

Нейрохимия, 2021, T. 38, № 2, стр. 186-192
Значимость окислительного повреждения белков и ДНК в крови пациентов с болезнью Паркинсона в оценке тяжести заболевания
Т. Н. Федорова 1, А. А. Логвиненко 1, Д. С. Бережной 1, В. В. Полещук 1, О. А. Музычук 1, А. А. Шабалина 1, С. Н. Иллариошкин 1
1 Федеральное государственное бюджетное научное учреждение “Научный центр неврологии”
Москва, Россия
Поступила в редакцию 31.07.2020
После доработки 16.12.2020
Принята к публикации 23.01.2021
Аннотация
Окислительный стресс (ОС) играет важную роль в каскаде событий, приводящих к дегенерации дофаминергических нейронов при болезни Паркинсона (БП). Определенный вклад в этот процесс вносят окислительные повреждения белков и нуклеиновых кислот. Активные формы кислорода и азота подвергают нитрованию белки с образованием стабильного соединения 3-нитротирозина (3-НТ), характеризующего развитие нитрозильного стресса, а также нуклеиновые кислоты с образованием продукта окисления ДНК 8-гидрокси-2-дезоксигуанозина (8-OH-dG). Продукты окисления белков и ДНК регистрируются в биологических жидкостях пациентов с БП, однако данные об их количественном содержании в зависимости от тяжести заболевания, являются противоречивыми. Целью данной работы явилось сопоставление уровня продуктов окислительного повреждения белков и ДНК в крови пациентов с тяжестью заболевания и оценка их диагностического значения. В периферической крови 134 пациентов с БП, находящихся на 1-4 стадиях заболевания по функциональной шкале Hoehn-Yahr, было измерено содержание 3-нитротирозина (3-НТ) и 8-гидрокси-2-дезоксигуанозина (8‑OH-dG). Повышение уровня 3-НТ в плазме крови отмечается у всех обследованных пациентов. При этом у пациентов, находящихся на 2-й, 3-й и 4-й стадиях заболевания повышение уровня 3-НТ относительно контроля составило в среднем 58%, а у пациентов, находящихся на 1-й стадии заболевания (с впервые уставленным диагнозом и не получавших лечения) – на 30%, что статистически значимо отличается от данных, полученных у пациентов на более поздних этапах заболевания. Следовательно, повышение продукта окислительного метаболизма белков 3-НТ в плазме крови пациентов с БП является весьма ранним и достаточно универсальным биомаркером БП, степень выраженности которого нарастает по мере прогрессирования нейродегенеративного процесса. Повышение уровня 8-гидрокси-2-дезоксигуанозина относительно нормы регистрируется в сыворотке крови у всех обследованных пациентов. При этом у пациентов, находящихся на 1-й, 2-й и 3-й стадиях заболевания это повышение было в среднем на 60%, а у наиболее тяжелых пациентов, находящихся на 4-й стадии заболевания, содержание 8-гидрокси-2-дезоксигуанозина было повышено относительно нормы уже на 183%, что в три раза превышало соответствующие значения в других подгруппах больных. Таким образом, в данном исследовании обнаружено системное увеличение содержания как 3-нитротирозина, так и 8-гидрокси-2-дезоксигуанозина в крови пациентов. При этом особую значимость представляют данные о повышении продуктов окисления белков и ДНК у пациентов с впервые поставленным диагнозом, находящихся на первой стадии заболевания и не получавших ранее патогенетической терапии, что может быть важным фактором в ранней диагностике заболевания. Идентификация биомаркеров окислительного повреждения белков и нуклеиновых кислот на ранних стадиях БП является важным шагом в направлении улучшения существующих диагностических критериев, а также выявления лиц, подверженных риску заболевания. Значительное повышение содержания 8-гидрокси-2-дезоксигуанозина в сыворотке крови пациентов, находящихся на 4-й стадии заболевания, свидетельствует о связи данного показателя с тяжестью заболевания и может представить дополнительную ценность с точки зрения объективной оценки прогрессирования заболевания. В целом, понимание патогенетических факторов, ответственных за гибель дофаминергических нейронов, включающих окислительные повреждения липидов, белков и нуклеиновых кислот может иметь большое значение для разработки комплексных нейропротекторных подходов к лечению БП и оценке эффективности проводимого лечения.
