие этого различным образом будет модулироваться активность нейронов в этих ядрах. Нейромодуляторы, выделяемые из аксонных окончаний С-Н и С-П клеток, могут способствовать взаимозависимой активности нейронов в прямом и непрямом пути через БГ за счет конвергенции этих аксонов на холинергических интернейронах и паллидостриатных клетках [164]. Проведенный нами ранее анализ механизмов влияния дофамина, ацетилхолина, опиоидов и каннабиноидов на взаимозависимую активность нейронов в разных ядрах БГ привел к заключению, что поскольку на шипиковых нейронах стриатума и их аксонных терминалях располагаются однотипные рецепторы, эффекты действия нейромодуляторов в различных ядрах могут суммироваться [164].
ЗАКЛЮЧЕНИЕ
Предложен гипотетический механизм появления ночных кошмаров, характерных для ПТСР. Механизм базируется на том, что в основе ночных кошмаров, как и в основе возникновения обычных сновидений, лежит модификация синаптической передачи в параллельных нейронных цепях, каждая из которых содержит одну из следующих структур: область зрительной коры, ПфК, БЛМ, ГП, а также связанные с ними области таламуса и ядер БГ. Появлению сновидений способствует индукция ДП эффективности возбудительных входов к С-Н клеткам и ДД эффективности входов к С-П клеткам, дающим начало соответственно прямому и непрямому пути через БГ. Это приводят к синергичному растормаживанию со стороны выходных ядер БГ нейронных паттернов в таламусе и увеличению активности связанных с ними областей коры (включая зрительные), а также ГП. Появлению ночных кошмаров способствует характерное для ПТСР повышение активности БЛМ, сигналы от которой суммируются с сигналами из ГП и ПфК на нейронах вентрального стриатума. В результате в сновидениях появляются фрагменты негативно окрашенных эпизодов. Из предлагаемого механизма следует, что предотвращению ночных кошмаров могут способствовать индукция ДД на С-Н клетках и ДП на С-П клетках, а также снижение активности нейронов лимбических структур за счет индукции ДД на их возбудительных входах. С учетом известных данных о расположении рецепторов разных типов на нейронах стриатума, из сформулированных нами ранее правил модуляции синаптической передачи следует, что депрессии входов к С-Н нейронам должны способствовать антагонисты дофаминовых Д1 рецепторов и альфа1 адренорецепторов, а также агонисты глюкокортикоидных, серотониновых 5-НТ1В и каннабиноидных СВ1 рецепторов. Нежелательно воздействовать на рецепторы, расположенные на С-П нейронах, поскольку побочным эффектом может являться потенциация возбудительных входов к нейронам ПфК, БЛМ и ГП, что облегчит появление ночных кошмаров. В частности, не следует использовать агонисты минералокортикоидных и серотониновых 5-НТ2А рецепторов, антагонисты дофаминовых Д2 рецепторов и альфа2 адренорецепторов (некоторые антипсихотические препараты и антидепрессанты). Используемые вещества должны обладать избирательным действием во избежание побочных эффектов. Препараты следует принимать только перед сном, и они не должны оказывать пролонгированного действия, чтобы не ухудшать двигательную активность и сенсорное восприятие в состоянии бодрствования.
Следует подчеркнуть, что известные из литературных источников данные об эффективности препаратов, используемых в клинике для лечения ночных кошмаров при ПТСР, согласуются с теми рекомендациями, которые вытекают из предлагаемого механизма. Это обстоятельство свидетельствует в пользу его применимости для направленной разработки лекарственных препаратов, являющихся избирательными агонистами или антагонистами только определенных типов рецепторов. Создание и использование таких препаратов может позволить избежать различных побочных эффектов при лечении пациентов с ПТСР, у которых возникают ночные кошмары.
Список литературы
El-Solh A.A. // Nat. Sci. Sleep. 2018. V. 10. P. 409–420.
Maher M.J., Rego S.A., Asnis G.M. // CNS Drugs. 2006. V. 20. № 7. P. 567–590.
Taylor H.R., Freeman M.K., Cates M.E. // Am. J. Health Syst. Pharm. 2008. V. 65. № 8. P. 716–722.
