

Нейрохимия, 2021, T. 38, № 4, стр. 329-338
Использование геномного редактирования для терапии болезни Альцгеймера: от эксперимента к клинике
М. Ю. Степаничев
Федеральное государственное бюджетное учреждение науки
Институт высшей нервной деятельности и нейрофизиологии РАН
Москва, Россия
Поступила в редакцию 13.05.2021
После доработки 22.06.2021
Принята к публикации 23.06.2021
Аннотация
В представленном обзоре рассмотрены современные достижения в области использования методов геномного редактирования для экспериментальной терапии нейродегенеративных заболеваний, в первую очередь болезни Альцгеймера. Кратко рассмотрены основные подходы, используемые для геномного редактирования, и системы редактирования, рассмотрены их преимущества и недостатки. Приведены конкретные примеры использования геномного редактирования для коррекции мутаций в генах, ассоциированных с болезнью Альцгеймера. Проанализированы имеющиеся проблемы, которые необходимо решить для внедрения геномного редактирования в практику лечения нейродегенеративных заболеваний.
ВВЕДЕНИЕ
Болезнь Альцгеймера (БА) – одна из наиболее распространенных причин возникновения деменции у пациентов старшей возрастной группы – представляет собой серьезную проблему для медицины и современного общества в целом. По данным клинико-эпидемиологического исследования репрезентативных групп населения г. Москвы, БА страдает около 4.5% населения в возрасте 60 лет и старше. Экстраполяция этих показателей на половозрастную структуру населения страны в целом показывает, что от этого заболевания страдает около 1% всего населения России [1]. Патогенез БА остается достаточно слабо изученным, несмотря на предпринимаемые усилия. Вероятно, это связано с его комплексностью и манифестацией симптомов после продолжительного латентного периода с момента старта заболевания. Риск развития БА обусловлен действием как моногенных, так и полигенных факторов, а также воздействием факторов окружающей среды. Менее 5% случаев БА, которые связаны с наличием мутаций в генах пресенилинов PSEN1 и PSEN2 и белка-предшественника β-амилоида (APP), проявляются достаточно рано, в возрасте до 60 лет. Мутация в гене, кодирующем аполипопротеин Е (APOE), считается фактором риска развития БА с поздним началом.
Проведенные в последние годы полногеномные исследования позволили связать функции более чем 25 генов с патогенезом БА. Установлена явная связь семейных форм БА с ранним проявлением симптоматики с мутациями в генах APP, PSEN1 и PSEN2, по крайней мере, в 10% случаев. Кроме того, около 25% пациентов с поздней манифестацией БА являются носителями мутантного аллеля APOE ε4. Наряду с этим была продемонстрирована ассоциация с генами, кодирующими белки, вовлеченные в разные патофизиологические процессы патогенеза БА (табл. 1). Все это указывает на существенную роль генетического компонента в патогенезе этого нейродегенеративного заболевания.
Таблица 1.
Возможные гены-мишени для терапевтического воздействия с использованием методов геномного редактирования
| Патофизиологические процессы | Гены |
|---|---|
| Метаболизм APP/β-амилоида | APP, PSEN1, PSEN2, ApoEε4, Clusterin, BACE |
| Метаболизм/фосфорилирование τ-белка | GSK-3β, P25/Cdk5, Bin-1 |
| Холинергическая трансмиссия | ChAT, nAChR, mAChR |
| Убиквитинирование | USP7, USP47 |
| Нейровоспаление/иммунитет | CX3CR1, CD33, TREM2, GMF, NLRP3 |
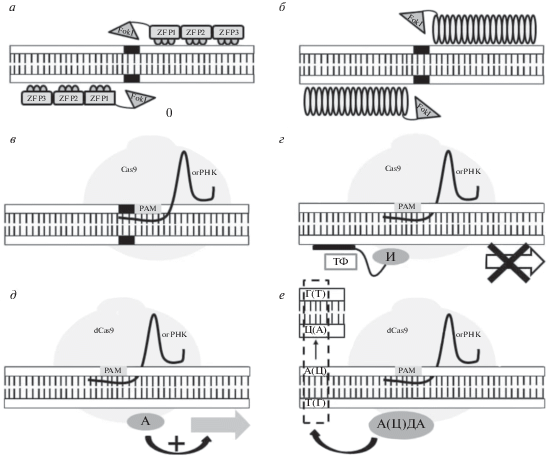
Системы геномного редактирования на основе дизайнерских нуклеаз (нуклеазы цинковых пальцев (zink finger nucleases, ZFN), подобные транскрипционному активатору эндонуклеазы (transcription activator-like endonuclease, TALEN), CRISPR–Cas9 (clustered regularly interspaced short palindromic repeats-(CRISPR)-associated protein (Cas9)) и другие представляют собой мощный инструмент для манипуляций с генами. Эти системы позволяют осуществлять не только канонические манипуляции с ДНК, такие как делеции или инсерции участков генов (рис. 1а–в), но и неканонические, а именно: репрессия или активация транскрипции и замена отдельных оснований (рис. 1г–е). Это дает возможность необратимо инактивировать гены, корректировать их последовательность, модулировать их активность. Разработка методов геномного редактирования для терапии таких моногенных заболеваний, как серповидноклеточная анемия или мышечная дистрофия, является удачным примером использования технологии для лечения моногенных заболеваний. Результаты этой работы с высокой вероятностью будут внедрены в клинику [2]. Успешность применения систем редактирования для модификации генома в нейронах в культуре и в мозге дает надежду на возможность их использования и для терапии заболеваний нервной системы. В этом обзоре мы рассмотрим существующие экспериментальные подходы к коррекции патогенетических механизмов БА, созданные на основе геномного редактирования. В заключении будут кратко проанализированы возможные проблемы, связанные с использованием этих методов, как возможного средства для лечения БА и других нейродегенеративных патологий.
Рис. 1.