Болезнь Паркинсона (БП) является распространенным нейродегенеративным заболеванием, характеризующимся прогрессирующей дегенерацией дофаминергических нейронов в области компактной части черной субстанции (ЧС) и наличием внутринейрональных включений, состоящих из неправильно свернутых и агрегированных форм пресинаптического белка α-синуклеина [1, 2].
Решающую роль в каскаде событий, приводящих к дегенерации дофаминергических нейронов, играет окислительный стресс (ОС), возникающий в условиях нарушения окислительно-восстановительного баланса, необходимого для нормального функционирования клетки. [3–7]. Гибель нейронов в области ЧС обусловлена не только развитием ОС, определенный вклад в этот процесс вносят окислительные повреждения белков и нуклеиновых кислот [2]. Уязвимость мозга к активным формам кислорода (АФК) и азота в этой области обусловлена наличием нейронов, чувствительных к окислительным повреждениям [8] за счет особенностей метаболизма, низкой активности антиоксидантных (АО) ферментов и более высокого уровня белков, ДНК и окисленных липидов [9].
АФК, преимущественно О2–, генерируемые в условиях ОС, реагируют с оксидом азота (NO) с образованием пероксинитрита (OONO–), одного из наиболее разрушительных реактивных радикалов [10]. Предполагается, что изменения метаболизма дофамина при БП могут быть результатом пероксинитрит-опосредованной инактивации фермента тирозин-гидроксилазы, участвующего в синтезе дофамина [11].
Поскольку ONOO– является нестабильным промежуточным соединением, он атакует наиболее уязвимую аминокислоту тирозин (Тир), в основном в составе белков, образующих нейрональный цитоскелет, а также α-синуклеина [12, 13], что обуславливает нарушение их структуры и функции [14]. Прогрессирующее нитрование белков приводит к образованию стабильного соединения 3-нитротирозина (3-НТ), характеризующего развитие нитрозильного стресса [15, 16].
Было обнаружено, что уровень 3-НТ увеличивается в мозге пациентов с БП [17]. По данным Eve et al. [18] повышение 3-НТ коррелирует с гиперэкспрессией нейрональной nNOS в мозге (преимущественно в базальных ганглиях) пациентов с БП [19]. Посттрансляционная модификация α-синуклеина 3-НТ значительно изменяет его биохимические, биофизические и клеточные свойства, приводит к олигомеризации и влияет на клиренс агрегатов α-синуклеина [20], и этот процесс вовлечен в возникновение и прогрессирование нейродегенеративных заболеваний [21–24]. Протеомное исследование показало, что нитрование N-концевого остатка Тир-39, присутствующего в белке α-синуклеина, избирательно увеличивалось в 7 раз [20].
Активные формы кислорода (ОН– радикал) и азота также могут подвергать повреждению нуклеиновые кислоты с образованием продукта окисления ДНК 8-гидрокси-2-дезоксигуанозина (8-OH-dG) [25–28]. В исследованиях post mortem показано увеличение содержания 8-OH-dG не только в структурах ЧС, но и в других областях мозга пациентов с БП [29–31].
Учитывая, что ключевой этиологический фактор БП до сих пор не установлен, а его развитие связывают с биохимическими нарушениями как в ЦНС, так и на периферии [3, 6], отдельный интерес представляют маркеры системных окислительных повреждений. Наличие 3-НТ и 8-OH-dG регистрируются не только в ткани мозга, но и в цереброспинальной жидкости (ЦСЖ), крови и моче пациентов с БП [32, 33]. Однако данные литературы, характеризующие показатели окисляемости белков и ДНК, измеряемые в биологических жидкостях пациентов с БП, являются противоречивыми. В ряде исследований показано, что повышение содержания 3-НТ в сыворотке крови пациентов с БП отмечается только у больных, находящихся на ранних стадиях заболевания (1 и 2), в то время как у пациентов на более поздних стадиях (3 и 4) содержание 3-НТ было сопоставимо с контролем [34, 35]. В другом исследовании авторы регистрировали повышенный уровень 3-НТ в плазме крови без учета стадии заболевания в общей популяции пациентов [36].
