

Нейрохимия, 2021, T. 38, № 4, стр. 313-319
Создание и исследование клеточных моделей наследственных нейродегенеративных заболеваний с помощью направленного редактирования геномов
С. П. Медведев 1, 2, 3, Т. Б. Маланханова 1, 2, 3, К. Р. Валетдинова 1, 3, С. М. Закиян 1, 2, 3
1 ФГБНУ “Федеральный исследовательский центр Институт цитологии и генетики СО РАН”
Новосибирск, Россия
2 ФГБУН “Институт химической биологии и фундаментальной медицины СО РАН”
Новосибирск, Россия
3 ФГБУ “Национальный медицинский исследовательский центр имени академика Е.Н. Мешалкина”
Минздрава России
Новосибирск, Россия
Поступила в редакцию 19.05.2021
После доработки 20.05.2021
Принята к публикации 21.05.2021
Аннотация
Нейродегенеративные заболевания занимают значительную долю в структуре заболеваемости по всему миру. В частности, в связи с увеличением среднего возраста населения в большинстве развитых стран, резко взрастает доля пациентов с такими диагнозами, как болезнь Альцгеймера и болезнь Паркинсона. Существующие методы лечения носят преимущественно симптоматический характер и, позволяя в ряде случаев добиться выраженного клинического эффекта, не могут пока предотвратить продолжающуюся гибель нейронов или обратить вспять нейродегенеративный процесс. Во многом эти проблемы связаны и с недостаточным пониманием молекулярно-генетических механизмов, которые лежат в основе патогенеза, а также с отсутствием моделей, позволяющих получать не только качественные, но и количественные данные о процессах, происходящих в нейронах пациентов. Развитие технологий индуцированной плюрипотентности, направленной дифференцировки плюрипотентных клеток, а также редактирования генов с помощью программируемых нуклеаз, позволяет существенно расширить арсенал исследовательских инструментов, особенно в области поиска новых мишеней для лекарственной и генной терапии.
ВВЕДЕНИЕ
Клеточные технологии являются одним из основных и наиболее динамично развивающихся направлений современной биомедицины и имеют огромный потенциал применения в фармакологии, токсикологии и регенеративной медицине. Клетки, культивируемые в лабораторных условиях, активно используются для тестирования новых лекарственных соединений на ранних этапах их разработки [1, 2]. На сегодняшний день в фармакологических исследованиях используются первичные культуры соматических клеток человека, трансгенные соматические клетки, моделирующие то или иное заболевание в условиях in vitro, а также многочисленные модельные линии клеток лабораторных животных и сами животные [3–7]. Однако перечисленные выше модели имеют значительные ограничения в использовании. Животные и их клетки, используемые в исследованиях свойств лекарственных веществ, значительно отличаются по своей генетике, физиологии, морфологии от человека и клеток его тела [8]. Трансгенные клетки человека, получаемые на основе таких перевиваемых линий как HEK293 или HeLa, как правило, также не позволяют полноценно смоделировать патогенез заболевания, существующего in vivo [9–11]. Кроме того, существует серьезная проблема получения культур специфических типов соматических клеток, то есть именно тех типов клеток, на которых проявляется основной патологический эффект при том или ином заболевании. Например, получение культур дофаминэргических нейронов (гибнут при болезни Паркинсона), моторных нейронов (спинальная мышечная атрофия, боковой амиотрофический склероз), кардиомиоцитов (синдром удлиненной QT-фазы сердечного ритма и другие сердечно-сосудистые заболевания) является процедурой высокоинвазивной, а иногда просто невозможной. Кроме того, соматические клетки часто имеют ограниченный потенциал выживания и пролиферации в культуре, что затрудняет получение значительной массы клеток для тестирования библиотек потенциальных лекарственных соединений, которые могут состоять из десятков и сотен тысяч веществ [12].
Все перечисленные выше проблемы снижают уровень достоверности результатов исследований, что, в свою очередь, способствует росту ложноположительных или ложноотрицательных результатов всего цикла тестирования конкретного лекарственного соединения. Рост ошибочных результатов в фармакологии, как правило, связан со значительными экономическими потерями. Выходом из данной ситуации может стать использование универсальных или плюрипотентных клеток, которые способны неограниченно культивироваться и давать в результате дифференцировки необходимые для исследований клеточные производные. Создание клеточных платформ, которые совмещаю в себе трансгенные клеточные линии для проведения направленного нокаута генов, выявления роли однонуклеотидных полиморфизмов (SNPs) и количественного измерения происходящих в клетках патологических процессов, может существенно расширить наши представления о молекулярно-генетических механизмах патогенеза заболеваний, найти новые мишени для лекарственной и генной терапии, а также проводить скрининговые исследования перспективных лекарственных соединений.