Канонические и неканонические подходы к использованию дизайнерских нуклеаз. (а–в), использование ZFN, TALEN и CRISPR–Cas9 для получения двухцепочечных разрывов ДНК; (г), применение CRISPR–dCas9 интерференции для репрессии гена; (д), применение CRISPR–dCas9 для активации транскрипции; (е), применение CRISPR–nCas9 для редактирования отдельных оснований. ZFP, белок цинкового пальца; Fok1, эндонуклеаза; PAM, protospacer adjacent motif; И, ингибитор транскрипции; ТФ, транскрипционный фактор; А, активатор транскрипции; А(Ц)ДА, аденин(цитозин)дезаминаза.
ПОДХОДЫ К РЕДАКТИРОВАНИЮ ГЕНОМА И ИХ ПРИМЕНЕНИЕ ДЛЯ ТЕРАПЕВТИЧЕСКИХ ЦЕЛЕЙ
При разработке терапевтических подходов наиболее часто рассматривают три основных типа манипуляций, а именно: инактивация гена, внедрение нового гена и редактирование или модификация гена [3]. Методы геномного редактирования, которые активно разрабатываются в последние десятилетия, способны обеспечить изменение генома в определенном (целевом) участке за счет использования активности направленных нуклеаз. Технически, необходимо обеспечить разрыв двухцепочечной ДНК в участке, где расположен целевой ген, после чего происходит репарация поврежденного участка. В качестве ферментов, осуществляющих разрезание ДНК, выступают эндонуклеазы, которые катализируют расщепление фосфодиэфирных связей внутри полинуклеотидных цепей. В настоящее время наиболее распространенными как в экспериментальных, так и в клинических исследованиях являются три типа нуклеаз (рис. 1а–в): ZFN [4], TALEN [5] и CRISPR/Cas9 [6].
Основными характеристиками каждого из этих инструментов являются наличие эндонуклеазы и ДНК-связывающего домена, который обеспечивает связывание нуклеазы с ДНК и определяет специфичность положения, в котором это связывание происходит [3]. В случае ZFN (рис. 1а) точность связывания с целевым участком ДНК определяется наличием минимум трех белков цинковых пальцев, а увеличение числа ДНК-связывающих цинковых пальцев в составе комплекса повышает аффинность связывания, хотя это в свою очередь снижает активность конструкции в целом [7]. Для точного разрезания двух цепей ДНК необходимо, чтобы нуклеаза образовала димер. Поэтому для редактирования определенного участка нужно использовать пару белков, отличающихся по составу цинковых пальцев.
Другой тип конструкций, применяемый для редактирования, использует для связывания с ДНК белки TALE (рис. 1б). В отличие от ZFN, в которой каждый домен взаимодействует с тремя нуклеотидами, один домен белка TALE связывается с одним нуклеотидом. Поэтому для обеспечения специфичности связывания TALEN с целевыми участками ДНК необходимо создать достаточно крупный белок, имеющий множество доменов [8]. Разрезание двух цепочек ДНК происходит после образования димера, что так же, как и в случае ZFN, обусловливает использование одновременно двух белков для редактирования одного гена. Обе упомянутые системы редактирования генома обладают сходной эффективностью, но при этом цитотоксичность и сложность получения конструкций ниже в случае использования TALEN [3].
CRISPR–Сas9 является, пожалуй, наиболее распространенным инструментом редактирования генома, основанным на использовании механизма адаптивного иммунитета прокариот. Эта система представлена CRISPR-кассетой и геном, кодирующим белок Cas9. CRISPR–Cas система способна обеспечивать устойчивость клеток прокариот к бактериофагам и плазмидам, которые содержат протоспейсеры, последовательности, комплементарные спейсерам CRISPR-кассеты [9, 10]. В этой системе нуклеаза Cas9 обеспечивает образование двухцепочечного разрыва ДНК, а место этого разрыва определяется последовательностью одноцепочечной направляющей (гидовой) РНК (огРНК). Преимуществом CRISPR–Cas9 является относительная простота конструкции, высокая эффективность редактирования, низкая цитотоксичность, что способствует ее широкому внедрению в исследовательскую практику, а в ближайшем будущем и в терапию ряда обусловленных наследственностью заболеваний [2, 11].
В отличие от описанных выше нуклеаз ZFN и TALEN, белок Cas9 для взаимодействия с ДНК использует CRISPR-кассету, состоящую из CRISPR-ассоциированной РНК и трансактивирующей РНК (рис. 1в) [12]. Эти две РНК обычно объединены в одну химерную молекулу огРНК. Помимо двух функциональных доменов огРНК должна содержать последовательность из 20 нуклеотидов, комплементарную целевому участку ДНК. Для обеспечения связывания CRISPR–Cas9 в ДНК должны содержаться последовательности из трех нуклеотидов вида NGG (protospacer adjacent motif, PAM), непосредственно прилежащие к целевому участку со стороны 3'-конца. После взаимодействия с комплементарным огРНК участком ДНК нуклеаза Cas9 разрезает обе цепи, образуя разрыв на расстоянии в три нуклеотида от PAM. Образовавшийся разрыв репарируется путем негомологичного соединения концов (non-homologous end joining, NHEJ), при котором свободные концы ДНК сшиваются друг с другом с образованием делеции или вставкой одного или нескольких нуклеотидов. При гомологичной репарации (homologous dependent repair, HDR) разрыв репарируется с использованием дополнительной матрицы, с которой происходит копирование участка вокруг образованного разрыва. В качестве матрицы может служить последовательность нуклеотидов неповрежденной парной хромосомы или последовательность нуклеотидов, внесенная извне с плазмидой или олигонуклеотидом.
