

Нейрохимия, 2021, T. 38, № 4, стр. 339-354
Ген SLC6A1 и психоневрологические заболевания: роль мутаций, перспективы лечения при помощи систем редактирования генома
Е. С. Букина 1, 2, Н. В. Кондратьев 3, С. В. Козин 1, В. Е. Голимбет 3, А. С. Артюхов 2, Э. Б. Дашинимаев 2, 4
1 ФГАОУ ВО Первый МГМУ им. И.М. Сеченова Минздрава России
Москва, Россия
2 Центр высокоточного геномного редактирования и генетических технологий для биомедицины,
РНИМУ им. Н.И. Пирогова
Москва, Россия
3 ФГБНУ Научный центр психического здоровья
Москва, Россия
4 ФГБУН Института биологии развития им. Н.К. Кольцова РАН
Москва, Россия
Поступила в редакцию 21.05.2021
После доработки 08.06.2021
Принята к публикации 11.06.2021
Аннотация
SLC6A1 (solute carrier family 6 member 1) – ген, кодирующий белок-транспортер гамма-аминомасляной кислоты (ГАМК) GAT1. GAT1 отвечает за обратный захват ГАМК из синаптической щели и межклеточного пространства. Мутации в гене SLC6A1 могут приводить к нарушению ГАМК-регуляции и связаны с эпилепсией и рядом психических заболеваний. В этом обзоре мы рассматриваем роль белка GAT1 в ГАМКергической регуляции и связь мутаций в гене SLC6A1 с эпилепсией, расстройствами аутического спектра, умственной отсталостью и шизофренией, а также перспективы их лечения при помощи систем редактирования генома.
ВВЕДЕНИЕ
SLC6A1 (The Solute Carrier Family 6 Member 1) – ген, кодирующий белок-транспортер GAT1 (GABA transporter 1). Функция белка GAT1 состоит в удалении избытка гамма-аминомасляной кислоты (ГАМК) из синаптической щели и прекращении ГАМК-нейротрансмиссии [1]. ГАМК является главным тормозным нейромедиатором в центральной нервной системе (ЦНС). В свою очередь транспортер GAT1 играет важную роль в контроле вне-синаптической концентрации ГАМК, модулируя как фазовое, так и тоническое торможение [2–5]. Кроме того, ГАМК является нейротрофическим фактором и играет важную роль на ранних этапах развития мозга и пролиферации нейрональных стволовых клеток [6, 7].
Клинические проявления мутаций в SLC6A1 включают в себя широкий спектр заболеваний [8, 9]. Основным заболеванием, для которого характерны мутации в гене SLC6A1, является эпилепсия. Эти мутации имеют тенденцию встречаться в более тяжелых случаях эпилепсии [10, 11]. Эпилепсия часто сопровождается расстройствами аутического спектра (РАС), умственной отсталости (УО) и различных неврологических симптомов (атаксия или неустойчивая походка, тремор и нарушения мелкой моторики) [10]. Поиск редких и de novo мутаций у больных с РАС показал связь мутаций в гене SLC6A1 с аутизмом [12, 13]. Непосредственную связь мутаций с РАС пока сложно оценить, поскольку аутизм коморбиден с эпилепсией [14] и для этой болезни также характерны нарушения в работе ГАМКергической системы [15]. Мутации в гене SLC6A1 оказались основным результатом исследования, посвященного поиску de novo мутаций у больных шизофренией, причем все носители мутаций не имели симптомов, связанных с эпилепсией и РАС, что говорит о независимой роли SLC6A1 в патогенезе шизофрении [16]. Было показано, что для больных шизофренией характерно нарушение функционирования белка GAT1 в ряде областей головного мозга (в префронтальной коре [17], лимбической системе [18] и мозжечке [19]). Согласно базе “GWAS Catalog” (www.ebi.ac.uk/gwas/genes/SLC6A1) на момент написания этого обзора, в гене SLC6A1 были найдены полиморфизмы, ассоциированные с рядом заболеваний, среди которых миопия [20], рак матки [21], алкоголизм [22] В исследовании генетической предрасположенности к алкогольной зависимости полиморфизмы в гене SLC6A1 были обнаружены как второй по значимости сигнал среди полученных результатов, что неудивительно, учитывая, что этанол действует на организм в том числе через рецепторы ГАМК. Кроме того, ассоциации гена SLC6A1 с синдром дефицита внимания/гиперактивности (СДВГ) были найдены в крупном кандидатном исследовании [23] и раннем GWAS [24], однако, в более поздних GWAS эти ассоциации не были реплицированы [25].
Таким образом, изучение роли генетических особенностей, связанных с мутациями в гене SLC6A1, в патогенезе различных заболеваний может позволить глубже понять механизмы их развития и разработать новые подходы к их диагностике и, возможно, генной терапии.
ГЕН SLC6A1
Ген SLC6A1 принадлежит к семейству, включающему 19 паралогичных генов, кодирующих переносчики простых химических веществ. У человека он локализован на 3 хромосоме (3p25.3) и согласно геномному браузеру Ensembl (http:// www.ensembl.org, v103) его размер составляет 47061 п.н. Ген SLC6A1 содержит 18 экзонов, имеет несколько аннотированных альтернативных промоторов, с которых может экспрессироваться 41 альтернативный транскрипт. Экспрессия различных вариантов гена SLC6A1 изучена недостаточно. Есть исследования, связывающие различные альтернативные транскрипты с эпилепсией [26] и развитием СДВГ [27]. На момент написания обзора для SLC6A1 было известно 277 ортологов. Значительный объем генетических исследований GAT1 сделан на генах-ортологах у лабораторных животных.