ИНДУЦИРОВАННЫЕ ПЛЮРИПОТЕНТНЫЕ СТВОЛОВЫЕ КЛЕТКИ
В 2006 году произошло революционное событие для всей клеточной биологии. Группой японских исследователей было обнаружено, что соматические клетки мыши могут быть подвергнуты перепрограммированию путем сверхэкспрессии набора определенных факторов (Oct4, Sox2, Klf4 и c-Myc) [13]. Позже несколькими группами ученых были получены подобные клетки человека [14–16]. Полученные клетки, которые по своим свойствам были очень сходны с эмбриональными стволовыми клетками, были названы индуцированными плюрипотентными стволовыми клетками (ИПСК). Данное открытие привлекло огромное внимание мирового научного сообщества, так как в руках исследователей появился уникальный инструмент, который может быть использован в различных фундаментальных и прикладных исследованиях, включающих изучение биологии развития человека, моделирование заболеваний и доклинические испытания в лабораторных условиях, а также применение ИПСК в заместительной терапии.
ИПСК человека имеют ряд существенных преимуществ в контексте применения в доклинических испытаниях потенциальных лекарственных соединений:
• ИПСК могут быть получены из различных типов соматических клеток человека (от фибробластов кожи до клеток периферической крови). Методики получения соматических клеток, пригодных для получения ИПСК, как правило, являются малоинвазивными [14, 17–19];
• по своим свойствам ИПСК очень сходны с эмбриональными стволовыми клетками человека, являются плюрипотентными, то есть могут давать при дифференцировке практически все производные, из которых состоит тело взрослого человека [14–17, 20];
• ИПСК обладают неограниченным потенциалом пролиферации в культуре и при необходимости могут быть получены в масштабе, необходимом для проведения скрининга библиотек лекарственных соединений [21];
• ИПСК аутологичны по отношению к донору соматических клеток. Они могут быть получены от конкретного пациента в любой период его жизни, что дает возможность поиска лекарств и исследования их возможных токсических свойств на клетках пациентов с конкретным генетическим фоном. Данное свойство ИПСК может сделать их одной из основ медицины нового поколения, так называемой персонифицированной медицины [22];
• Применение ИПСК дает новые возможности исследования патогенеза и поиска лекарств для терапии комплексных заболеваний, которые захватывают не один тип клеток. Например, из ИПСК, полученных из клеток одного пациента, могут быть получены нейроны, кардиомиоциты, гепатоциты и другие типы дифференцированных клеток. Таким образом, становится возможным исследование действия лекарственных соединений на клетки с разными молекулярно-генетическими, физиологическими и биохимическими характеристиками, в то же время полученными от одного пациента [22–26].
Несмотря на значительный прогресс в создании клеточных моделей заболеваний на основе ИПСК, в исследованиях пациент-специфичных ИПСК ученые сталкиваются с несколькими проблемами. Во-первых, невозможно подобрать полностью адекватный “здоровый” контроль для исследования дифференцировки и оценки влияния различных воздействий на клетки. Контрольные клетки, полученные от здоровых людей, имеют другой общий генетический фон. Во-вторых, линии клеток могут отличаться друг от друга по спектру дифференцировки, иметь разную скорость деления и другие индивидуальные особенности, даже если они получены одним методом и в одной лаборатории. Безусловно, данные особенности могут сказываться на точности результатов исследований в целом. В-третьих, существует большая проблема, связанная с наличием пациентов, которые могли бы стать донорами соматических клеток. Особенно остро эта проблема стоит, если речь идет о редких наследственных заболеваниях или редких разновидностях мутаций [27]. Кроме того, для работы с пациентами необходим определенный набор разрешений и лицензий. В последние годы активно развиваются технологии направленного редактирования геномов, которые открывают новые, еще более широкие, возможности применения ИПСК в биомедицинских исследованиях и позволяют решить вышеперечисленные проблемы.