Все три типа нуклеаз были апробированы в исследованиях для модификации генома в нейронах и клетках глии [13–16], включая нокаут гена, встраивание нового гена в геном или редактирование имеющихся модификаций гена, препятствующих его нормальной работе. Однако, наряду с этими “каноническими” возможностями, перспективным представляется использование систем редактирования для изменения уровня транскрипции за счет модуляции активности регуляторов транскрипции, изменения уровня метилирования промоторных участков или репрессии транскрипции [17]. В частности, при использовании CRISPR-интерференции деактивированная нуклеаза Cas9 (dCas9) в комплексе с ингибитором транскрипции может быть применена для репрессии гена (рис. 1г). Такой подход может быть перспективен для ингибирования избыточной экспрессии, например, α-синуклеина при болезни Паркинсона или APP при БА. Аналогично этому комплекс CRISPR–dCas9 с активаторами транскрипции может быть использован для реализации альтернативной стратегии (рис. 1д), т.е. CRISPR-активации.
Еще одним важным подходом к использованию возможностей CRISPR–Cas является редактирование однонуклеотидных замен [18]. Присоединение к CRISPR–nCas9 (nickase Cas9) аденин- или цитозин-дезаминазы предоставляет возможность замены А:Т на Г:Ц и Ц:Г на Т:А соответственно (рис. 1е). Такой подход максимально подходит в тех ситуациях, когда PAM расположен вблизи нуклеотида, который необходимо отредактировать, но при этом изменение соседних нуклеотидов нежелательно.
К неканоническим подходам относится и редактирование РНК, которое позволяет изменить экспрессию гена на уровне транскрипта. Несмотря на то, что антисмысловые нуклеотиды известны давно и уже применяются, например, для лечения спинальной мышечной атрофии [19, 20], их использование имеет ряд ограничений. Поэтому использование Cas13d, фермента, который связывается с РНК в клетках и расщепляет ее, может служить альтернативой с большей спецификой в отношении мишени по сравнению с антисмысловыми олигонуклеотидами [21].
В следующих разделах мы рассмотрим применение этих технологий для экспериментальной терапии БА.
КОРРЕКЦИЯ APP/β-АМИЛОИДНОГО КАСКАДА
Увеличение продукции и секреции β-амилоидного пептида (Аβ) и образование внеклеточных амилоидных бляшек является характерной чертой БА. Начальным шагом для отложения бляшек является расщепление трансмембранного белка APP с образованием Аβ, который при определенных условиях агрегирует, способствуя проявлению его нейротоксичных свойств. В патологических условиях агрегаты Аβ вызывают гибель нейронов и активацию микроглиального ответа, что приводит к дальнейшему прогрессированию заболевания. Образование Аβ происходит в результате протеолиза APP по амилоидогенному пути, который инициируется расщеплением APP β-секретазой BACE-1 с освобождением растворимого N-терминального фрагмента sAPPβ. Оставшийся в мембране С-терминальный фрагмент подвергается дальнейшему расщеплению γ-секретазным ферментативным комплексом с образованием Аβ40/42 и пептидом AICD, причем оба пептида способны вызывать гибель нейронов [22].
В настоящее время известно более 60 мутаций гена APP, из которых 27 являются патогенными, и лишь одна защищает от БА (https://www.alzforum.org/mutations). Практически все мутации сопровождаются нарушением метаболизма APP, который приводит к сдвигу соотношения образования Аβ40/42 в сторону преобладания более склонного к агрегации и токсичного Аβ42 [22, 23]. Интересно, что большинство патогенных мутаций расположены возле мест расщепления APP β- или γ-секретазами [24]. Центральными белками мембранного γ-секретазного комплекса являются PSEN1 и PSEN2. Известно 309 мутаций гена PSEN1 и 59 мутаций гена PSEN2, из которых 300 и 15, соответственно, являются патогенными. Мутации этих трех генов приводят к ранней манифестации БА.
Наличие мутаций в этих генах предоставляет возможность применения генно-инженерных технологий для их коррекции. Так, Gyorgy et al. [25] использовали CRISPR–Cas9 для коррекции шведской мутации APP в фибробластах, полученных от пациентов с БА. Трансфекция клеток плазмидами CRISPR и огРНК, нацеленными на мутантный аллель, приводила к его разрушению и 60%-му снижению секреции Aβ. Трансдукция первичной культуры нейронов, полученных от трансгенных мышей линии Tg2576, с помощью AAV векторов, несущих соответствующие мутации APP(SWE), огРНК и CRISPR–Cas9, также приводила к значимому снижению продукции Aβ [26]. В этой же работе авторы вводили конструкции в гиппокамп, но единственным результатом, который был опубликован является то, что в ДНК имелись индел мутации в 1.3% мутантных APP(SWE) аллелей.
Активность β-секретазы BACE1 определяет продукцию Aβ при БА. Стратегия, основанная на CRISPR–Cas9, была применена в индуцированных плюрипотентных стволовых клетках (ИПСК) человека и в мозге мыши для редактирования участка гена APP, близкого к С-концу, кодирующему сайт расщепления β-секретазой BACE-1. Это позволило модифицировать механизм расщепления APP по амилоидогенному пути, ослабляя продукцию Aβ и, в то же время, повышая образование APP-α, обладающего нейропротекторными свойствами [27]. При этом не наблюдалось видимого влияния на нейрофизиологию клеток in vitro. По всей видимости, использование такой стратегии препятствует сближению APP и BACE-1, которое необходимо для ферментативной реакции. Несмотря на то, что в настоящее время нет данных о связи мутаций гена, кодирующего этого фермент, с БА, недавно была обнаружена ассоциация полиморфизма BACE1 C786G с диабетом второго типа и умеренным когнитивным снижением [28]. Park et al. [29] изучили, могут ли нанокомплексы, загруженные CRISPR–Cas9, эффективно нацеливаться на Bace1 в постмитотических нейронах мозга взрослых мышей линии 5XFAD. Для этого Cas9 и огРНК были загружены в амфифильный нанокомплекс, что позволило получить эффективное нацеливание генов в постмитотических нейронах in vivo. Результатом этого было снижение уровня Aβ и когнитивного дефицита как у мышей 5XFAD, так и у мышей с повышенной экспрессией APP.