SLC6A1 в основном экспрессируется центральной нервной системе. В головном мозге GAT1 локализован преимущественно на пресинаптической мембране аксонов ГАМКергических нейронов [28]. В исследованиях на модельных животных также показано присутствие транспортера на мембранах астроцитов [29], олигодендроцитов [30] и клетках микроглии [31]. Вне мозга заметная экспрессия SLC6A1 наблюдается в тканях печени, однако, согласно базе Human Protein Atlas (www.proteinatlas.org), самого белка GAT1 в печени почти нет [32]. Кроме того, GAT1 обнаруживается в мужской половой системе, где белок, по-видимому, необходим для нормального сперматогенеза [33].
РОЛЬ БЕЛКА GAT1 В ГАМКЕРГИЧЕСКОЙ РЕГУЛЯЦИИ
GAT1 осуществляет перенос ГАМК вместе с ионами натрия и хлора из синаптической щели в терминали пресинаптических нейронов и близлежащие глиальные клетки, тем самым способствуя прекращению ГАМК-сигнализации (рис. 1) [34].
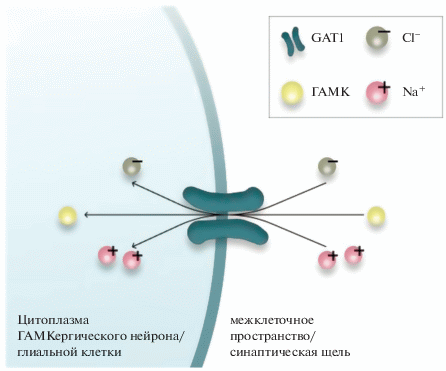
Транспорт ГАМК – это активный процесс, требующий наличия электрохимического градиента для Na+, создаваемого Na+/K+ АТФазой. ГАМК-транспортеры осуществляют совместный перенос ГАМК, Na+ и Cl– в соотношении 1ГАМК : : 2Na+ : 1Cl– по градиенту концентрации ионов Na+ и Cl– [35, 36]. Обратный захват нейромедиатора происходит в течение миллисекунд после его высвобождения и, таким образом, не позволяет ГАМК активировать соседние синапсы [37]. Однако GAT1, являясь ионным каналом, участвует в генерации еще как минимум двух токов, которые стехиометрически не связаны с переносом ГАМК, Na+ и Cl– через мембрану: (1) ГАМК-опосредованный ток ионов Na+ во внутриклеточное пространство [38]; (2) ГАМК-независимый катионный ток утечки [39]. Благодаря этим токам активация GAT1 может создавать локальные изменения мембранного потенциала [37].
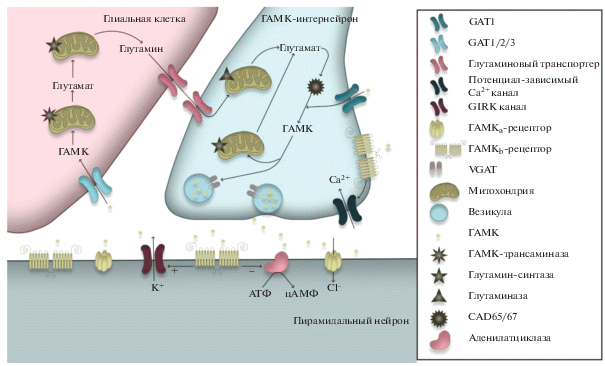
Основная функция ГАМКергической системы состоит в модуляции текущей активности нейронных сетей. Воздействуя на ионотропные и метаботропные рецепторы, ГАМК контролирует генерацию потенциалов действия и временную структуру паттернов активности, создаваемых целыми популяциями нейронов [40–43]. Это требует тонкого контроля времени активации ГАМК-рецепторов, что, в свою очередь, зависит от точного времени высвобождения ГАМК из пресинаптических терминалей и клиренса ГАМК из внеклеточного пространства. Для нормальной работы ГАМК-рецепторов, обеспечивающих высокое отношение сигнала к шуму, концентрация нейромедиатора в окружающей внеклеточной жидкости должна поддерживаться на низком уровне. Это может быть достигнуто только путем обратного захвата, поскольку метаболизм ГАМК осуществляется внутриклеточно в нейронах и глиальных клетках (рис. 2) [44–46]. Кроме того, работа ГАМК-транспортеров ограничивает выход ГАМК из активных синапсов (т.е. перелив) и, следовательно, контролирует пространственную специфичность ГАМКергической передачи [47–49].
Рис. 2.
Метаболизм ГАМК. ГАМКергические нейроны осуществляют высвобождение ГАМК, которая воздействует на постсинаптические рецепторы ГАМКa и ГАМКb. Избыток нейромедиатора транспортируется в клетки глии преимущественно обратным транспортером GAT2 и в пресинаптическую терминаль преимущественно транспортером GAT1. Метаболизм ГАМК происходит внутриклеточно с участием митохондрий. В клетках глии ГАМК превращается в глутамат, затем в глутамин, который далее переносится глутаматными транспортерами в ГАМКергический синапс. Там глутамин претерпевает обратное превращение через глутамат в ГАМК и упаковывается в везикулы. При возбуждении ГАМКергического нейрона вновь происходит релиз ГАМК. Таким образом, цикл замыкается. Внесинаптические ГАМКa и ГАМКb рецепторы активируются ГАМК, постоянно присутствующей в межклеточном пространстве в малых концентрациях.
В ряде экспериментов с нокаутированием SLC6A1 у мышей были выявлены такие проявления, как снижение агрессивности [50], тревоги, депрессии [51], гипалгезия [52] и снижение поведенческих реакций по отношению к этанолу [53]. Данные результаты могут быть ассоциированы с повышенным внеклеточным уровнем ГАМК в связи с дисфункцией GAT1 и, соответственно, усилением торможения, поскольку GAT1 считается основным транспортером ГАМК в ЦНС. Однако в работах Chiu и др. у нокаутированных по гену SLC6A1 мышей наблюдались тремор, атаксия, нервозность и повышенное ГАМК-индуцированное возбуждение в мозжечке [54, 55]. Этот фенотип напоминает побочные эффекты лечения высокоселективным ингибитором GAT1 – тиагабином (противоэпилептический препарат) [56].