CRISPR-ОПОСРЕДОВАННЫЕ СИСТЕМЫ – ИНСТРУМЕНТ ДЛЯ ИССЛЕДОВАНИЯ КЛЕТОЧНЫХ МОДЕЛЕЙ ЗАБОЛЕВАНИЙ
В начале 2013 г. были опубликованы работы, в которых описывалось использование компонентов адаптивной иммунной системы бактерий CRISPR–Cas9 для редактирования геномов млекопитающих [28, 29]. Данная система, обладает высокой эффективностью и специфичностью работы, но при этом отличается простотой создания генетических конструкций. Для создания конструкции, опознающей конкретную последовательность ДНК в геноме, достаточно поместить синтетический фрагмент ДНК длиной около 20 нуклеотидов (спейсер) в специальный вектор, экспрессирующий направляющую РНК (нРНК) и нуклеазу Cas9. Спейсер является частью нРНК, и его последовательность определяет специфичность связывания нРНК с ДНК-мишенью. Связывание осуществляется по принципу комплементарности оснований РНК–ДНК. При этом, нуклеаза Cas9, находящаяся в комплексе с нРНК производит двунитевой разрыв ДНК. Разрывы ДНК, которые были внесены нуклеазой, репарируются по двум основным общим механизмам. Это негомологичное сшивание концов и гомологичная рекомбинация. При негомологичном сшивании концов ДНК, как правило, в месте разрыва образуются небольшие делеции или/и инсерции нуклеотидов, что может приводить к сдвигу рамки считывания генов (если разрыв находится в кодирующей области) и, как следствие, к нокауту гена. Если же репарация идет по механизму гомологичной рекомбинации, а для этого необходима донорная молекула ДНК, то в конкретное место генома может быть помещен трансген или заменен единственный нуклеотид. Этот механизм может с успехом применяться для внесения или исправления генных мутаций, внесения репортерных генов и так далее [28, 29].
С момента первых работ по применению CRISPR–Cas9 для модификации геномов в клетках животных и человека, эта технология претерпела значительное совершенствование. В частности, были созданы новые, генноинженерные варианты белка Cas9, которые практически не обладают нецелевой активностью, а также найдены альтернативные системы, например, CRISPR–Cpf1 (Cas12a) [30–32].
Прогресс в области создания и применения CRISPR-опосредованных систем открывает широкие перспективы их применения для изучения полигенных нейродегенеративных заболеваний, например, таких как болезнь Паркинсона, болезнь Альцгеймера или боковой амиотрофический склероз. При исследовании клеточных моделей нейродегенеративных заболеваний данные системы редактирования можно использовать в следующих направлениях:
• Нокаут генов для поиска тех из них, которые задействованы в патогенезе заболеваний напрямую или модифицируют его [33];
• Создание изогенных клеточных моделей [26, 34–36];
• Создание трансгенных клеточных линий, которые предназначены для мониторинга направленной дифференцировки в релевантные типы нейронов, а также получения качественных и количественных данных о происходящих в клетках патологических процессах (апоптоз, оксидативный стресс, дисфункция эндоплазматического ретикулума) [37, 38].