Значительное повышение 8-OH-dG выявлено в моче пациентов с БП относительно группы контроля [37, 39, 36 ]. Мета-анализ, проведенный Zexu Wei [7],также показал, что содержание в крови больных 8-OH-dG и нитрита было повышено относительно контрольной группы; однако в ЦСЖ повышения уровня 8-OH-dG выявлено не было, что по предположению авторов связано с небольшим размером выборки.
В работе Chiung-Mei Chen et al. [40] повышение уровня 8-OH-dG, коррелирующее со стадией заболевания, регистрировали в лейкоцитах крови пациентов с БП. Однако в другом исследовании уровень 8-OH-dG в крови был увеличен относительно контроля, стадия заболевания при этом не учитывалась [38]. В работе Kikuchi A. [41] окисляемость нуклеиновых кислот в сыворотке крови характеризовалась только повышением соотношения продуктов окисления ДНК/РНК (8-OHdG/ 8-OHG). Представленные данные литературы свидетельствуют о том, что оценка содержания продуктов окислительного повреждения белков и нуклеиновых кислот в крови пациентов как биомаркеров, коррелирующих с тяжестью заболевания, остается актуальным вопросом.
Исходя из вышеизложенного, целью данной работы явилось сопоставление уровня продуктов окислительного повреждения белков и ДНК в крови пациентов с БП с тяжестью заболевания по неврологической шкале, и оценка их диагностического значения.
МАТЕРИАЛЫ И МЕТОДЫ
Всего в исследовании приняли участие 134 пациента с БП. Оценка степени тяжести БП проводилась по функциональной шкале Hoehn M., Yahr M., в соответствии с которой выделяли 4 стадии заболевания: 1 – гемипаркинсонизм, 2 – умеренные симптомы двустороннего паркинсонизма, 3 – присоединение постуральной неустойчивости, 4 – выраженная постуральная неустойчивость. В соответствии с данной шкалой среди пациентов было выделено 25 человек с 1-й стадией заболевания, 38 – со 2-й, 51 – с 3-й и 20– с 4-й стадией заболевания. Следует отдельно подчеркнуть, что в группу с 1-й стадией заболевания вошли пациенты с впервые уставленным диагнозом, не получавшие ранее противопаркинсонической терапии. Все пациенты на более тяжелых стадиях получали стандартное противопаркинсоническое лечение – препараты с действующим веществом Леводопа + Бенсеразид. Среди пациентов мужчин было 61 и женщин –73, возраст больных составил от 31 до 78 лет, средний возраст – 58.03 ± 0.88 лет. Все пациенты находились на стационарном или амбулаторном обследовании и лечении в отделении нейродегенеративных и наследственных заболеваний нервной системы и в научно-консультативном отделении ФГБНУ НЦН. Диагноз БП установлен на основании признаков для критериев Банка головного мозга Великобритании/QSBB. Контрольную группу составили 25 практически здоровых лиц, соответствующих основной группе по полу и возрасту.
Содержание 8-гидрокси-2-дезоксигуанозина определяли в сыворотке крови 71 пациента иммуноферментным методом с использованием стандартного набора реагентов Human 8-Hydroxydeoxy-guanosine ELISA Kit (CSB-E10140h) фирмы Cusabio. Определение 8-OH-dG данным набором ИФА основано на методе конкурентного связывания тестируемого соединения.
Содержание 3-нитротирозина определяли в плазме крови 63 пациентов иммуноферментным методом с использованием стандартного набора реагентов Nitrotyrosine ELISA TEST KIT (HK501) фирмы Hycultbiotech, в основе которого лежит “сэндвич” метод твердофазного ИФА.
Кровь у пациентов брали утром натощак при помощи вакуумной системы для забора крови в пробирки с антикоагулянтом ЭДТА, рекомендованным производителем набора для определения 3-НТ, и в пробирки для последующего получения сыворотки для определения 8-OH-dG. После центрифугирования при 3000 об./мин в течение 10 мин образцы плазмы переносили в пробирки Eppendorf и помещали в морозильную камеру при –80°С до проведения анализа.