Для понимания подобных фенотипических проявлений рассмотрим возможные последствия дисфункции GAT1 в контексте строения ГАМКергической системы.
Семейство ГАМК-транспортеров. Транспортеры гамма-аминомасляной кислоты (ГАМК-транспортеры) относятся к семейству переносчиков SLC6 (solute carrier 6 transporter family). Семейство SLC6 состоит из 19 белков и, в зависимости от характера переносимых веществ, подразделяется на четыре группы: ГАМК-транспортеры, транспортеры аминокислот, транспортеры моноаминов и транспортеры незаменимых аминокислот (рис. 3) [57]. Эти транспортеры поддерживают внеклеточный уровень ГАМК и возбуждающих аминокислот на низком уровне и регулируют таким образом их функциональную активность и внутриклеточный метаболизм. Транспортеры обладают различными свойствами как в отношении их транспортных функций (обратного захвата нейромедиаторов), так и в отношении их способности действовать в качестве ионных каналов [58].
Рис. 3.
Молекулярно-филогенетический анализ семейства обратных транспортеров SLC6. Семейство SLC6 делится на четыре группы: переносчики ГАМК (зеленый), аминокислот (голубой), моноаминов (розовый) и незаменимых аминокислот (серый). В скобках указаны вещества, которые также могут транспортироваться переносчиками ГАМК.
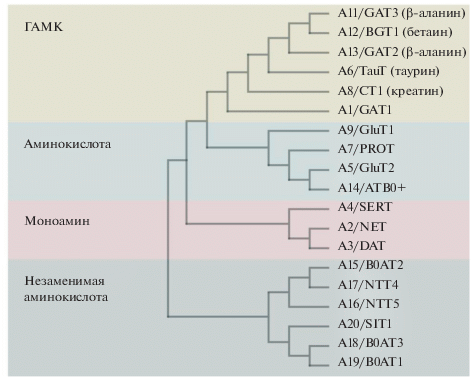
Группа ГАМК-транспортеров состоит из шести белков: A1/GAT1, A13/GAT2, A11/GAT3, A12/BGT1, A8/CT1 и A6/TauT, каждый из которых способен транспортировать ГАМК и другие молекулы: β‑аланин (GAT2 и GAT3), таурин (TauT), креатин (CT1) и бетаин (BGT1) [59]. Среди указанных транспортеров наибольшую роль в процессах обратного захвата ГАМК играют белки GAT1, GAT2 и GAT3 (в особенности GAT1) [8, 45, 59, 60]. GAT1 принадлежит к высокоселективным, натрий- и хлорид- зависимым ГАМК-транспортерам и локализуется преимущественно на пресинаптической мембране ГАМКергических нейронов и в меньшей степени на мембранах астроцитов, окружающих синапс [36, 45, 58].
Результаты иммуногистохимических анализов у крыс отражают слабую экспрессию GAT2 в ЦНС по сравнению с периферическими органами, и наоборот, широкое распространение GAT1 и GAT3 по всему мозгу [61]. Самые высокие уровни экспрессии GAT1 наблюдаются в гиппокампе, обонятельных луковицах, молекулярном слое коры больших полушарий (L1), пириформной (грушевидной) коре, верхних холмиках четверохолмия, межножковом ядре и ядре спинномозгового пути тройничного нерва. Самые высокие уровни экспрессии GAT3 наблюдаются в обонятельных луковицах, таламусе, гипоталамусе, варолиевом мосту и продолговатом мозге, бледном шаре, базальных ганглиях, черной субстанции, ядрах мозжечка и ядре спинномозгового пути тройничного нерва [61]. Уровень экспрессии GAT1-3 в коре больших полушарий меняется в зависимости от слоев. GAT1 экспрессируется в основном в L2–L4 слоях (наружном зернистом, наружном пирамидном и внутреннем зернистом), GAT2 обнаруживается преимущественно в молекулярном слое, а GAT3 – в L3 слое (наружном пирамидном) и верхней части L5 слоя (внутреннего пирамидного) [61, 62].
Если сравнивать экспрессию ГАМК-транспортеров в ЦНС на клеточном и субклеточном уровнях, то экспрессия GAT1 наиболее выражена в пресинаптических мембранах ГАМКергических нейронов и менее выражена в глиальных клетках [62]. Степень экспрессии GAT3 в ЦНС в целом ниже, чем GAT1, и наблюдается преимущественно в клетках глии [28]. Экспрессия GAT2 в ЦНС также ниже по сравнению с GAT1, и он, как и BGT1, по большей части экспрессируется за пределами ЦНС: в печени и почках [45, 63]. Таким образом, GAT1 является главным транспортером ГАМК в ЦНС.
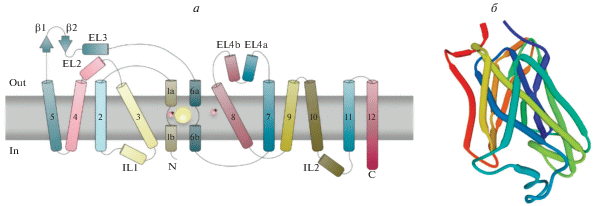
Структура белка GAT1. ГАМК-переносчики семейства SLC6 имеют общую структуру, представляющую собой 12 трансмембранных доменов (рис. 4). Каждый транспортер имеет различное сродство к ГАМК, различную локализацию в тканях и различную субстратную специфичность [45]. Белок GAT1, молекулярная масса которого составляет 67074 Да, состоит из 599 аминокислот [64]. Среди всех ГАМК-транспортеров, экспрессия GAT1 в ЦНС выражена наиболее сильно [45]. Перечисленные особенности GAT1 сделали его мишенью для разработки селективных противоэпилептических лекарственных средств [65].
Рис. 4.