ГЕНЕТИЧЕСКИ КОДИРУЕМЫЕ СЕНСОРЫ ПАТОЛОГИЧЕСКИХ КЛЕТОЧНЫХ ПРОЦЕССОВ
Одним из перспективных подходов для исследования молекулярно-генетических механизмов нейродегенерации in vitro может быть совмещение технологий создания линий пациент-специфичных ИПСК, редактирования геномов, генетически кодируемых биосенсоров и направленной дифференцировки ИПСК в релевантные типы нейронов. Для этого могут быть применены генетические конструкции, которые могут быть интегрированы в локус AAVS1 ИПСК пациентов и здоровых доноров с применением системы CRISPR-Cas9 [38]. Использование направленной интеграции трансгенов в локус AAVS1, который относится к так называемым “safe harbor” локусам, позволяет контролировать число копий трансгенов на геном, а также обеспечивает более стабильную экспрессию тарнсгенов [39]. Генетически кодируемые сенсоры представляют собой искусственно созданные генноинженерные конструкции, которые позволяют визуализировать и получать количественные данные о процессах, происходящих в живых клетках, например, в нейронах, получаемых при направленной дифференцировке ИПСК пациентов с нейродегенеративными заболеваниями [40, 41]. В частности, с помощью сенсоров можно изучать такие процессы, как оксидативный стресс и дисфункция эндоплазматического ретикулума. Высокий уровень метаболизма нейронов подразумевает высокие показатели образования активных форм кислорода (АФК), и соответственно, нейроны должны обладать эффективной системой защиты от них, однако ее становится недостаточно при оксидативном стрессе. Известно, что глутатион является внутриклеточным антиоксидантом, и при развитии патологического процесса показана значительная роль нарушения гомеостаза глутатиона [42]. Например, снижение концентрации глутатиона в митохондриях влечет за собой активацию нейрональной 12-липоксигеназы, что приводит к образованию пероксидов, притоку Са2+ и, в конечном счете, к гибели клеток. Например, при болезни Паркинсона происходит потеря дофаминергических нейронов в черной субстанции головного мозга, а дополнительными источниками окислительного стресса является автоокисление дофамина, которое приводит к образованию АФК, и ферментативное окисление, катализируемое моноаминоксидазой, которое приводит к образованию перекиси водорода. Обычно H2O2 инактивируется каталазой или глутатион-S-пероксидазой в реакции, в которой глутатион используется в качестве кофактора. Было показано, что с помощью генетически кодируемых сенсоров возможно получить достаточно чувствительную систему для оценки окислительно-восстановительного потенциала глутатиона в живых клетках. Фермент Grx1 в конструкции обеспечивает реакции окисления и восстановления белка roGFP2 после взаимодействия с глутатионом. Оценка окислительно-восстановительного потенциала при этом проводится с помощью ратиометрических расчетов уровней возбуждения светом определенной длины волны (405/488 нм) белка roGFP2. Еще одним преимуществом данного подхода является то, что можно применить два типа конструкций (Cyto и Mito), которые позволяют производить независимые измерения в цитоплазме и митохондриях клеток (за счет наличия сигнального пептида, который перенаправляет белок-сенсор в митохондрии). Данные конструкции будут интегрированы под доксициклин-зависимый промотор, чтобы систему можно было активировать на разных стадиях дифференцировки ИПСК в зрелые дофаминергические нейроны. Помимо этого, данная система может быть применена для выявления и тестирования химических соединений, направленных на нормализацию уровня АФК.
Следующий, не менее важный момент, – явление стресса эндоплазматического ретикулума (ЭПР), который вызван накоплением несвернутых, дефектных белков в люмене ЭПР [43]. Данный патофизиологический процесс характерен для большинства нейродегенеративных заболеваний и может быть одной из мишеней для терапевтического воздействия [44–49]. Для формирования третичной структуры белка важно образование дисульфидных связей, что опосредуется ферментом протеиндисульфид-изомераза, который в свою очередь активируется окислением. Данный процесс тесно связан с образованием перекиси водорода, в пространстве в которым оказываются вновь синтезированные белки. В ЭПР при этом накапливаются денатурированные или частично свернутые белки в таком случае активируется система Unfolded Protein Response (UPR). UPR может способствовать повышению активности шаперонов и снижению синтеза белка. Этот механизм представлен белками, заякоренными в мембране ЭПР – PERK, IRE1 и ATF6. Показано, что мутация белка паркина приводит к нарушению функции протеасом, и соответственно, к перегрузке ЭПР. Было показано на мышах и на культуре клеток, что можно визуализировать и стресс ЭПР с помощью генетически кодируемых сенсоров.
Для изучения процессов связанных со стрессом ЭПР может быть применена следующая тестовая модель: в локус AAVS1, под контролем доксициклин-управляемого промотора производится встройка конструкции XBP1-флуоресцентный белок [50], функционирование которой зависит от активации UPR с через белок IRE1 (в XBP1 эндорибонуклеаза IRE1 будет вырезать 26-нуклеотидный интрон сдвигая рамку считывания белка, тем самым обеспечивая экспрессию той части конструкции, которая кодирует флуоресцентный белок), что позволит визуализировать клетки с активированным белком IRE1. Клетки с активированной системой UPR (имеющие красное свечение) могут быть отсортированы и использованы для более подробного молекулярно-генетического и биохимического исследования. Кроме того, данная система может работать в обратном направлении. Выделенные с помощью сортировки клетки могут применятся для тестирования препаратов, которые направлены на коррекцию нарушений фолдинга белков в ЭПР (рис. 1).