Статистический анализ полученных результатов проводился с использованием пакета программ Statistica 7.0. Проверка на нормальность распределения оcуществлялась при помощи критерия Колмогорова–Смирнова. Дальнейший анализ осуществляли с помощью непараметрических критериев: дисперсионный анализ – тест Краскела–Уоллиса, оценка достоверности межгрупповых различий – тест Данна. Все данные в статье представлены в виде среднего и ошибки среднего.
РЕЗУЛЬТАТЫ
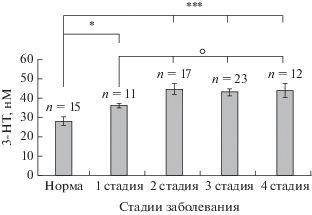
Уровень 3-нитротирозина в плазме крови у пациентов, находящихся на 2-й (44.6 ± 2.6 нМ), 3-й (43.1 ± 1.9 нМ) и 4-й (44.2 ± 3.6 нМ) стадиях заболевания, был статистически значимо повышен относительно нормы (27.8 ± 2.2 нМ), соответственно, на 60.4%, 55%, 61%, что в среднем по всем группам составило 58% (рис. 1). У пациентов на 1-й стадии заболевания, т.е. у больных с впервые установленным диагнозом и не получавших лечения, уровень 3-нитротирозина (36.1 ± 1.1 нМ) был также статистически значимо повышен относительно нормы – на 30%, что, однако, оказалось существенно (p < 0.05) ниже, чем в остальных сопоставляемых группах пациентов с более развернутыми стадиями БП (рис. 1).
Рис. 1.
Содержание 3-нитротирозина (3-НТ, нM) в плазме крови пациентов с 1-й, 2-й, 3-й и 4-й стадиями БП. *** p < 0.001, * p < 0.05: достоверность различий по сравнению с группой нормы, ⚪ p < 0.05: достоверность различий по сравнению с 1-й стадией.
Содержание 8-гидрокси-2-дезоксигуанозина в сыворотке крови у пациентов, находящихся на 1-й (73.7 ± 2.8 нг/мл), 2-й (66.8 ± 3.07 нг/мл) и 3-й (74.7 ± 2.3 нг/мл) стадиях заболевания, было статистически значимо повышено относительно нормы (44.7 ± 2.6 нг/мл), соответственно, на 64.9%, 49.4% и 66.9% (рис. 2). Содержание 8-гидрокси-2-дезоксигуанозина у больных, находящихся на 4-й стадии заболевания, было также статистически значимо повышено (126.6 ± 12.9 нг/мл) относительно нормы – на 183%, но при этом уровень этого повышения был значимо больше (на 72%, 89% и 70%) относительно пациентов, находящихся на 1-й, 2-й и 3-й стадиях, соответственно (рис. 2).
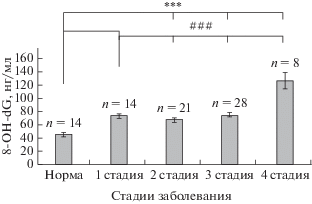
ОБСУЖДЕНИЕ
В настоящее время ОС рассматривается как основной патогенетический механизм гибели нейронов при БП. Вместе с тем, окислительные повреждения затрагивают не только липидные компоненты клеточных мембран, но и белки и нуклеиновые кислоты. По данным литературы, стабильным маркером окислительного повреждения белков является 3-нитротирозин [15, 16]. Устойчивое нарушение окислительного баланса нуклеиновых кислот характеризует продукт окисления ДНК – 8-гидрокси-2-дезоксигуанозин [27, 28]. Окислительные повреждения липидов, белков и ДНК могут приводить к прогрессирующему повреждению нейронов при БП. Так генерация 3-нитротирозина в белках α-синуклеина является одним из патологических факторов, вызывающих гибель дофаминергических нейронов по пути апоптоза при БП [24, 42, 43]. При этом нарушение окислительного гомеостаза, приводящее повреждению белков и ДНК, может затрагивать не только нервную ткань, но и носить системный характер [44]. Согласно “прионной гипотезе” БП, начальные стадии заболевания связаны с возникновением агрегатов α-синуклеина и повреждениями как раз на периферии – в кишечнике и периферической нервной системе [44]. В связи с этим, изменение маркеров ОС в крови может отображать патогенетические каскады, как первичные, так и вторичные по отношению к патологии в мозге.