Строение белка GAT1. (а) Схематичное строение белка GAT1 Aquifex aeolicus LeuTAa. Транспортер состоит из 12 трансмембранных доменов. N- и С-концевые участки белка располагаются в цитоплазме. В структуре GAT1 имеются две внеклеточные β-цепи (серо-зеленые стрелки), четыре внеклеточные (EL2, EL3, EL4a, EL4b) и две внутриклеточные спирали (IL1, IL2). Ионы Na+ изображены в виде двух розовых сфер. Молекула субстрата (Leu) изображена в виде большой желтой сферы. Модифицировано из [59]. (б) Модель 3D-структуры белка GAT1 на основе транспортера 4xp9.1.A. Рентгеновская структура дофаминового транспортера дрозофилы [66].
Для понимания процессов, происходящих при нарушении обратного захвата ГАМК, рассмотрим типы ГАМКергических нейронов, строение ГАМК-рецепторов и формы торможения в центральной нервной системе.
ГАМКергические интернейроны (типы, локализация, участие в поддержании баланса между возбуждением и торможением). Гамма-аминомасляная кислота (ГАМК) осуществляет функцию главного тормозного нейромедиатора в центральной нервной системе. В префронтальной коре ГАМКергические интернейроны оказывают тормозные влияния на пирамидные нейроны контролируя таким образом входящее возбуждение от афферентных структур и генерацию потенциалов действия [41, 67]. ГАМКергические интернейроны можно классифицировать по их морфологии, электрофизиологическим свойствам или гистологическим маркерам [68, 69]. Согласно наиболее распространенной классификации выделяют следующие типы интернейронов по экспрессии характерных маркеров: парвальбуминовые (экспрессирующие Са2+-связывающий белок парвальбумин, PV) и интернейроны, экспрессирующие ионотропный серотониновый рецептор 5HT3a (5HT3aR) [70]. PV интернейроны (наиболее распространенный тип ГАМКергических интернейронов) могут, в свою очередь, быть разделены на два подтипа: корзинчатые клетки (иннервируют сому и проксимальные дендриты) и клетки-канделябры (образуют синаптические контакты с начальным сегментом аксона) [71, 72].
В ряде работ было показано, что корзинчатые интернейроны участвуют в генерации гамма-колебаний (30–80 Гц), связанных с когнитивными функциями и обработкой информации у разных видов [73–75]. Наличие значительных изменений в гамма-колебаниях было обнаружено при заболеваниях, связанных с когнитивными нарушениями, таких как шизофрения и расстройства аутистического спектра (РАС) [76, 77]. Для того чтобы данные процессы протекали эффективно, уровни торможения и возбуждения должны поддерживаться в определенном соотношении, которое для префронтальной коры составляет примерно 20/80% (возбуждение/торможение) [78, 79]. Баланс между процессами возбуждения и торможения начинает формироваться на начальных этапах развития нервной системы и продолжает активно поддерживается гомеостатическими механизмами на протяжении всей жизни [80, 81]. Накопленные данные свидетельствуют о том, что дисбаланс возбуждения и торможения вносит вклад в этиологию и симптоматику целого ряда заболеваний, связанных с нейрональным развитием, включая шизофрению, расстройства аутистического спектра (РАС) и эпилепсию [82].
У больных шизофренией нарушения рабочей памяти связаны с дисфункцией дорсолатеральной префронтальной коры [83]. Для шизофрении характерен дефицит декарбоксилазы глутаминовой кислоты (GAD67), фермента, синтезирующего ГАМК [84–86]. Хотя у пациентов большинство ГАМК-интернейронов префронтальной коры экспрессируют нормальный уровень мРНК GAD67, приблизительно 25–35% ГАМК-интернейронов (преимущественно PV-нейроны [87]) не имеют обнаруживаемого уровня этого транскрипта. Однако нарушения только в PV-нейронах не могут полностью объяснить дефицит экспрессии мРНК GAD67 поскольку такие изменения также наблюдались в I, II и V слоях коры, где количество PV-нейронов невелико [88, 89]. Таким образом, эта дисфункция, по крайней мере частично, связана с нарушениями процессов фазового (синаптического) и тонического (внесинаптического) торможения входных сигналов пирамидальных клеток SST-содержащими и CCK-содержащими типами ГАМК-интернейронов [83].
ГАМК-рецепторы. До настоящего времени было описано три типа ГАМК-рецепторов, два из которых, ГАМКА и ГАМКC, являются лиганд-зависимыми ионными каналами, а ГАМКB – метаботропным рецептором, связанным с G-белком [90].
Ионотропные рецепторы состоят из 5 субъединиц. В структуру ГАМКC-рецепторов входят ρ1–3 субъединицы [91]. Данный тип рецепторов первоначально относили к подтипу ГАМКА. Их функции связаны со зрительной обработкой информации, регуляцией ритмов сна–бодрствования, восприятием боли, памятью, обучением, гормональной регуляцией и желудочно-кишечной секрецией. Эти рецепторы локализованы в сетчатке, таламусе, гиппокампе, гипофизе и желудочно-кишечном тракте [92]. ГАМКА-рецепторы представляют больший интерес в контексте развития психических и неврологических заболеваний. Они образуют множество изоформ из различной комбинации 16-ти типов субъединиц (α1–6, β1–3, γ1–3, δ, ε, π, и θ) [93, 94]. От субъединичного состава зависят различные свойства ГАМКА-рецепторов: кинетика активации (влияет на их десенситизацию [95]), локализация (рецепторы ГАМК, содержащие субъединицы α1 и γ2, сосредоточены в постсинаптических участках, где они опосредуют фазное торможение [96, 97]), в то время как ГАМК-рецепторы, содержащие субъединицы α4 и δ, в переднем мозге избирательно локализованы в внесинаптических участках [96–98]. Внесинаптические рецепторы обладают высокой чувствительностью к ГАМК и опосредуют тоническое торможение [83]). Активация пресинаптических ГАМКА-рецепторов подавляет высвобождение нейромедиатора путем ингибирования потенциал-зависимых Ca2+-каналов. В свою очередь, постсинаптические ГАМКА-рецепторы отвечают за генерацию тормозного постсинаптического потенциала (ТПСП) и развитие быстрой гиперполяризации постсинаптической мембраны [99]. Однако, было показано, что длительная активация постсинаптических ГАМКА-рецепторов способна приводить к развитию длительного деполяризационного постсинаптического потенциала [100]. Субъединичный состав ГАМКА-рецепторов может иметь значение в патогенезе заболеваний, например, композиция субъединиц ГАМКА-рецепторов в нейронах может меняться во время эпилептогенеза; эти изменения отражаются в фармакодинамике лекарственных препаратов [101, 102].