Murkar A.L.A., De Koninck J. // Sleep Med. Rev. 2018. V. 41. P. 173–184.
Onton J.A., Matthews S.C., Kang D.Y., Coleman T.P. // Front. Hum. Neurosci. 2018. V. 12. Article 196.
Phelps A.J., Kanaan R.A.A., Worsnop C., Redston S., Ralph N., Forbes D. // Sleep. 2018. V. 41. № 1. doi: 10.1093/sleep/zsx188
da Silva Rocha-Lopes J., Machado R.B., Suchecki D. // Mol. Neurobiol. 2018. V. 55. № 4. P. 2884–2896.
Mellman T.A., Kobayashi I., Lavela J., Wilson B., Hall Brown T.S. // Sleep. 2014. V. 37. № 8. P. 1321–1326.
Machida M., Wellman L.L., Fitzpatrick Bs.M.E., Hallum Bs.O., Sutton Bs.A.M., Lonart G., Sanford L.D. // Sleep. 2017. V. 40. № 4. P. 1–13.
Wellman L.L., Fitzpatrick M.E., Hallum O.Y., Sutton A.M., Williams B.L., Sanford L.D. // Sleep. 2016. V. 39. № 6. P. 1293–1303.
Walker M.P. // Prog. Brain Res. 2010. V. 185. P. 49–68.
van der Helm E., Yao J., Dutt S., Rao V., Saletin J.M., Walker M.P. // Curr. Biol. 2011. V. 21. № 23. P. 2029–2032.
Mellman T.A., Bustamante V., Fins A.I., Pigeon W.R., Nolan B. // Am. J. Psychiatry. 2002. V. 159. № 10. P. 1696–1701.
Mellman T.A., Pigeon W.R., Nowell P.D., Nolan B. // J. Trauma Stress. 2007. V. 20. № 5. P. 893–901.
Menz M.M., Rihm J.S., Salari N., Born J., Kalisch R., Pape H.C., Marshall L., Büchel C. // Neuroimage. 2013. V. 75. P. 87–96.
van Liempt S. // Eur. J. Psychotraumatol. 2012. V. 3. P. 19142.
Pace-Schott E.F., Germain A., Milad M.R. // Biol. Mood Anxiety Disord. 2015. V. 5. Article 3.
Germain A. // Am. J. Psychiatry. 2013. V. 170. № 4. P. 372–382.
Waltman S.H., Shearer D., Moore B.A. // Curr. Psychiatry Rep. 2018. V. 20. № 12. P. 108.
Miller K.E., Brownlow J.A., Woodward S., Gehrman P.R. // Curr. Psychiatry Rep. 2017. V. 19. № 10. P. 71.
Силькис И.Г. // Журн. высш. нерв. деят. 2006. Т. 56. № 1. С. 5–21.
Friedman M.J., Bernardy N.C. // Neurosci. Lett. 2017. V. 649. P. 181–185.
Kelmendi B., Adams T.G., Yarnell S., Southwick S., Abdallah C.G., Krystal J.H. // Eur. J. Psychotraumatol. 2016. V. 7. P. 31858.
Silkis I. // Biosystems. 2001. V. 59. № 1. P. 7–14.
Силькис И.Г. // Рос. физиол. журн. им. И.М. Сеченова 2002. Т. 88. № 2. С. 144–157.
Силькис И.Г. // Успехи физиол. наук. 2003. Т. 34. № 4. С. 54–74.
Силькис И.Г. // Рос. физиол. журн. им. И. М. Сеченова 2004. Т. 90. № 3. С. 282–293.
Силькис И.Г. // Нейрохимия. 2013. Т. 30. № 4. С. 305–313.
Силькис И.Г. // Нейрохимия. 2014. Т. 31. № 3. С. 185–199.
Силькис И.Г. // Нейрохимия. 2014. Т. 31. № 4. С. 287–299.
Mellman T.A., Kumar A., Kulick-Bell R., Kumar M., Nolan B. // Biol. Psychiatry. 1995. V. 38. № 3. P. 174–179.
Silkis I. // Biosystems. 2000. V. 57. № 3. P. 187–196.
Силькис И.Г. // Рос. физиол. журн. им. И.М. Сеченова. 2000. Т. 86. № 5. С. 519–531.