Интересно, что в исландской популяции наличие мутации A673T в гене APP снижает расщепление белка APP β-секретазой на 40%. Введение этой мутации в ген в клетках линии HEK293T и нейробластомы SH-SY5Y с помощью nCas9-дезаминазного комплекса приводило к замене остатка аланина на треонин в последовательности белка, которая сопровождалась уменьшением накопления Aβ [30]. Этот механизм может рассматриваться как потенциальный подход к лечению БА.
Наряду с активностью β-секретазы патологическая активность γ-секретазы, возникающая в результате мутаций генов PSEN1 и PSEN2, также способствует продукции токсичного Аβ42. Может ли коррекция этих мутаций приводить к нормализации функции клеток? Возможность этого была продемонстрирована в культуре холинергических нейронов, полученных из ИПСК здоровых субъектов и больных БА. Для этого ИПСК направленно дифференцировали в холинергические нейроны и, используя CRISPR–Cas9, исправляли мутацию PSEN2-N141I, связанную с семейной формой БА [31]. Это приводило к нормализации соотношения Aβ40/Aβ42, а также улучшало электрофизиологические характеристики нейронов [31] и Ca2+ гомеостаз [32].
КОРРЕКЦИЯ ПРОЦЕССОВ, СВЯЗАННЫХ С МЕТАБОЛИЗМОМ БЕЛКА ТАУ
Участие фосфорилирования белка τ в образовании парных спиральных филаментов – основного компонента нейрофибриллярных клубков, не вызывает сомнения, хотя генетические данные не указывают на наличие каких-либо мутаций в гене MAPT, кодирующем этот белок, которые приводили бы к развитию БА. В то же время некоторые из 15 известных мутаций (https://www.alzforum.org/mutations) могут повышать риск или вызвать развитие фронто-темпоральной деменции. Несмотря на это, было предпринято несколько попыток исправить ряд известных мутаций в гене MAPT и изучить последствия этого в ИПСК человека. В частности, Fong et al. [33] использовали ZFN для коррекции мутации A152T. После дифференцировки ИПСК в нейроны наблюдали снижение иммунореактивности гиперфосфорилированного белка τ и улучшение фенотипических характеристик нейронов. Другая группа авторов с помощью плазмидных векторов трансфицировала ИПСК CRISPR–Cas9 и олигонуклеотидной матрицей для коррекции мутации N279K гена MAPT [34], но не смогла увидеть существенных изменений в транскриптоме исходных и исправленных клеток. В отличие от этой работы авторы исследования [35] использовали CRISPR–Cas9 для коррекции мутации R406W в ИПСК, полученных от больного фронто-темпоральной деменцией с паркинсонизмом. Авторы успешно репарировали мутантный аллель без внесения дополнительных индел мутаций, а клетки при этом сохранили свою плюрипотентность.
Гиперфосфорилирование белка τ зависит от активности целого ряда протеинкиназ, воздействуя на которые можно регулировать этот процесс. Так, повышенная активность циклин-зависимой киназы Cdk5 может приводить к развитию таупатии [36]. Это становится следствием накопления активатора Cdk5 белка p25, который образуется в результате расщепления более крупного регуляторного белка p35 цистеиновой протеазой кальпаином. В условиях патологии, в том числе и при БА, накопление p25 ведет к избыточной активации Cdk5 и способствует гиперфосфорилированию белка τ. Валидность этого механизма подтвердили Seo et al. [37], которые модифицировали ИПСК, полученные от пациента с фронто-темпоральной деменцией, воспользовавшись системой CRISPR–Cas9 для введения в геном этих клеток гена, кодирующего укороченный фрагмент Δp35KI, неспособный образовывать активатор Cdk5 белок p25. В органоидах, которые получили авторы этой работы отсутствовали признаки гиперфосфорилирования белка τ. Таким образом, замена гена, кодирующего p35, может быть новым подходом к лечению таупатии. Следует отметить, что попытки изучить влияние других генов, ассоциированных с БА, на метаболизм белка τ не увенчались существенным успехом. Так, CRISPR–Cas9-опосредованный нокаут 22 генов в клетках HEK293T не влиял на агрегацию белка τ или захват его агрегатов [38]. Несмотря на недостатки использованной экспериментальной модели, можно предположить, что гены, ассоциированные с развитием БА и вовлеченные в работу сигнальных каскадов, прежде всего связанных с иммунным ответом и липидным метаболизмом, слабо влияют на состояние белка τ.
КОРРЕКЦИЯ МУТАНТНОГО АЛЛЕЛЯ АПОЛИПОПРОТЕИНА APOE4
Ранее были приведены примеры коррекции мутаций, которые приводят к развитию ранних форм БА. Основным фактором, который связывают с предрасположенностью к развитию БА в позднем возрасте, является генотип APOE4. В мозге ApoЕ экспрессируется в астроцитах, олигодендроцитах, перицитах, клетках сосудистого сплетения и, в меньшей степени, в нейронах и участвует в транспорте холестерина и фосфолипидов (см. [39]). В отличие от аллелей ε2 и ε3 мутантный аллель ε4 кодирует последовательность белка ApoE4, который связывается с Aβ, но не способен полностью освободить мозг от этого патогенного пептида. Это приводит к усиленному образованию сенильных бляшек. В связи с этим возможность модификации мутантного аллеля APOE4 в нормальный APOE3 может рассматриваться как подход к лечению БА. Принципиально это можно реализовать с использованием редактора оснований, как это было сделано в работе [40]. Авторы этой работы использовали нуклеофекцию ДНК, кодирующей цитозиндезаминазу, для замены Ц → Г в кодоне 158 и соответствующей огРНК в культуре астроцитов. В результате до 75% ридов содержали исправленную последовательность ДНК. В другой работе [41] были использованы ИПСК пациентов с генотипом APOE4/E3, которые при культивировании демонстрировали повышенный протеолиз APP, фосфорилирование белка τ, нарушение Са2+ гомеостаза, по сравнению с клетками, полученными от добровольцев с генотипом APOE3/E3. После CRISPR-Cas9-опосредованного редактирования мутантного аллеля были получены изогенные линии клеток E4/E3 и E3/E3. Дифференцировка клеток Е3/Е3 в нейроны сопровождалась более выраженным отрастанием нейритов. Нейроны E3/E3 были более устойчивы к действию нейротоксинов и имели более низкий уровень фосфорилирования белка τ, но процессинг APP не нормализовался. Тем не менее, модификация мутантного аллеля APOE ε4 имеет терапевтический потенциал для лечения БА.