Метаботропные ГАМКB-рецепторы состоят из двух субъединиц (GBR1 и GBR2) [103] и могут располагаться как на пре-, так и на постсинаптической мембране [104, 105]. При взаимодействии ГАМКB-рецептороа с лигандом (ГАМК) запускается каскад реакций, приводящий к открытию связанных с G-белком калиевых каналов и, как следствие, развитию медленного ТПСП, длящегося сотни миллисекунд [106–108]. В результате развивается длительная гиперполяризация, наступающая после быстрой гиперполяризации, вызванной активацией ионотропных рецепторов [99]. Внесинаптические ГАМКB-рецепторы способны активироваться постоянно присутствующими в межклеточном пространстве малыми концентрациями ГАМК, а также ГАМК, вышедшей за пределы синаптической щели в результате перелива ГАМК [106]. В результате активации пресинаптических ГАМКB-рецепторов снижается ГАМК-нейротрансмиссия тормозных интернейронов [105].
Разнообразие форм ГАМКергического торможения. Выделяют разные формы ГАМКергического торможения. В зависимости от того, какой тип нейрона подвергается торможению, различают торможение ГАМКергических интернейронов (приводит к увеличению возбуждения) и торможение возбуждающих нейронов (приводит к снижению возбуждения) [109]. По локализации тормозных синаптических окончаний: торможение, развивающееся в аксо-дендритных синапсах (контроль входных токов, распространяющихся от дендритов к соме), торможение в аксо-соматических контактах и торможение начального сегмента аксона (контроль выходных токов - генерации потенциалов действия) [110]. В контексте патологий, связанных с нарушением обратного захвата ГАМК, подробнее остановимся на классификации торможения по типу синаптической передачи. Фазное торможение опосредуется воздействием высокой концентрации ГАМК на постсинаптические ГАМК-рецепторы в результате дискретного выброса нейромедиатора из пресинаптического окончания. Однако в межклеточном пространстве постоянно присутствует небольшое количество ГАМК, которое поступает туда в результате выхода нейротрансмиттера за пределы синаптической щели, а также в результате обратного функционирования ГАМК-транспортеров [111–113]. Малые концентрации ГАМК воздействуют на внесинаптические ГАМК-рецепторы, активация которых вызывает тоническое торможение [114, 115]. Данный вид торможения поддерживает определенное значение потенциала мембраны и модулирует возбудимость клетки путем снижения порога генерации потенциалов действия [114]. В ряде работ было показано, что длительная стимуляция внесинаптических ГАМК-рецепторов способна приводить к изменению их работы и развитию деполяризации в результате их активации (а не гиперполяризации) [116–118], что теоретически может происходить при нарушении обратного захвата ГАМК.
У мышей с дефицитом GAT1 усиливается тоническое торможение и удлиняется время затухания фазного торможения, опосредованного постсинаптическими ГАМК-рецепторами, тогда как частота, амплитуда и кинетика ГАМК-индуцированных постсинаптических токов не изменяются [55, 119]. Однако в других работах на GAT1-дефицитных мышах показано снижение частоты ТПСП, что может быть связано с увеличением экспрессии ферментов (GAD65/67), участвующих в синтезе ГАМК в пресинаптических терминалях тормозных нейронов [120, 121].
В совокупности эти результаты свидетельствуют о многочисленных молекулярных и клеточных функциях GAT1. Нарушение работы ГАМК-транспортера обусловливает сложный патогенез заболеваний у пациентов с мутациями в гене SLC6A1.
ГАМК и нейрогенез. Дифференцировка, миграция и интеграция нейронов регулируются рядом молекулярных процессов.
ГАМК является нейротрофическим фактором [122]. В эмбриональном периоде ГАМК является возбуждающим нейромедиатором [123, 124]. Тоническое возбуждение, опосредованное активацией ГАМК-рецепторов, стимулирует и направляет миграцию проекционных нейронов [122]. Стимуляция ГАМКB и ГАМКC-рецепторов способствует миграции нейронов через кортикальную пластинку [125], тогда как активация ГАМКА-рецепторов данный процесс прекращает [126].
Нарушение ГАМК-сигнализации на ранних стадиях развития изменяет клеточную миграцию и архитектуру коры [127–132].
Кроме того, ГАМК участвует в нейрогенезе и во взрослом возрасте. Хотя остальные нейромедиаторы также регулируют различные процессы созревания нейронов из нейрональных стволовых клеток, среди них ГАМК является наиболее важным. Так, развитие и функции клеток-зерен во взрослом мозге контролируются ГАМКергической системой [133].
Нейротрофический фактор мозга (BDNF) и другие нейротрофины способствуют миграции клеток, синаптогенезу, росту дендритов и поддержанию функционирования синапсов на протяжении всей жизни [125, 134–136]. Изменение уровня BDNF в развивающемся мозге может нарушить миграцию ГАМКергических интернейронов [137–139]. Кроме того функции BDNF остаются критическими и на более поздних этапах развития мозга (в подростковом возрасте), когда синаптические контакты укрепляются или подвергаются прунингу [140, 141]. В данный период предрасположенность к развитию психических заболеваний является наиболее высокой [142].