Рис. 1.
Схема функционирования генетически кодируемых биосенсоров стресса эндоплазматического ретикулума. Схема работы биосенсора XBP1-TagRFP (а). Схема работы биосенсора XBP1-luc2-hRluc (б). Схема работы биосенсора EGFP-ATF6 (в). XBP1 – фрагмент гена XBP1, содержащий стресс-чувствительный интрон размером 26 п.н., TagRFP – ген красного флуоресцентного белка, EGFP – ген зеленого флуоресцентного белка, luc2 – ген люциферазы светлячка Photinus pyralis, Rluc – ген люциферазы кораллового полипа Renilla reniformis, Puro – ген устойчивости к пуромицину, STOP – стоп-кодон, F – FLAG-эпитоп.
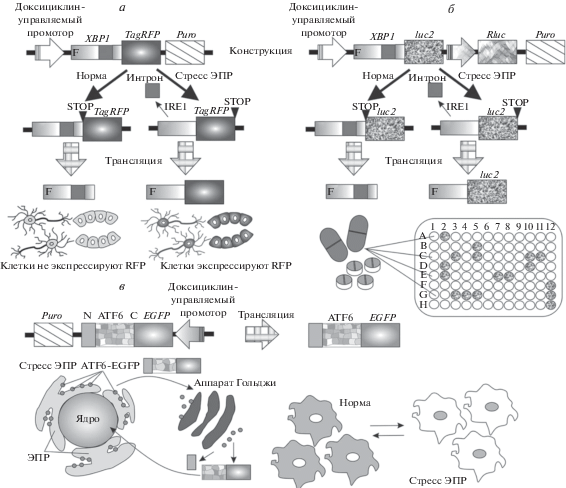
Помимо этого, может быть применена система, в которой ген, кодирующий флуоресцентный белок, заменен геном люциферазы. Это позволит проводить измерения уровня активности люциферазы (который будет прямо пропорционален уровню активации IRE1 и стресс-зависимого сплайсинга мРНК XBP1) в ответ на различные воздействия, в том числе, на действие низкомолекулярных химических соединений. Кроме того, данный вариант сенсора позволяет масштабировать измерения до 96-луночного формата, что существенно повышает производительность и дает возможность скрининга библиотек потенциальных лекарственных препаратов (рис. 1).
Помимо использования взаимодействия IRE1 и XBP1 существуют и другие сенсоры стресса ЭПР, например, основанные на визуализации активации транскрипционного фактора ATF6 (Activating transcription factor 6) [43]. В норме данный белок заякорен в мембране ЭПР. В ответ на стресс он перемещается в аппарат Гольджи, где подвергается протеолизу, активируется и перемещается в ядро клетки. Принцип действия сенсора основан на том, что к белку ATF6 с N-конца присоединен флуоресцентный белок EGFP, который не мешает протеолизу, активации и перемещению ATF6. При этом мы можем наблюдать распространение флуоресцентного сигнала в ядро клетки. Подсчитывая ядра с сигналами на микроизображениях и статистически обрабатывая эту информацию, можно судить о степени выраженности стресса ЭПР (рис. 1).
ЗАКЛЮЧЕНИЕ
Поиск новых, эффективных и безопасных средств терапии нейродегеративных заболеваний является одним из вызовов современной медицины и фармакологии. Последние достижения клеточной и молекулярной биологии позволяют по-новому взглянуть на создание модельных систем для исследований in vitro. Эти системы, на основе трансгенных индуцированных плюрипотентных стволовых клеток, позволяют производить широкий спектр исследований, выявлять тонкие молекулярные механизмы патогенеза, искать и находить мишени для терапевтического воздействия и тестировать потенциальные лекарственные препараты.
Список литературы
Tralau T., Luch A. // Trends Pharmacol. Sci. 2012. V. 33. P. 353–364.
Niu N.,Wang L. // Pharmacogenomics. 2015. V. 16. P. 273–285.
Feany M.B., Bender W.W. // Nature. 2000. V. 404. P. 394–398.
Liu Z., Wang X., Yu Y., Li X., Wang T., Jiang H., Ren Q., Jiao Y., Sawa A., Moran T., Ross C.A., Montell C., Smith W.W. // Proc. Natl. Acad. Sci. USA. 2008. V. 105. P. 2693–2698.