НЕТРАДИЦИОННЫЕ МИШЕНИ ДЛЯ ГЕНОМНОГО РЕДАКТИРОВАНИЯ ПРИ БОЛЕЗНИ АЛЬЦГЕЙМЕРА
Гены, кодирующие функциональные элементы холинергической системы. Холинергический дефицит лежит в основе мнестических нарушений, являющихся отличительной чертой БА, по крайней мере, на ранних этапах патогенеза. Несмотря на это, данные о возможной связи мутаций в генах, кодирующих ферменты синтеза ацетилхолина, или его рецепторы, практически отсутствуют. В опубликованной недавно статье из Чехии [42] говорится о полиморфизме A120T в гене CHAT, который при моделировании in silico показал ассоциацию с риском развития БА в исследованной популяции. Не исключено, что более глубокое изучение различных компонентов холинергической системы позволит выявить новые мутации и продемонстрировать их связь с БА. В этом случае можно будет рассматривать ген CHAT, а возможно и другие гены, как потенциальную мишень для применения геномного редактирования.
Гены, связанные с воспалением/фагоцитозом. В последние годы уделяется большое внимание участию микроглии в патогенезе БА. Микроглия отвечает за удаление Аβ из мозга и, в значительной степени, за протекание иммунных и воспалительных процессов. Поэтому мутации генов, определяющих активность микроглии, могут оказывать влияние на механизмы развития БА. Одним из таких генов является TREM2, кодирующий триггерный рецептор типа 2, экспрессирующийся на миелоидных клетках (Triggering receptor expressed on myeloid cells 2). Трансмембранный белок Trem2 модулирует активность микроглиоцитов, связанную с поддержанием энергетического и липидного гомеостаза микроглиоцитов в процессе развития и старения мозга [43, 44]. Trem2 рецептор обеспечивает реакцию микроглии на повреждение и концентрацию клеток вблизи амилоидных бляшек [44]. Усиливая активность микроглии в отношении гиперфосфорилированного белка τ в присутствии Аβ, Trem2 способствует нейропротекции [45]. Мутации в гене TREM2, а их насчитывается 48 (https://www.alzforum.org/mutations), снижают функциональную активность рецептора и тем самым повышают риск развития БА и других нейродегенеративных заболеваний [45]. Хотя до настоящего времени не было предпринято попыток коррекции известных патогенных мутаций TREM2 в клетках микроглии, эта мишень представляется интересной с точки зрения разработки подхода к нормализации микроглального ответа в условиях БА.
Поверхностный рецептор CD33, экспрессируется фагоцитирующими клетками, включая микроглию [46]. Функции этого рецептора до конца не известны, но считают, что он участвует в фагоцитозе, секреции цитокинов, апоптозе. Полногеномные исследования установили, что экспрессия гена, кодирующего укороченную форму этого белка в клетках микроглии, может иметь протекторный эффект при БА, усиливая фагоцитоз Аβ [47]. Можно ли использовать эту мишень для проведения генной терапии еще необходимо исследовать.
Недавно был открыт редкий вариант гена PLCG2 (p.P522R), кодирующий фосфолипазу С‑гамма-2. Этот фермент экспрессируется в миелоидных клетках и снижает риск развития БА [48]. Введение этого гена в геном мыши при помощи CRISPR–Cas9 повышало острый воспалительный ответ макрофагов, полученных от этих животных, но не приводило к увеличению числа микроглиальных клеток или изменению их морфологии в коре больших полушарий мозга [49]. Вместе с тем функциональная активность такой модифицированной микроглии возрастала, что указывает на возможность использования этого подхода для улучшения устойчивости мозга к развитию БА.
Для других генов, ассоциация функции или дисфункции которых с патогенезом БА была выявлена в недавних полногеномных исследованиях, роль в развитии этого заболевания только выясняется. Поэтому представляется преждевременным говорить о значении их модификации для терапии БА даже в эксперименте.
ПРАКТИЧЕСКИЕ АСПЕКТЫ ИСПОЛЬЗОВАНИЯ ГЕНОМНОГО РЕДАКТИРОВАНИЯ ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ АЛЬЦГЕЙМЕРА
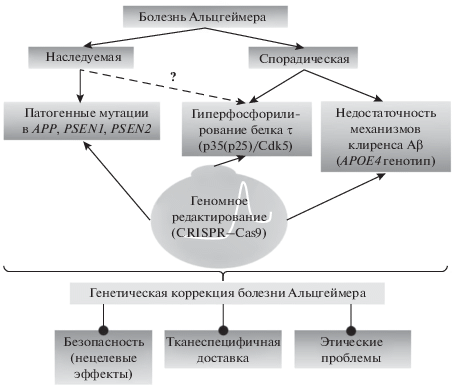
Достижения функциональной геномики существенно продвинули понимание работы мозга на клеточном уровне в норме и при патологии, предоставив целый ряд инструментов, которые позволяют исследовать роль отдельных генов в моделях на клетках и живых организмах. Так, генетические факторы, связанные с развитием целого ряда неврологических заболеваний, включая БА, болезнь Паркинсона, болезнь Гентингтона и другие, были введены в геном животных и исследованы (см. например [50–52]. CRISPR–Cas9, по-видимому, существенно упрощает и облегчает модификацию генов по сравнению с другими имеющимися средствами и может быть использована для разработки подходов к терапии как наследуемой, так и ненаследуемой БА (рис. 2). Однако имеющиеся в теории возможности ставят перед исследователями и врачами целый ряд вопросов, которые необходимо уточнить до принятия решения об их практическом использовании. Условно их можно разделить на вопросы безопасности, доставки и этические проблемы.