Фактор роста BDNF, рецепторы к которому присутствуют в различных типах ГАМК-интернейронов, синтезируется и секретируется пирамидальными нейронами [143]. ГАМК регулирует активность BDNF путем переключения активации на ингибирование гена BDNF во время изменения ГАМК-сигнализации с возбуждающей на тормозную [144]. В процессе развития ГАМК-индуцированная мембранная деполяризация приводит к открытию кальциевых каналов L-типа и высвобождению BDNF, что, в свою очередь, способствует дифференцировке нейронов [145]. Увеличение экспрессии хлорид-калиевого котранспортера 2-го типа (KCC2) примерно во время рождения изменяет градиент ионов хлора, и результат активации ГАМК рецепторов с деполяризующего на гиперполяризующий [146, 147]. По мере развития гиперполяризации [148] кальциевые каналы L-типа перестают активироваться и индуцировать синтез и высвобождение BDNF [144].
Таким образом, BDNF и ГАМК тесно взаимодействуют, и нарушения обратного захвата ГАМК могут привести к снижению нейротрофических процессов как на начальных этапах развития, так и в зрелом мозге и, следовательно, к развитию ряда неврологических заболеваний.
ГЕНЕТИКА SLC6A1 И ПСИХОНЕВРОЛОГИЧЕСКИЕ ЗАБОЛЕВАНИЯ
Мутации в белок-кодирующих генах являются существенным генетическим фактором риска развития психических и психоневрологических заболеваний. Рассмотрим некоторые фенотипические проявления мутаций в гене SLC6A1.
Эпилепсия. Эпилепсия – хроническое неврологическое заболевание, характеризующееся наличием судорожных приступов [149]. Мутации в гене SLC6A1 изначально были найдены у пациентов с миоклоническими-атоническими судорогами [10]. Однако спектр фенотипических проявлений мутаций в этом гене оказался шире. В работе Johannesen et al. были проанализированы 34 случая (24 пробанда, 6 членов их семей и клинические данные от 4 ранее описанных испытуемых) с мутациями в гене SLC6A1 [8]. В 31 из 34 случаев у испытуемых диагностирована эпилепсия, представленная в следующих формах: абсансы, атипичные абсансы, атонические судороги и миоклонические или миоклонические-атонические судороги, реже (5/31) – генерализованные тонико-клонические судороги. Важно отметить, что общим для всех пациентов стало наличие в разной степени выраженной умственной отсталости (от легкой до средней) с нарушением речевой функции. Притом, у 3 из 34 испытуемых, даже в отсутствие эпилепсии, наблюдалась умственная отсталость в легкой форме. Также, у половины пациентов имелись поведенческие расстройства (гиперактивность, агрессивное поведение, синдром дефицита внимания и аутизм в различных комбинациях). Подобные проявления наблюдались ранее на животных моделях: у мышей с мутациями в slc6a1 [150].
Как обсуждалось ранее, дисфункция GAT1 может влиять на динамику процессов возбуждения и торможения через множество механизмов. Нарушения процессов обратного захвата, приводят к повышению концентрации ГАМК как в синаптической щели, так и вне синапса [119]. Это может привести к гиперстимуляции внесинаптических ГАМКА и ГАМКB-рецепторов, которые отвечают за развитие тонического торможения [151]. В работе Cope и др. было показано усиление ГАМКА-опосредованного тонического торможения в таламо-кортикальных нейронах у мышей с нокаутом GAT1, приводящее к гиперполяризации таламо-кортикальных нейронов и нарушению генерации потенциалов действия [151]. Кроме того, длительная активация ГАМКB-рецепторов, индуцирующая открытие потенциал-зависимых Ca2+ каналов Т‑типа, способна вызвать циркулирующее возбуждение в таламо-кортикальной системе через генерацию последовательных потенциалов действия [152, 153]. Предыдущие исследования показали, что активация ГАМКB-рецепторов вызывает судороги у мышей и крыс и что предварительное введение антагониста ГАМКB-рецепторов может уменьшить их продолжительность или привести к их отсутствию [154, 155]. Снижение функции GAT1 может также привести к снижению концентрации ГАМК в синаптических терминалях, и, следовательно, к нарушению высвобождения ГАМК и нарушению фазного торможения. Кроме того, фазное торможение, опосредованное ГАМКА-рецепторами, может быть нарушено вследствие изменения их субъединичного состава [156]. Результаты in vivo исследований на нокаутированных по GAT1 мышах показали значительное увеличение тонического торможения в гиппокампе, при неизменной или сниженной амплитуде ТПСП на постсинаптических мембранах [119, 157]. Таким образом, снижение функции GAT1 может привести к развитию эпилепсии как через чрезмерную активацию внесинаптических ГАМКА- и ГАМКB-рецепторов, так и через снижение синаптической ГАМКА-опосредованной сигнализации [158].
GAT1 является одной из основных мишеней для лечения эпилепсии [26]. Например, препарат тиагабин (селективный ингибитор GAT1) широко используется при фокальной эпилепсии [26]. Встает закономерный вопрос: если мутации в гене SLC6A1 и нарушение функционирования белка GAT1 могут являться одной из причин эпилепсии, почему для лечения эпилепсии используют ингибиторы GAT1? Объяснение этому может быть в том, что мутации в GAT1, которые приводят к эпилепсии, не обязательно проявляются в снижении его концентрации, а также тем, что на GAT1 нокаутных моделях нет части проявлений эпилепсии (например, нет судорог).
Таким образом, крайне важно выяснить, как мутации в гене SLC6A1 влияют на ГАМКергическую сигнализацию и развитие мозга, а также последствия терапии ингибиторами GAT1.