Li T., Yang D., Sushchky S., Liu Z., Smith W.W. // Parkinsons Dis. 2011. V. 2011. P. 942412.
Yao C., El Khoury R., Wang W., Byrd T.A., Pehek E.A., Thacker C., Zhu X., Smith M.A., Wilson-Delfosse A.L., Chen S.G. // Neurobiol. Dis. 2010. V. 40. P. 73–81.
McGurk L., Berson A., Bonini N.M. // Genetics. 2015. V. 201. P. 377–402.
Rosen D.R., Martin-Morris L., Luo L.Q., White K. // Proc. Natl. Acad. Sci. USA. 1989. V. 86. P. 2478–2482.
Bungeroth M., Appenzeller S., Regulin A., Volker W., Lorenzen I., Grotzinger J., Pendziwiat M., Kuhlenbaumer G. // Neurobiol. Aging. 2014. V. 35. P. 1913–1919.
Dansithong W., Paul S., Scoles D.R., Pulst S.M., Huynh D.P. // PLoS One. 2015. V. 10. P. e0136930.
Eckermann K., Kugler S., Bahr M. // Biochim. Biophys. Acta. 2015. V. 1852. P. 1658–1664.
Ebert A.D.,Svendsen C.N. // Nat. Rev. Drug Discov. 2010. V. 9. P. 367–372.
Takahashi K.,Yamanaka S. // Cell. 2006. V. 126. P. 663–676.
Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. // Cell. 2007. V. 131. P. 861–872.
Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R., Slukvin, II, Thomson J.A. // Science. 2007. V. 318. P. 1917–1920.
Lowry W.E., Richter L., Yachechko R., Pyle A.D., Tchieu J., Sridharan R., Clark A.T., Plath K. // Proc. Natl. Acad. Sci. USA. 2008. V. 105. P. 2883–2888.
Dimos J.T., Rodolfa K.T., Niakan K.K., Weisenthal L.M., Mitsumoto H., Chung W., Croft G.F., Saphier G., Leibel R., Goland R., Wichterle H., Henderson C.E., Eggan K. // Science. 2008. V. 321. P. 1218–1221.
Aasen T., Raya A., Barrero M.J., Garreta E., Consiglio A., Gonzalez F., Vassena R., Bilic J., Pekarik V., Tiscornia G., Edel M., Boue S., Izpisua Belmonte J.C. // Nat. Biotechnol. 2008. V. 26. P. 1276–1284.
Loh Y.H., Hartung O., Li H., Guo C.G., Sahalie J.M., Manos P.D., Urbach A., Heffner G.C., Grskovic M., Vigneault F., Lensch M.W., Park I.H., Agarwal S., Church G.M., Collins J.J., Irion S., Daley G.Q. // Cell Stem Cell. 2010. V. 7. P. 15–19.
Medvedev S.P., Grigor’eva E.V., Shevchenko A.I., Malakhova A.A., Dementyeva E.V., Shilov A.A., Pokushalov E.A., Zaidman A.M., Aleksandrova M.A., Plotnikov E.Y., Sukhikh G.T., Zakian S.M. // Stem Cells Dev. 2011. V. 20. P. 1099–1112.
Elitt M.S., Barbar L., Tesar P.J. // Hum Mol Genet. 2018. V. 27. P. R89–R98.
Soldner F., Hockemeyer D., Beard C., Gao Q., Bell G.W., Cook E.G., Hargus G., Blak A., Cooper O., Mitalipova M., Isacson O., Jaenisch R. // Cell. 2009. V. 136. P. 964–977.
Sanchez-Danes A., Richaud-Patin Y., Carballo-Carbajal I., Jimenez-Delgado S., Caig C., Mora S., Di Guglielmo C., Ezquerra M., Patel B., Giralt A., Canals J.M., Memo M., Alberch J., Lopez-Barneo J., Vila M., Cuervo A.M., Tolosa E., Consiglio A., Raya A. // EMBO Mol. Med. 2012. V. 4. P. 380–395.
Sareen D., O’Rourke J.G., Meera P., Muhammad A.K., Grant S., Simpkinson M., Bell S., Carmona S., Ornelas L., Sahabian A., Gendron T., Petrucelli L., Baughn M., Ravits J., Harms M.B., Rigo F., Bennett C.F., Otis T.S., Svendsen C.N., Baloh R.H. // Sci. Transl. Med. 2013. V. 5. P. 208ra149.