Рис. 2.
Возможности использования редактирования генома для терапии наследуемой и ненаследуемой БА и проблемы, которые нуждаются в решении. Расшифровку аббревиатур см. в тексте.
Одним из важнейших вопросов остается безопасность использования инструментов редактирования, в первую очередь на основе CRISPR–Cas9. Это обусловлено небольшим размером огРНК, которая направляет нуклеазу к целевому участку гена. Известно, что генетически детерминированные формы БА наследуются по аутосомно-доминантному типу, проявляясь при наличии даже одного мутантного аллеля [53]. Поэтому редактирование должно затрагивать только мутантный аллель, оставляя здоровый аллель или гомологичные участки генома интактными, что не всегда удается на практике. Например, использование упоминавшегося ранее конъюгированного с CRISPR-Cas9 редактора оснований для внесения патогенной мутации в ген PSEN1 вызывало изменения и в структуре гена PSEN2 [54]. В то же время авторы большинства рассмотренных работ, которые проводили секвенирование генома клеток после их модификации, сообщают об отсутствии существенных изменений за пределами целевого участка редактирования.
Введение в мозг конструкций, несущих элементы систем редактирования, представляет собой существенную проблему. Хотя имеющиеся в распоряжении исследователей плазмиды способны доставлять генетический материал огРНК и Cas9 в клетки разных тканей, их использование для трансфекции зрелого мозга затруднено. Наиболее вероятным средством доставки систем редактирования в мозг являются векторы на основе рекомбинантных лентивирусов (rLV) или аденоассоциированных вирусов (rAAV). Размеры плазмиды, несущей ген, кодирующий наиболее используемую нуклеазу Cas9 из Streptococcus pyogenes, составляют более 10 kb. Поэтому упаковать одновременно огРНК и CRISPR–Cas9 можно только в вектор на основе rLV. Однако такие вирусы интегрируются в ДНК хозяина, что может приводить к нежелательным последствиям. Перспективным представляется использование интеграза-дефицитных LV (IDLV), которые лишены способности встраиваться в геном хозяина и экспрессируются временно [55]. Использование отдельных rAAV вирусов для огРНК и CRISPR–Cas9 требует заражения одних и тех же клеток двумя векторами одновременно, что влияет на конечную эффективность трансдукции и редактирования. Вместе с тем в успешных экспериментах уровень коэкспрессии таких конструкций в мозге достигает 60% [56]. Следует отметить, что разрабатываются и другие, не содержащие вирусов, векторные системы, такие как наночастицы, наноструктурные ДНК ячейки с полиэтиленимином, микровезикулы [57].
Помимо проблемы побочных эффектов воздействия и доставки необходимо решить задачу выбора стратегии применения методов геномного редактирования. В известных клинических испытаниях по восполнению синтеза фактора роста нервов в мозге пациентов с БА использовали либо аутологичные клетки, которые после ex vivo трансдукции AAV вектором, несущим ген NGF, трансплантировали в мозг, либо прямые инъекции вектора [58, 59]. В идущих клинических испытаниях генной терапии векторные конструкции вводят внутривенно, внутрицистернально или внутритекально (см. [60]). Методы геномного редактрования, которые апробируются для лечения серповидноклеточной анемии, предполагают ex vivo редактирование аутологичных гематопоэтических или прекурсорных клеток. В случае, когда аутологичные клетки определенных тканей проблематично получить для ex vivo редактирования как, например, при мышечной дистрофии, предлагается проводить терапию in vivo [2]. Ткань мозга скорее относится ко второму типу, который требует непосредственного введения конструкций, необходимых для редактирования. Такой тип воздействия будет лимитирован способностью вектора распространяться в паренхиме мозга и редактированием генома в ограниченном количестве клеток. Будет ли этого достаточно для восстановления нарушенных функций нужно выяснить в будущих исследованиях. Наконец, для применения геномного редактирования с целью терапии БА необходимо решить, когда и/или на какой стадии развития патологического процесса будет возможно применять такой подход.
ЗАКЛЮЧЕНИЕ
Генная инженерия начиналась в 1970-е с манипуляций с нуклеиновыми кислотами, а сегодня находится на переднем крае науки, позволяя редактировать отдельные гены. По-видимому, наиболее перспективным со многих точек зрения средством редактирования будет в ближайшие годы система CRISPR–Cas9. Использование методов геномного редактирования на основе этой системы для коррекции некоторых моногенных заболеваний уже является реальностью. Нейродегенеративные заболевания, в том числе и БА, с их прогрессирующим течением, поздней манифестацией и в большинстве случаев полигенной природой представляют серьезный вызов для нейробиологии и медицины. В то же время налицо определенные успехи использования геномного редактирования для исправления на молекулярном уровне нарушений, обусловленных мутациями в генах APP, PSEN1, PSEN2, MAPT и APOE4, в экспериментальных условиях. Несмотря на то, что предстоит преодолеть еще множество проблем, развитие систем редактирования и векторной доставки, оценки рисков вместе с прозрачностью исследований и кооперацией исследователей безусловно позволит начать новую эру в лечении болезни Альцгеймера.
Список литературы
Калын Я.Б., Гаврилова С.И. // Современная терапия в психиатрии и неврологии. 2014. № 2. С. 10–15.