Расстройства аутистического спектра. Аутизм или, шире, расстройства аутистического спектра (РАС) – генетически обусловленные заболевания, характеризующиеся нарушением нервно-психического развития, манифестирующие, как правило, в детском возрасте. Генетическая архитектура РАС включает редкие вариации в сотнях генов (возникшие de novo или унаследованные от родителей) и полиморфизмы в тысячах локусов. Генетические исследования как редкой, так и распространенной изменчивости, связанной с РАС, показывают, что в основном мутации, вовлеченные в развитие аутизма, влияют на начальные этапы развития мозга, в том числе на процессы дифференцировки клеток коры больших полушарий [159–161]. Нарушения в работе ГАМКергической системы связаны с РАС, что хорошо видно в физиологических исследованиях и в исследованиях на животных моделях аутизма [162]. Часто гены, мутации в которых связаны с развитием РАС и эпилепсии, играют роль в функционировании ГАМКергической системы [163, 164]. Важно отметить, что РАС и эпилепсия нередко сопутствуют друг другу, что свидетельствует об общности патогенеза данных заболеваний [165]. Имеется ряд исследований, в которых изучались мутации в гене SLC6A1 в контексте развития аутизма [8, 9, 166–168]. Дисбаланс процессов возбуждения и торможения, одной из причин которого является нарушение функции переносчика GAT1, также является одной из гипотез развития РАС [169]; [15, 82]. Кроме того, ГАМК является нейротрофическим фактором и играет важную роль в пролиферации нейронных стволовых клеток на ранних этапах развития мозга [6, 7]. Нарушение этого процесса может являться одной из причин развития РАС.
Умственная отсталость. Умственная отсталость (УО) – заболевание, характеризующееся задержкой умственного развития, при котором наблюдается нарушение познавательных способностей, речи и моторики, что приводит к потере социальной дееспособности [170]. У большинства пациентов с мутациями в гене SLC6A1 наблюдается развитие умственной отсталости, которая может развиваться как сама по себе, так и на фоне другого психического или физического нарушения [8]. В более ранних работах упоминалось о том, что одним из самых характерных проявлений мутации в SLC6A1 является предшествующая эпилепсии умственная отсталость в легкой или умеренной форме, как правило с нарушениями речи [10]. В исследовании Gong и др. проводились поведенческие тесты на нокаутированных по гену SLC6A1 мышах [5]. В результате была обнаружена взаимосвязь между изменением ГАМКергической регуляции в гиппокампе и нарушениями процессов обучения, запоминания и синаптической пластичности. Кроме того, поскольку ГАМК играет ключевую роль в процессах эмбрионального развития нервной ткани [7], нарушение этого процесса из-за дисфункции белка GAT1 также может являться одной из причин умственной отсталости.
Шизофрения. Шизофрения – это полигенное полиморфное психическое расстройство, характеризующееся утратой целостности психических процессов [171]. Под диагнозом “шизофрения” объединен ряд отдельных синдромов, отличающихся генетическим бэкграундом и выраженностью позитивных, негативных и когнитивных симптомов [169, 171]. На данный момент наиболее успешным подходом к лечению шизофрении является медикаментозная терапия антипсихотиками, которая не всегда дает желаемый эффект в связи с клинической гетерогенностью заболевания. Когнитивные симптомы (снижение концентрации, ухудшение кратковременной памяти, нарушение ассоциативных связей, отсутствие цельного и последовательного мышления) плохо поддаются лечению антипсихотическими препаратами [172–174]. Они возникают при всех формах шизофрении и обнаруживаются до появления позитивных (галлюцинации, бред, неорганизованная речь и поведение) и негативных (эмоциональное обеднение, социальная аутизация, угасание желаний, ангедония, скудность речи) симптомов [175].
Вероятно, клиническая гетерогенность данного заболевания обусловлена генетической гетерогенностью. Генетическая архитектура шизофрении чрезвычайно сложна [176, 177]. Большую роль в развитии данного заболевания играет наследственная предрасположенность. Она может быть обусловлена определенным набором однонуклеотидных полиморфизмов (single-nucleotide polymorphism, SNP), обнаруживаемых методом полногеномного анализа ассоциаций (Genome-Wide Association Studies, GWAS) и отражающим уровень полигенного риска (polygenic risk score, PRS) развития заболевания [98, 178–185]. Наследственная предрасположенность может также быть обусловлена наличием редких мутаций, унаследованных от родителей и выявляемых различными методами секвенирования [16, 186–188]. Особый интерес представляют мутации de novo, поскольку их наличие в генах, ассоциированных с развитием шизофрении, может быть обнаружено у пациентов с низкой наследственной предрасположенностью, вычисляемой по показателям полигенного риска [16, 189]. В недавнем исследовании Rees и др. были проанализированы данные экзомного секвенирования для поиска мутаций de novo в 613 тройках (больные шизофренией пациенты и их родители) и объединены с уже опубликованными данными (в общей сложности 3444 тройки). Это исследование дает дополнительные доказательства того, что определенные классы мутаций de novo (в том числе в гене SLC6A1) могут быть причиной повышенного риска развития шизофрении. В результате объединения данных экзомного секвенирования и GWAS было показано, что носители мутаций de novo, ассоциированных с развитием шизофрении, имеют меньший полигенный риск развития заболевания, чем пациенты, не являющиеся носителями. Эти статистически независимые результаты доказывают возможность развития заболевания при отсутствии наследственной предрасположенности [16]. Кроме того, связь de novo мутаций в гене SLC6A1 с развитием шизофрении была независимо обнаружена в исследовании Schizophrenia exome meta-analysis consortium (SCHEMA) [190].
Как обсуждалось ранее, мутации в гене SLC6A1 способны приводить к дисфункции белка GAT1 и, следовательно, к нарушению регуляции количества ГАМК в синаптической щели [8]. Избыток медиатора, воздействующего на ГАМКА-рецепторы, может привести к гиперполяризации мембраны постсинаптического нейрона и его рефрактерности [157]. В свою очередь активация избытком ГАМК ГАМКB-рецепторов опосредуется открытием медленных вольтаж-зависимых кальциевых каналов Т-типа, и, соответственно, циклическими возбуждениями в таламо-кортикальной системе [191]. Таламо-кортикальная система участвует в регуляции когнитивных процессов и кратковременной памяти [192]. Кроме того, поскольку префронтальная кора посылает прямые и косвенные возбуждающие проекции на дофаминергические нейроны среднего мозга, нарушение ГАМКергической регуляции в префронтальной коре может привести к гипердофаминергии и, в свою очередь, к развитию психоза [174].