Juopperi T.A., Kim W.R., Chiang C.H., Yu H., Margolis R.L., Ross C.A., Ming G.L., Song H. // Mol. Brain. 2012. V. 5. P. 17.
Malankhanova T., Suldina L., Grigor’eva E., Medvedev S., Minina J., Morozova K., Kiseleva E., Zakian S., Malakhova A. // J. Pers. Med. 2020. V. 10.
Soldner F., Laganiere J., Cheng A.W., Hockemeyer D., Gao Q., Alagappan R., Khurana V., Golbe L.I., Myers R.H., Lindquist S., Zhang L., Guschin D., Fong L.K., Vu B.J., Meng X., Urnov F.D., Rebar E.J., Gregory P.D., Zhang H.S., Jaenisch R. // Cell. 2011. V. 146. P. 318–331.
Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. // Science. 2013. V. 339. P. 819–823.
Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. // Science. 2013. V. 339. P. 823–826.
Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A., Koonin E.V., Zhang F. // Cell. 2015. V. 163. P. 759–771.
Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. // Nature. 2016. V. 529. P. 490–495.
Schmid-Burgk J.L., Gao L., Li D., Gardner Z., Strecker J., Lash B., Zhang F. // Mol. Cell. 2020. V. 78. P. 794–800. e798.
Rehbach K., Fernando M.B., Brennand K.J. // J. Neurosci. 2020. V. 40. P. 1176–1185.
Schuster S., Saravanakumar S., Schols L., Hauser S. // Stem Cell Res. 2019. V. 34. P. 101378.
Malankhanova T., Sorokin M., Medvedev S., Zakian S., Malakhova A. // Curr. Protoc. Hum. Genet. 2020. V. 106. P. e100.
Nagel M., Mussig S., Hoflinger P., Schols L., Hauser S., Schule R. // Stem Cell Res. 2020. V. 49. P. 102059.
Calatayud C., Carola G., Fernandez-Carasa I., Valtorta M., Jimenez-Delgado S., Diaz M., Soriano-Fradera J., Cappelletti G., Garcia-Sancho J., Raya A., Consiglio A. // Sci. Rep. 2019. V. 9. P. 6811.
Ustyantseva E.I., Medvedev S.P., Vetchinova A.S., Minina J.M., Illarioshkin S.N., Zakian S.M. // Biochemistry (Mosc.). 2019. V. 84. P. 299–309.
Sadelain M., Papapetrou E.P., Bushman F.D. // Nat. Rev. Cancer. 2011. V. 12. P. 51–58.
Kim H., Ju J., Lee H.N., Chun H., Seong J. // Sensors (Basel). 2021. V. 21.
Bi X., Beck C., Gong Y. // Biosensors (Basel). 2021. V. 11.
Schulz J.B., Lindenau J., Seyfried J., Dichgans J. // Eur. J. Biochem. 2000. V. 267. P. 4904–4911.
Samali A., Fitzgerald U., Deegan S., Gupta S. // Int. J. Cell Biol. 2010. V. 2010. P. 830307.
Lee J.H., Han J.H., Kim H., Park S.M., Joe E.H., Jou I. // Acta Neuropathol. Commun. 2019. V. 7. P. 68.
Colla E. // Front. Neurosci. 2019. V. 13. P. 560.
Dafinca R., Barbagallo P., Talbot K. // Front. Cell. Neurosci. 2021. V. 15. P. 653688.
Bella E.D., Bersano E., Antonini G., Borghero G., Capasso M., Caponnetto C., Chio A., Corbo M., Filosto M., Giannini F., Spataro R., Lunetta C., Mandrioli J., Messina S., Monsurro M.R., Mora G., Riva N., Rizzi R., Siciliano G., Silani V., Simone I., Soraru G., Tugnoli V., Verriello L., Volanti P., Furlan R., Nolan J.M., Abgueguen E., Tramacere I., Lauria G. // Brain. 2021. https://doi.org/10.1093/brain/awab167
Shacham T., Patel C., Lederkremer G.Z. // Biomolecules. 2021. V. 11.
Ghemrawi R.,Khair M. // Int. J. Mol. Sci. 2020. V. 21.
Iwawaki T., Akai R., Kohno K., Miura M. // Nat Med. 2004. V. 10. P. 98–102.
Дополнительные материалы отсутствуют.