Doudna J.A. // Nature. 2020. V. 578. P. 229–236.
Chugunova A.A., Dontsova O.A., Sergiev P.V. // Biochemistry (Mosc.). 2016. V. 81. P. 662–677.
Kim Y.G., Cha J., Chandrasegaran S. // Proc. Natl. Acad. Sci. USA. 1996. V. 93. P. 1156–1160.
Miller J.C., Tan S., Qiao G., Barlow K.A., Wang J., Xia D.F., Meng X., Paschon D.E., Leung E., Hinkley S.J., Dulay G.P., Hua K.L., Ankoudinova I., Cost G.J., Urnov F.D., Zhang H.S., Holmes M.C., Zhang L., Gregory P.D., Rebar E.J. // Nat. Biotechnol. 2011. V. 29. P. 143–150.
Doudna J.A., Charpentier E. // Science. 2014. V. 346. P. 1258096.
Shimizu Y., Şöllü C., Meckler J.F., Adriaenssens A., Zykovich A., Cathomen T., Segal D.J. // Biochemistry. 2011. V. 50. P. 5033–5041.
Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. // Nucleic Acids Res. 2011. V. 39. P. e82.
Smirnov A.V., Yunusova A.M., Lukyanchikova V.A., Battulin N.R. // Vavilov J. Genet. Breed. 2016. V. 20. P. 493–510.
Savitskaya E.E., Musharova O.S., Severinov K.V. // Biochemistry (Mosc.). 2016. V. 81. P. 653–661.
Bannikov A.V., Lavrov A.V. // Mol. Biol. (Mosc.). 2017. V. 51. P. 582–594.
Wright A.V., Nuñez J.K., Doudna J.A. // Cell. 2016. V. 164. P. 29–44.
Hsu P.D., Zhang F. // ACS Chem. Neurosci. 2012. V. 3. P. 603–610.
Powell S.K., Gregory J., Akbarian S., Brennand K.J. // Mol. Cell. Neurosci. 2017. V. 82. V. 157–166.
Savell K.E., Bach S.V., Zipperly M.E., Revanna J.S., Goska N.A., Tuscher J.J., Duke C.G., Sultan F.A., Burke J.N., Williams D., Ianov L., Day J.J. // eNeuro. 2019. V. 6. P. ENEURO.0495-18.2019.
Feng W., Liu H.K., Kawauchi D. // Prog. Neuro-Psychopharmacology Biol. Psychiatry. 2018. V. 81. P. 459–467.
Savell K.E., Day J.J. // Yale J. Biol. Med. 2017. V. 90. P. 567–581.
Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. // Nature. 2017. V. 551. P. 464–471.
Talbot K., Wood M.J.A. // Sci. Transl. Med. 2019. V. 11. P. eaay2069.
Rinaldi C., Wood M.J.A. // Nat. Rev. Neurol. 2018. V. 14. P. 9–22.
Abudayyeh O.O., Gootenberg J.S., Franklin B., Koob J., Kellner M.J., Ladha A., Joung J., Kirchgatterer P., Cox D.B.T., Zhang F. // Science. 2019. V. 365. P. 382–386.
Nhan H.S., Chiang K., Koo E.H. // Acta Neuropathol. 2015. V. 129. P. 1–19.
Tcw J., Goate A.M. // Cold Spring Harb. Perspect. Med. 2017. V. 7. P. a024539.
Dai M.H., Zheng H., Zeng L.D., Zhang Y. // Oncotarget. 2018. V. 9. P. 15132–15143.
Gyorgy B., Ingelsson M., Lööv C., Takeda S., Lannfelt L., Hyman B.T., Breakefield X.O. // Mol. Ther. 2016. V. 24. P. S226–S227.
György B., Lööv C., Zaborowski M.P., Takeda S., Kleinstiver B.P., Commins C., Kastanenka K., Mu D., Volak A., Giedraitis V., Lannfelt L., Maguire C.A., Joung J.K., Hyman B.T., Breakefield X.O., Ingelsson M. // Mol. Ther. Nucleic Acids. 2018. V. 11. P. 429–440.
Sun J., Carlson-Stevermer J., Das U., Shen M., Delenclos M., Snead A.M., Koo S.Y., Wang L., Qiao D., Loi J., Petersen A.J., Stockton M., Bhattacharyya A., Jones M.V., Zhao X., McLean P.J., Sproul A.A., Saha K., Roy S. // Nat. Commun. 2019. V. 10. P. 53.
Tian S., Huang R., Guo D., Lin H., Wang J., An K., Wang S. // Curr. Alzheimer Res. 2020. V. 17. P. 355–364.
Park H., Oh J., Shim G., Cho B., Chang Y., Kim S., Baek S., Kim H., Shin J., Choi H., Yoo J., Kim J., Jun W., Lee M., Lengner C.J., Oh Y.K., Kim J. // Nat. Neurosci. 2019. V. 22. P. 524–528.
Guyon A., Rousseau J., Bégin F.G., Bertin T., Lamothe G., Tremblay J.P. // Mol. Ther. Nucleic Acids. 2021. V. 24. P. 253–263.
Ortiz-Virumbrales M., Moreno C.L., Kruglikov I., Marazuela P., Sproul A., Jacob S., Zimmer M., Paull D., Zhang B., Schadt E.E., Ehrlich M.E., Tanzi R.E., Arancio O., Noggle S., Gandy S. // Acta Neuropathol. Commun. 2017. V. 5. P. 77.
Moreno C.L., Della Guardia L., Shnyder V., Ortiz-Virumbrales M., Kruglikov I., Zhang B., Schadt E.E., Tanzi R.E., Noggle S., Buettner C., Gandy S. // Mol. Neurodegener. 2018. V. 13. P. 33.