Данные литературы, отражающие количественное содержание как 3-нитротирозина, так и 8-гидрокси-2-дезоксигуанозина в крови пациентов с БП, являются противоречивыми. В связи с этим в нашей работе мы оценили изменение содержания продуктов окислительного повреждения белков и ДНК крови пациентов с БП в зависимости от тяжести заболевания.
В полученных нами результатах повышение уровня 3-НТ в плазме крови относительно нормы отмечается у всех обследованных пациентов, находящихся на 1-й, 2-й, 3-й и 4-й стадиях заболевания. Следует добавить, что у пациентов на 2-й, 3-й и 4-й стадиях заболевания повышение 3-НТ было выше в среднем на 58%, в то время как у пациентов, находящихся на 1-й стадии заболевания (с впервые уставленным диагнозом и не получавших лечения) – лишь на 30%, что статистически значимо отличается от данных, полученных у пациентов на более далеко зашедших этапах заболевания. Следовательно, повышение уровня 3-НТ в плазме крови пациентов является весьма ранним и достаточно универсальным биомаркером БП, степень выраженности которого нарастает по мере прогрессирования нейродегенеративного процесса. Наши данные частично согласуются с работой Bolner R. et al. [36], в которой повышенный уровень 3-НТ в плазме крови зарегистрирован в общей выборке пациентов с БП без учета стадии заболевания. В других исследованиях [12, 35] повышение содержания 3-НТ в сыворотке крови пациентов с БП авторы связывают только с ранними стадиями заболевания (1 и 2), в то время как у пациентов, находящихся на более поздних стадиях (3 и 4), содержание 3-НТ было сопоставимо с контролем. По нашим же данным, процесс окислительного повреждения белков сопутствует всем стадиям заболевания и усиливается параллельно с развитием патологии в мозге.
Полученные нами результаты свидетельствуют о том, что повышенный относительно нормы уровень 8-гидрокси-2-дезоксигуанозина регистрируется в сыворотке крови у всех обследованных пациентов. При этом у пациентов, находящихся на 1-й, 2-й и 3-й стадиях заболевания это повышение составляло в среднем 60%, а у пациентов на 4-й стадии заболевания содержание 8-гидрокси-2-дезоксигуанозина было повышено относительно нормы уже на 183%, что в три раза превышало соответствующие значения в других подгруппах больных. Следовательно, процесс окислительного повреждения ДНК был значительно более выражен у пациентов с наиболее тяжелой стадией заболевания. Наши данные согласуются с работой Zexu Wei [7], в которой показано повышение содержания 8-OH-dG в крови при БП, однако это исследование было проведено в общей популяции больных без учета стадии заболевания. В работе Chen et al. [40] повышение уровня 8-OH-dG в лейкоцитах крови пациентов с БП коррелировало со стадией заболевания, что совпадает с нашими данными по сыворотке крови. Сопоставление этих данных позволяет предполагать наличие системных окислительных повреждений ДНК при развитии БП.
Таким образом, результаты проведенного нами исследования свидетельствуют о том, что окислительные повреждения белков и нуклеиновых кислот в крови пациентов с БП проявляются на всех стадиях заболевания, начиная с 1-й. Эти результаты существенно дополняют данные о постепенном снижении активности антиоксидантной системы при БП, полученные нами ранее [45], и свидетельствуют в пользу самостоятельной роли системных повреждений в патогенезе данного заболевания. В этом исследовании мы обнаружили системное увеличение содержания как 3-нитротирозина, так и 8-гидрокси-2-дезоксигуанозина в крови пациентов. Отдельный интерес представляют данные о наличии продуктов окисления белков и ДНК в крови пациентов с впервые поставленным диагнозом на 1-й стадии заболевания, не получавших ранее патогенетической терапии, поскольку они показывают значение этих параметров в ранней диагностике заболевания. Идентификация биомаркеров окислительного повреждения белков и нуклеиновых кислот на ранних стадиях БП является важным шагом в направлении улучшения существующих диагностических критериев, а также выявления лиц, подверженных риску заболевания. Значительное повышение содержания 8-гидрокси-2-дезоксигуанозина в сыворотке крови пациентов, находящихся на 4-й стадии, свидетельствует о связи данного показателя с тяжестью заболевания и может представить дополнительную ценность с точки зрения прогноза его развития.