Gottesmann C. // Psychiatry. Clin. Neurosci. 2002. V. 56. № 4. P. 345–354.
Gottesmann C. // Front. Neurol. 2011. V. 2. Article 81.
Braun A.R., Balkin T.J., Wesensten N.J., Gwadry F., Carson R.E., Varga M., Baldwin P., Belenky G., Herscovitch P. // Science. 1998. V. 279. № 5347. P. 91–95.
Maquet P. // J. Psychopharmacol. 1999. V. 13. № 4. Suppl. 1. P. S25–S28.
Igawa M., Atsumi Y., Takahashi K., Shiotsuka S., Hirasawa H., Yamamoto R., Maki A., Yamashita Y., Koizumi H. // Psychiatry. Clin. Neurosci. 2001. V. 55. № 3. P. 187–188.
Силькис И.Г. // Журн. высш. нерв. деят. 2008. Т. 58. № 5. С. 521–539.
Силькис И.Г. // Журн. высш. нерв. деят. 2010. Т. 60. № 6. С. 645–663.
Corsi-Cabrera M., Miro E., del-Rio-Portilla Y., Perez-Garci E., Villanueva Y., Guevara M.A. // Brain Cogn. 2003. V. 51. № 3. P. 337–345.
Walker M.P., Liston C., Hobson J.A., Stickgold R. // Cogn. Brain Res. 2002. V. 14. № 3. P. 317–324.
Sesack S.R., Grace A.A. // Neuropsychopharmacology. 2010. V. 35. № 1. P. 27–47.
Germain A., Buysse D.J., Nofzinger E. // Sleep Med. Rev. 2008. V. 12. № 3. P. 185–195.
Sharp B.M. // Transl. Psychiatry. 2017. V. 7. № 8. e1194.
Силькис И.Г. // Журн. высш. нерв. деят. 2014. V. 64. № 1. С. 82–100.
O’Donnell P., Grace A.A. // J. Neurosci. 1995. V. 15. № 5. P. 3622–3639.
French S.J., Totterdell S. // J. Comp. Neurol. 2002. V. 446. № 2. P. 151–165.
Gill K.M., Grace A.A. // Int. J. Neuropsychopharmacol. 2011. V. 14. № 10. P. 1301–1314.
Floresco S.B., Blaha C.D., Yang C.R., Phillips A.G. // J. Neurosci. 2001. V. 21. № 8. P. 2851–2860.
Mulder A.B., Hodenpijl M.G., Lopes da Silva F.H. // J. Neurosci. 1998. V. 18. № 13. P. 5095–5102.
McGinty V.B., Grace A.A. // J. Neurophysiol. 2009. V. 101. № 4. P. 1823–1835.
Muñoz-Torres Z., Velasco F., Velasco A.L., Del Río-Portilla Y., Corsi-Cabrera M. // Clin. Neurophysiol. 2018. V. 129. № 10. P. 2118–2126.
Genzel L., Spoormaker V.I., Konrad B.N., Dresler M. // Neurobiol. Learn. Mem. 2015. V. 122. P. 110–121.
Lowery-Gionta E.G., Crowley N.A., Bukalo O., Silverstein S., Holmes A., Kash T.L. // Neuropharmacology. 2018. V. 139. P. 68–75.
Zhang J.Y., Liu T.H., He Y., Pan H.Q., Zhang W.H., Yin X.P., Tian X.L., Li B.M., Wang X.D., Holmes A., Yuan T.F., Pan B.X. // Biol. Psychiatry. 2019 V. 85. № 3. P. 189–201.
Khazaie H., Ghadami M.R., Masoudi M.1. // J. Inj. Violence. Res. 2016. V. 8. № 2. P. 99–107.
Esposito K., Benitez A., Barza L., Mellman T. // J. Trauma Stress. 1999. V. 12. № 4. P. 681–687.
Schreuder B.J., van Egmond M., Kleijn W.C., Visser A.T. // J. Anxiety Disord. 1998. V. 12. № 6. P. 511–524.
Myskiw J.C., Izquierdo I., Furini C.R. // Brain Res. Bull. 2014 V. 105. P. 61–69.