Fong H., Wang C., Knoferle J., Walker D., Balestra M.E., Tong L.M., Leung L., Ring K.L., Seeley W.W., Karydas A., Kshirsagar M.A., Boxer A.L., Kosik K.S., Miller B.L., Huang Y. // Stem Cell Reports. 2013. V. 1. P. 226–234.
Hallmann A.L., Araúzo-Bravo M.J., Mavrommatis L., Ehrlich M., Röpke A., Brockhaus J., Missler M., Sterneckert J., Schöler H.R., Kuhlmann T., Zaehres H., Hargus G. // Sci. Rep. 2017. V. 7. P. 42991.
Nimsanor N., Kitiyanant N., Poulsen U., Rasmussen M.A., Clausen C., Mau-Holzmann U.A., Nielsen J.E., Nielsen T.T., Hyttel P., Holst B., Schmid B. // Stem Cell Res. 2016. V. 17. P. 556–559.
Patrick G.N., Zukerberg L., Nikolic M., de la Monte S., Dikkes P., Tsai L.H. // Nature. 1999. V. 402. P. 615–622.
Seo J., Kritskiy O., Ashley Watson L., Barker S.J., Dey D., Raja W.K., Lin Y.T., Ko T., Cho S., Penney J., Silva M.C., Sheridan S.D., Lucente D., Gusella J.F., Dickerson B.C., Haggarty S.J., Tsai. LH. // J. Neurosci. 2017. V. 37. P. 9917–9924.
Kolay S., Diamond M.I. // FEBS Open Bio. 2020. V. 10. P. 1912–1920.
Husain M.A., Laurent B., Plourde M. // Front. Neurosci. 2021. V. 15. P. 630502.
Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. // Nature. 2016. V. 533. P. 420–424.
Wadhwani A.R., Affaneh A., Van Gulden S., Kessler J.A. // Ann. Neurol. 2019. V. 85. P. 726–739.
Hálová A., Janoutová J., Ewerlingová L., Janout V., Bonczek O., Zeman T., Gerguri T., Balcar V.J., Šerý O. // J. Biomed. Sci. 2018. V. 25. P. 41.
Konishi H., Kiyama H. // Neurochem. Int. 2020. V. 141. P. 104878.
Rueda-Carrasco J., Hong S. // Trends Neurosci. 2020. V. 43. P. 739–740.
Haass C. // Neuron. 2021. V. 109. P. 1243–1245.
Zhao L. // Gerontology. 2019. V. 65. P. 323–331.
Bhattacherjee A., Jung J., Zia S., Ho M., Eskandari-Sedighi G., St Laurent C.D., McCord K.A., Bains A., Sidhu G., Sarkar S., Plemel J.R., Macauley M.S. // Mol. Neurodegener. 2021. V. 16. P. 19.
Magno L., Lessard C.B., Martins M., Lang V., Cruz P., Asi Y., Katan M., Bilsland J., Lashley T., Chakrabarty P., Golde T.E., Whiting P.J. // Alzheimer’s Res. Ther. 2019. V. 11. P. 16.
Takalo M., Wittrahm R., Wefers B., Parhizkar S., Jokivarsi K., Kuulasmaa T., Mäkinen P., Martiskainen H., Wurst W., Xiang X., Marttinen M., Poutiainen P., Haapasalo A., Hiltunen M., Haass C. // Mol. Neurodegener. 2020. V. 15. P. 52.
Stepanichev M.Y. // Adv. Gerontol. 2019. V. 9. P. 154–163.
Stepanichev M. // Front. Genome Ed. 2020. V. 2. P. 4.
van Giau V., Lee H., Shim K.H., Bagyinszky E., An S.S.A. // Clin. Interv. Aging. 2018. V. 13. P. 221–233.
Van Cauwenberghe C., Van Broeckhoven C., Sleegers K. // Genet. Med. 2016. V. 18. P. 421–430.
Sasaguri H., Nagata K., Sekiguchi M., Fujioka R., Matsuba Y., Hashimoto S., Sato K., Kurup D., Yokota T., Saido T.C. // Nat. Commun. 2018. V. 9. P. 2892.
Wang X., Wang Y., Wu X., Wang J., Wang Y., Qiu Z., Chang T., Huang H., Lin R.J., Yee J.K. // Nat. Biotechnol. 2015. V. 33. P. 175–179.
Swiech L., Heidenreich M., Banerjee A., Habib N., Li Y., Trombetta J., Sur M., Zhang F. // Nat. Biotechnol. 2015. V. 33. P. 102–106.
Hanafy A.S., Schoch S., Lamprecht A. // Pharmaceutics. 2020. V. 12. P. 1–14.
Tuszynski M.H., Yang J.H., Barba D., U H.S., Bakay R.A., Pay M.M., Masliah E., Conner J.M., Kobalka P., Roy S., Nagahara A.H. // JAMA Neurol. 2015. V. 72. P. 1139–1147.
Tuszynski M.H., Thal L., Pay M., Salmon D.P., U H.S., Bakay R., Patel P., Blesch A., Vahlsing H.L., Ho G., Tong G., Potkin S.G., Fallon J., Hansen L., Mufson E.J., Kordower J.H., Gall C., Conner J. // Nat. Med. 2005. V. 11. P. 551–555.
Sun J., Roy S. // Nat. Neurosci. 2021. V. 24. P. 297–311.
Robinson M., Lee B.Y., Hane F.T. // J. Alzheimers. Dis. 2017. V. 57. P. 317–330.
Raikwar S.P., Kikkeri N.S., Sakuru R., Saeed D., Zahoor H., Premkumar K., Mentor S., Thangavel R., Dubova I., Ahmed M.E., Selvakumar G.P., Kempuraj D., Zaheer S., Iyer S.S., Zaheer A. // J. Neuroimmune Pharmacol. 2019. V. 14. P. 608–641.
Sims R., Hill M., Williams J. // Nat. Neurosci. 2020. V. 23. P. 311–322.
Дополнительные материалы отсутствуют.