В целом, понимание патогенетических факторов, ответственных как за развитие нейродегенерации, так и системных повреждений при БП, включая окислительные повреждения липидов, белков и нуклеиновых кислот, имеет большое значение для разработки комплексных протекторных подходов к лечению БП и оценки эффективности проводимого лечения.
Список литературы
Spillantini M.G., Crowther R.A., Jakes R., Hasegawa M., Goedert M. // Proc. Natl. Acad. Sci. USA. 1998. V. 95(11). P. 6469–6473.
Olanow C.W. // Mov. Disord. 2015. V. 30(1). P. 37–44.
Lin M.T., Beal M.F. // Nature. 2006. V. 443. P. 787.
Blesa J., Trigo-Damas I., Quiroga-Varela A., Jackson-Lewis V.R. // Front. Neuroanat. 2015. V. 9. P. 91.
Kim G.H., Kim J.E., Rhie S.J., Yoon S. // Exp. Neurobiol. 2015. V. 24. P. 325–340.
Paloczi J., Varga Z.V., Hasko G., Pacher P. // Antioxid. Redox Signal. 2018. V. 29. P. 75–108.
Wei Z., Li X., Li X., Liu Q., Cheng Y. // Front. Mol. Neurosci. 2018. V. 11. P. 236.
Niedzielska E., Smaga I., Gawlik M., Moniczewski A., Stankowicz P., Pera J., Filip M. // Mol. Neurobiol. 2016. V. 53. P. 4094–4125.
Jami M.-S., Salehi-Najafabadi Z., Ahmadinejad F., Hoedt E., Chaleshtori M.H., Ghatrehsamani M., Neubert T.A., Larsen J.P., Møller S.G. // Neurochem. International. 2015. V. 90. P. 134–141.
Fernández E., García-Moreno J.-M., Martín de Pablos A., Chacón J. // Antioxid. Redox Signal. 2013. V. 19. P. 912–918.
Ara J., Przedborski S., Naini A. B., Jackson-Lewis V., Trifiletti R.R., Horwitz J., Ischiropoulos H. // Proc. Natl. Acad. Sci. USA. 1998. V. 95. P. 7659–7663.
Fernandez E., Garcia–Moreno J.M., Martin De Pablos A., Chacon J. // Antioxid. Redox Signal. 2014. V. 21(15). P. 2143–2148.
Sengupta S., Bhattacharjee A. // J. Proteomics Genomics. 2016. V. 1. P. 105.
Zhan X., Wang X., Desiderio D.M. // Mass Spectrometry Reviews. 2015. V. 34. P. 423–448.
Seeley K.W., Fertig A.R., Dufresne C.P., Pinho J.P. // Int. J. Mol. Sci. 2014. V. 15. P. 6265–6285.
Cruz D.F., Fardilha M. // Porto Biomed. J. 2016. V. 1. P. 129–135.
Danielson S.R., Andersen J.K. // Free Radic. Biol. Med. 2008. V. 44(10). P. 1787–1794.
Eve D.J., Nisbet A.P., Kingsbury A.E., Hewson E.L., Daniel S.E., Lees A.J., Marsden C.D., Foster O.J. // Brain Res. Mol. Brain Res. 1998. V. 63. P. 62–71.
Gatto E.M., Riobó N.A., Carreras M.C., Cherñavsky A., Rubio A., Satz M.L., Poderoso J.J. // Nitric Oxide: Biology and Chemistry. 2000. V. 4(5). P. 534–539.
Gonos E.S., Kapetanou M., Sereikaite J., Bartosz G., Naparło K., Grzesik M., Sadowska-Bartosz I. // Aging (Albany NY). 2018. V. 10(5). P. 868–901.
McCormack A.L., Mak S.K., Di Monte D.A. // Cell. Death Dis. 2012. V. 3. P. 315.
Duda J.E., Giasson B.I., Chen Q., Gur T.L., Hurtig H.I., Stern M.B., Gollomp S.M., Ischiropoulos H., Lee V.M., Trojanowski J.Q. // American J. Pathology. 2000. V. 157(5). P. 1439–1445.