Toyoda H., Li X.Y., Wu L.J., Zhao M.G., Descalzi G., Chen T., Koga K., Zhuo M. // Neural Plast. 2011. V. 2011. Article ID 813749.
Spoormaker V.I., Gvozdanovic G.A., Sämann P.G., Czisch M. // Exp. Brain Res. 2014. V. 232. № 5. P. 1547–1554.
Lerner I., Lupkin S.M., Sinha N., Tsai A., Gluck M.A. // J. Neurosci. 2017. V. 37. № 46. P. 11233–11244.
Nielsen T. // Front. Neurol. 2017. V. 8. Article 201.
Bennett M.R., Hatton S.N., Lagopoulos J. // Brain Struct. Funct. 2016. V. 221. № 5. P. 2401–2426.
Brown T.M., Boudewyns P.A. // J. Trauma Stress. 1996. V. 9. № 1. P. 129–136.
Baird T., McLeay S., Harvey W., Theal R., Law D., O’Sullivan R. // J. Clin. Sleep Med. 2018. V. 14. № 5. P. 745–752.
Mysliwiec V., O’Reilly B., Polchinski J., Kwon H.P., Germain A., Roth B.J. // J. Clin. Sleep Med. 2014. V. 10. № 10. P. 1143–1148.
Newton S.S., Thome J., Wallace T.L., Shirayama Y., Schlesinger L., Sakai N., Chen J., Neve R., Nestler E.J., Duman R.S. // J. Neurosci. 2002. V. 22. № 24. P. 10883–10890.
Otmakhova N.A., Lisman J.E. // J. Neurosci. 1996. V. 16. № 23. P. 7478–7486.
Zahrt J., Taylor J.R., Mathew R.G., Arnsten A.F. // J. Neurosci. 1997. V. 17. № 21. P. 8528–8535.
Di S., Itoga C.A., Fisher M.O., Solomonow J., Roltsch E.A., Gilpin N.W., Tasker J.G. // J. Neurosci. 2016. V. 36. № 32. P. 8461–8470.
Patel S., Hill M.N., Cheer J.F., Wotjak C.T., Holmes A. // Neurosci. Biobehav. Rev. 2017. V. 76. Pt. A. P. 56–66.
Ganon-Elazar E., Akirav I. // Psychoneuroendocrinology. 2013. V. 38. № 9. P. 1675–1687.
Azad S.C., Kurz J., Marsicano G., Lutz B., Zieglgänsberger W., Rammes G. // Learn. Mem. 2008. V. 15. № 3. P. 143–152.
Péterfi Z., Urbán G.M., Papp O.I., Németh B., Monyer H., Szabó G., Erdélyi. F, Mackie K., Freund T.F., Hájos N., Katona I. // J. Neurosci. 2012. V. 32. № 41. P. 14448–14463.
Xu J.Y., Chen R., Zhang J., Chen C. // PLoS One. 2010. V. 5. № 4. e10306.
Melis M., Gessa G.L., Diana M. // Prog. Neuropsychopharmacol. Biol. Psychiatry. 2000. V. 24. № 6. P. 993–1006.
Giuffrida A., Parsons L.H., Kerr T.M., Rodriguez de Fonseca F., Navarro M., Piomelli D. // Nat. Neurosci. 1999. V. 2. № 4. P. 358–363.
De Berardis D., Marini S., Serroni N., Iasevoli F., Tomasetti C., de Bartolomeis A., Mazza M., Tempesta D., Valchera A., Fornaro M., Pompili M., Sepede G., Vellante F., Orsolini L., Martinotti G., Di Giannantonio M. // Curr. Drug Targets. 2015. V. 16. № 10. P. 1094–1106.
Strawn J.R., Geracioti T.D. // Depress. Anxiety. 2008. V. 25. № 3. P. 260–271.
Mitrano D.A., Jackson K., Finley S., Seeley A. // Neuroscience. 2018. V. 371. P. 126–137.
Mitrano D.A., Pare J.F., Smith Y., Weinshenker D. // Neuroscience. 2014. V. 258. P. 90–100.
Ohta H., Kohno Y., Arake M., Tamura R., Yukawa S., Sato Y., Morimoto Y., Nishida Y., Yawo H. // Neurosci. Res. 2016. V. 112. P. 47–56.
Kobayashi M., Sasabe T., Shiohama Y., Koshikawa N. // Neurosci. Lett. 2008. V. 430. № 2. P. 175–180.
Izumi Y., Zorumski C.F. // Synapse. 1999. V. 31. № 3. P. 196–202.
Khanday M.A., Somarajan B.I., Mehta R., Mallick B.N. // eNeuro. 2016. V. 3. № 6. P. 1–19.
Jacobson S.M., Prus A.J. // Behav. Pharmacol. 2010. V. 21. № 7. P. 654–659.
Hertel P., Fagerquist M.V., Svensson T.H. // Science. 1999. V. 286. № 5437. P. 105–107.
Hara M., Fukui R., Hieda E., Kuroiwa M., Bateup H.S., Kano T., Greengard P., Nishi A. // J. Neurochem. 2010. V. 113. № 4. P. 1046–1459.
Zhu M.Y. // Neurotox. Res. 2018. V. 34. № 4. P. 848–859.
Park J.W., Bhimani R.V., Park J. // ACS Chem. Neurosci. 2017. V. 8. № 9. P. 1913–1924.
McCall J.G., Siuda E.R., Bhatti D.L., Lawson L.A., McElligott Z.A., Stuber G.D., Bruchas M.R. // eLife. 2017. V. 6. e18247.
Sarabdjitsingh R.A., Kofink D., Karst H., de Kloet E.R., Joëls M. // PLoS One. 2012. V. 7. № 8. e42143.
Fink A.E., LeDoux J.E. // J. Neurophysiol. 2018. V. 119. № 5. P. 1658–1664.
Dunwiddie T.V., Taylor M., Heginbotham L.R., Proctor W.R. // J. Neurosci. 1992. V. 12. № 2. P. 506–517.
Thomas M.J., Moody T.D., Makhinson M., O’Dell T.J. // Neuron. 1996. V. 17. № 3. P. 475–482.
Ramos B.P., Arnsten A.F. // Pharmacol. Ther. 2007. V. 113. № 3. P. 523–536.
Wang R.Y., Arvanov V.L. // Brain Res. 1998. V. 779. № 1–2. P. 309–313.
Marek G.J., Aghajanian G.K. // Neuroscience. 1998. V. 86. № 2. P. 485–497.
Ballanger B., Strafella A.P., van Eimeren T., Zurowski M., Rusjan P.M., Houle S., Fox S.H. // Arch. Neurol. 2010. V. 67. № 4. P. 416– 421.
Horn C.A., Pietrzak R.H., Corsi-Travali S., Neumeister A. // Psychoneuroendocrinology. 2014. V. 39. P. 88–93.
Russo M.F., Ah Loy S.R., Battle A.R., Johnson L.R. // Front. Cell Neurosci. 2016. V. 10. Article 161.
de Kloet E.R., Karst H., Joëls M. // Front. Neuroendocrinol. 2008. V. 29. P. 268–272.
Groeneweg F.L., Karst H., de Kloet E.R., Joëls M. // Mol. Cell. Endocrinol. 2012. V. 350. P. 299–309.
Pavlides C., Watanabe Y., Magarinos A.M., McEwen B.S. // Neuroscience. 1995 V. 68. № 2. P. 387–394.
Christ M., Wehling M., Kirsch E., Viengchareun S., Zennaro M.C., Lombes M. // Mol. Cell Endocrinol. 2005. V. 231. № 1–2. P. 23–31.
Shaqura M., Li X., Al-Khrasani M., Shakibaei M., Tafelski S., Fürst S., Beyer A., Kawata M., Schäfer M., Mousa S.A. // Neuropharmacology. 2016. V. 111. P. 1–13.
Sóvágó J., Makkai B., Gulyás B., Hall H. // Eur. J. Neurosci. 2005. V. 22. № 1. P. 65–71.
Alfarez D.N., Joels M., Krugers H.J. // Eur. J. Neurosci. 2003. V. 17. № 9. P. 1928–1934.
Pavlides C., McEwen B.S. // Brain Res. 1999. V. 851. № 1–2. P. 204–214.