Reynolds M.R., Berry R.W., Binder L.I. // Biochemistry. 2007. V. 46(25). P. 7325–7336.
Burai R., Ait-Bouziad N., Chiki A., Lashuel H.A. // J. Am. Chem. Soc. 2015. V. 137(15). P. 5041–5052.
Yermilov V., Rubio J., Ohshima H. // FEBS Lett. 1995. V. 376. P. 207–210.
Dizdaroglu M., Jaruga P., Birincioğlu M., Rodriguez H. // Free Radic. Biol. Med. 2002. V. 32. P. 1102–1115.
Collins A.R, Cadet J., Möller L., Poulsen E.H., Viña J. // Arch. Biochem. Biophys. 2004. V. 423. P. 57–65.
Surendran S., Rajasankar S. // Neurol. Sci. 2010. V. 31. P. 531–540.
Sanchez-Ramos J.R., Overvik E., Ames B.N. // Neurodegeneration. 1994. V. 3. P. 197–204.
Alam Z.I., Jenner A., Daniel S.E., Lees A.J., Cairns N., Marsden C.D., Jenner P., Halliwell B. // J. Neurochem. 1997. V. 69. P. 1196–1203.
Nakabeppu Y., Tsuchimoto D., Yamaguchi H., Sakumi K. // J. Neurosci. Res. 2007. V. 85. P. 919–934.
Zhang J., Perry G., Smith M.A., Robertson D., Olson S.J., Graham D.G, Montine T.J. // Am. J. Pathol. 1999. V. 154(5). P. 1423–1429.
Abe T., Isobe C., Murata T., Sato C., Tohgi H. // Neurosci. Let. 2003. V. 336(2). P. 105–108.
Fernandez E., Garcıa-Moreno J.M., Martın de Pablos A., Chacon J. // Antioxid. Redox Signal. 2013. V. 19. P. 912–918.
Garcıa-Moreno J.M., Martın de Pablos A, Garcıa-Sanchez M.I., Mendez-Lucena C., Damas-Hermoso F., Rus M., Chacon J., Fernandez E. // Antioxid. Redox Signal. 2013. V. 18. P. 1296–1302.
Bolner A., Micciolo R., Bosello O., Nordera G.P. // Clin. Lab. 2016. V. 62. P. 105–112.
Sato S., Mizuno Y., Hattori N. // Neurol. 2005. V. 64. P. 1081–1083.
Bogdanov M., Matson W.R., Wang L., Matson T., Saunders-Pullman R., Bressman S.S., Flint Beal M. // Brain. 2008. V. 131(2). P. 389–96.
Seet R.C.S., Lee C.Y. J., Lim E.C.H., Tan J.J.H., Quek A.M.L.,Chong W.L., Looi W.F., Huang S.H., Wang H., Chan Y.H., Halliwell B. // Free Radic. Biol. Med. 2010. V. 48(4). P. 560–566.
Chen C.-M., Liu J.-L., Wu Y.-R., Chen Y.-C., Cheng H.-S., Cheng M.-L., Chiu D.T.-Y. // Neurobiol. Dis. 2009. V. 33(3). P. 429–35.
Kikuchi A., Takeda A., Onodera H., Kimpara T., Hisanaga K., Sato N., Nunomura A., Castellani R.J., Perry G., Smith M.A., Itoyama Y. // Neurobiol. Dis. 2002. V. 9. P. 244–248.
Jenner P. // Ann. Neurol. 2003. V. 53(3). P. 26–36.
Blanchard-Fillion B., Prou D., Polydoro M., Spielberg D., Tsika E., Wang Z., Hazen S.L., Koval M., Przedborski S., Ischiropoulos H. // J. Neurosci. 2006. V. 26. P. 6124–6130.
Borghammer P, Van Den Berge N. // J. Parkinsons Dis. 2019. V. 9. P. 281–295.
Fedorova T.N., Logvinenko A.A., Poleshchuk V.V., Illarioshkin S.N. // Neurochem. J. 2017. V. 11. P. 340–345.
Дополнительные материалы отсутствуют.
Инструменты
Нейрохимия