Sánchez-Resendis O., Medina A.C., Serafín N., Prado-Alcalá R.A., Roozendaal B., Quirarte G.L. // Front. Behav. Neurosci. 2012. V. 6. Article 33.
Iasevoli F., Aloj L., Latte G., Avvisati L., Marmo F., Tomasetti C., Buonaguro E.F., Simeoli C., Pivonello R., Colao A., de Bartolomeis A. // Curr. Mol. Pharmacol. 2013. V. 6. № 3. P. 149–155.
de Oliveira A.R., Reimer A.E., Brandão M. // Psychoneuroendocrinology. 2014 V. 43: P. 114–25.
Hartmann J., Dedic N., Pöhlmann M.L., Häusl A., Karst H., Engelhardt C., Westerholz S., Wagner K.V., Labermaier C., Hoeijmakers L., Kertokarijo M., Chen A., Joëls M., Deussing J.M., Schmidt M.V. // Mol. Psychiatry. 2017. V. 22. № 3. P. 466–4475.
Prager E.M., Brielmaier J., Bergstrom H.C., McGuire J., Johnson L.R. // PLoS One. 2010. V. 5. № 12. e14344.
Segev A., Akirav I. // Neuropsychopharmacology. 2016. V. 41. № 4. P. 1066–1079.
Pavlides C., Ogawa S., Kimura A., McEwen B.S. // Brain Res. 1996. V. 738. № 2. P. 229–235.
Shavit Stein E., Itsekson Hayosh Z., Vlachos A., Maggio N. // Front. Cell. Neurosci. 2017. V. 11. Article 299.
Nguyen E.T., Streicher J., Berman S., Caldwell J.L., Ghisays V., Estrada C.M., Wulsin A.C., Solomon M.B. // Physiol. Behav. 2017. V. 178. P. 82–92.
Szeszko P.R., Lehrner A., Yehuda R. // Harv. Rev. Psychiatry. 2018. V. 26. № 3. P. 142–157.
Ganon-Elazar E., Akirav I. // Psychoneuroendocrinology. 2013. V. 38. № 9. P. 1675–1687.
Rimmele U., Besedovsky L., Lange T., Born J. // Neuropsychopharmacology. 2013. V. 38. № 5. P. 884–894.
Chen Y., Zheng X., Xie L., Huang L., Ke Z., Zheng J., Lu H., Hu J. // Brain Res. Bull. 2017. V. 131. P. 214–220.
Силькис И.Г. // Журн. высш. нерв. деят. 2009. Т. 59. № 6. С. 645–661.
Yang C.H., Huang C.C., Hsu K.S. // J. Neurosci. 2004 V. 24. № 49. P. 11029-11034.
Berger W., Mendlowicz M.V., Marques-Portella C., Kinrys G., Fontenelle L.F., Marmar C.R., Figueira I. // Prog. Neuropsychopharmacol. Biol. Psychiatry. 2009. V. 33. № 2. P. 169–180.
Dierks M.R., Jordan J.K., Sheehan A.H. // Ann. Pharmacother. 2007. V. 41. № 6. P. 1013–1017.
Kerbage H., Richa S.,1. // Curr. Clin. Pharmacol. 2015. V. 10. № 2. P. 116–125.
Simon P.Y., Rousseau P.F. // Can. J. Psychiatry. 2017. V. 62. № 3. P. 186–198.
Breen A., Blankley K., Fine J. // J. Am. Assoc. Nurse Pract. 2017. V. 29. № 2. P. 65–69.
Tawa J., Murphy S. // J. Am. Assoc. Nurse Pract. 2013. V. 25. № 8. P. 419–423.
Villain H., Benkahoul A., Birmes P., Ferry B., Roullet P. // PLoS One. 2018. V. 13. № 1. e0191563.
Zuckerman A., Ram O., Ifergane G., Matar M.A., Kaplan Z., Hoffman J.R., Sadot O., Cohen H. // J. Neurotrauma. 2019. V. 36. № 2. P. 380–394.
Jia M., Smerin S.E., Zhang L., Xing G., Li X., Benedek D., Ursano R., Li H. // J. Psychiatr. Res. 2015. V. 60. P. 29–39.
Rasmusson A.M., Marx C.E., Pineles S.L., Locci A., Scioli-Salter E.R., Nillni Y.I., Liang J.J., Pinna G. // Neurosci. Lett. 2017. V. 649. P. 156–163.
Oscroft N.S., Smith I.E. // Sleep Med. 2010. V. 11. № 6. P. 596.
Naß J., Efferth T. // Curr. Neuropharmacol. 2017. V. 15. № 6. P. 831–860.
Nappi C.M., Drummond S.P., Hall J.M. // Neuropharmacology. 2012. V. 62. № 2. P. 576–585.
Menon V., Madhavapuri P. // J. Pharmacol. Pharmacother. 2017. V. 8. № 4. P. 182–184.
Tribl G.G., Wetter T.C., Schredl M. // Sleep Med. Rev. 2013. V. 17. № 2. P. 133–142.
Parish J.M. // J. Clin. Sleep Med. 2007. V. 3. № 5. P. 529–531.
Morena M., Berardi A., Colucci P., Palmery M., Trezza V., Hill M.N., Campolongo P. // Neuropsychopharmacology. 2018. V. 43. № 6. P. 1284–1296.
Crippa J.A., Guimarães F.S., Campos A.C., Zuardi A.W. // Front. Immunol. 2018. V. 9. Article 2009.
Loflin M.J., Babson K.A., Bonn-Miller M.O. // Curr. Opin. Psychol. 2017. V. 14. P. 78–83.
Steenkamp M.M., Blessing E.M., Galatzer-Levy I.R., Hollahan L.C., Anderson W.T. // Depress. Anxiety. 2017. V. 34. № 3. P. 207–216.
Berardi A., Schelling G., Campolongo P. // Pharmacol. Res. 2016. V. 111. P. 668–678.
Krumm B.A. // Nurse Pract. 2016. V. 41. № 1. P. 50–54.
Lanius R.A., Boyd J.E., McKinnon M.C., Nicholson A.A., Frewen P., Vermetten E., Jetly R., Spiegel D. // Curr. Psychiatry Rep. 2018. V. 20. № 12. P. 118.
Mizrachi Zer-Aviv T., Segev A., Akirav I. // Behav. Pharmacol. 2016. V. 27. № 7. P. 561–569.
Korem N., Akirav I. // Neuropsychopharmacology. 2014. V. 39. № 12. P. 2709–2722.
Shishko I., Oliveira R., Moore T.A., Almeida K. // Ment. Health Clin. 2018. V. 8. № 2. P. 86–94.
Greer G.R., Grob C.S., Halberstadt A.L. // J. Psychoactive Drugs. 2014. V. 46. № 1. P. 73–77.
Bonn-Miller M.O., Boden M.T., Bucossi M.M., Babson K.A. // Am. J. Drug Alcohol Abuse. 2014. V. 40. № 1. P. 23–30.
Hill M.N., Campolongo P., Yehuda R., Patel S. // Neuropsychopharmacology. 2018. V. 43. № 1. P. 80–102.
Wilkinson S.T., Yarnell S., Radhakrishnan R., Ball S.A., D’Souza D.C. // Ann. Rev. Med. 2016. V. 67. P. 453–466.
Bitencourt R.M., Takahashi R.N. // Front. Neurosci. 2018. V. 12. Article 502.
Elms L., Shannon S., Hughes S., Lewis N. // J. Altern. Complement. Med. 2018. Dec 13.
Shannon S., Opila-Lehman J. // Perm. J. 2016. V. 20. № 4. P. 108–111.
Brownlow J.A., Harb G.C., Ross R.J. // Curr. Psychiatry Rep. 2015. V. 17. № 6. P. 41.
Силькис И.Г. // Журн. высш. нерв. деят. им. И.П. Павлова. 2002. Т. 52. № 4. С. 392–405.
Силькис И.Г., Маркевич В.А. // Успехи физиол. наук. 2016. Т. 47. № 4. С. 57–76.
Силькис И.Г. // Успехи физиологических наук. 2005. Т. 36. № 2. С. 66–83.
Силькис И.Г. // Рос. физиол. журн. им. И.М. Сеченова. 2004. Т. 90. № 3. С. 282–293.
Дополнительные материалы отсутствуют.