В работе [175] был проведен систематический анализ экспрессии связанных с ГАМК транскриптомов префронтальной коры больных шизофренией и контрольной группы. У субъектов с шизофренией наблюдался дефицит экспрессии GAT1 в PV нейронах, иннервирующих начальные сегменты аксонов пирамидальных нейронов. Наряду с этим, на постсинаптических мембранах начальных сегментов аксонов пирамидальных клеток в ГАМКА-рецепторах было зафиксировано увеличение количества субъединицы α2, в то время как увеличения количества самих рецепторов выявлено не было. Данные результаты свидетельствует об изменении субъединичного состава ГАМКА-рецепторов при увеличении количества нейромедиатора в синаптической щели. В норме субъединица α2 входит только в состав ГАМКА-рецепторов, располагающихся в сомато-дендритных синаптических контактах [193]. ГАМК-опосредованная регуляция дендритной области пирамидальных нейронов важна для фильтрации входных возбуждающих токов из различных областей коры и подкорки, в то время как ГАМК-регуляция в перисоматической области (начальный сегмент аксона и тело клетки) критически важна для контроля генерации потенциалов действия во времени и синхронизации пирамидальных нейронов [68, 194]. Изменение субъединичного состава может быть причиной нарушений фильтрации выходных сигналов [156]. Также в данном исследовании были обнаружены изменения в ГАМК-нейротрансмиссии, опосредованной нарушениями в работе SST-содержащих и CCK-содержащих корзинчатых ГАМК-интернейронов, которые образуют синаптические окончания преимущественно с дистальными дендритами и телами пирамидальных нейронов соответственно. В данных областях также было выявлено изменение субъединичного состава ГАМКА-рецепторов (снижение количества субъединиц α1 и γ2, присутствующих в постсинаптических рецепторах, опосредующих фазное торможение, и субъединиц α4 и δ, присутствующие во внесинаптических рецепторах, опосредующих тоническое торможение), и как следствие, изменение фазного и тонического торможения в дендритах пирамидальных нейронов.
Таким образом, в данной работе у пациентов с шизофренией были обнаружены изменения ГАМК-регуляции как входных, так и выходных участков пирамидальных нейронов префронтальной коры, что подтверждает результаты in vivo исследований, показавших, что в развитие когнитивной симптоматики при шизофрении вовлечены нарушения ГАМКергической сигнализации в ПФК [195] и дисбаланс процессов возбуждения и торможения [196]. Данные изменения влияют на обработку информации в префронтальной коре и, таким образом, вносят вклад в нарушение когнитивных функций у больных шизофренией [169].
В работе [157] у мышей с нокаутом GAT1 наблюдались многочисленные аномалии поведения, связанные с позитивными, негативными и когнитивными симптомами. Дефицит GAT1 не изменял уровень дофамина в стриатуме, но значительно усиливал тоническое торможение в префронтальной коре.
У пациентов с шизофренией наблюдаются значительные изменения в гамма-колебаниях, связанных с когнитивными функциями и обработкой информации, что коррелирует с уровнем функциональных нарушений в рабочей памяти [197].
Поскольку на ранних этапах развития мозга ГАМК является возбуждающим нейромедиатором и наряду с BDNF участвует в процессах нейрогенеза [122], дисфункция GAT1 и нарушение регуляции внесинаптической концентрации ГАМК также могут быть причинами когнитивных нарушений при шизофрении.
Таким образом, нарушение обратного захвата ГАМК способно приводить к различным нарушениям (изменения субъединичного состава ГАМКА-рецепторов, нарушения процессов фазового и тонического торможения, нейрогенеза), которые в совокупности способны приводить к развитию таких заболеваний, как эпилепсия, РАС, УО и шизофрения.
Перспективы лечения психоневрологических заболеваний, связанных с мутациями SLC6A1, при помощи систем редактирования генома. Поскольку мы предполагаем что мутации de novo в гене SLC6A1 напрямую связаны с целым спектром психоневрологических нарушений, встает вопрос – может ли человечество на данной стадии своего развития предложить методы коррекции данных патологий, причем желательно устраняя источник проблемы, а не применяя симптоматическое лечение?
Одним из подходов является генная терапия при помощи доставки в нейроны головного мозга целой “правильной” копии гена SLC6A1, что позволило бы вернуть утраченные функции белка-транспортера GAT1. В настоящий момент широкое распространение получил ряд технологий по доставке генов в целевые клетки при помощи в т.ч. AAV-векторов на основе адено-ассоциированных вирусов [198]. С применением данного метода был разработан ряд лекарственных средств для лечения спинально-мышечной атрофии [199], мукополисахаридоза [200], болезни Паркинсона [201] и т.д. AAV-векторы зарекомендовали себя как эффективное средство доставки, при этом достаточно безопасное в плане иммуногенности и онкогенности [202]. Значимым преимуществом AAV-векторов является то, что ДНК вектора не встраивается в геном клетки и персистирует в виде эписом, что позволяет обойти риск инсерционного мутагенеза, как, например, при использовании векторов на основе ленти- и ретро-вирусов. Однако есть вероятность, что данный подход будет неэффективен в терапии заболеваний связанных с мутациями в SLC6A1. Во-первых, большинство описанных мутаций в SLC6A1 существуют в гетерозиготном варианте [9]. То есть, в генотипе носителей изначально имеется немутантная аллель гена, что может обусловливать отсутствие недостатка в его экспрессии. Но несмотря на это, у больных все равно наблюдается развитие характерной симптоматики. Во-вторых, большинство описанных высокопенетрантных мутаций в SLC6A1 не являются нокаутирующими, поэтому не ясно, по какому механизму мутантный белок GAT1 нарушает работу нервной системы, и соответственно нет понимания, чем может помочь доставка здоровой копии гена в данных случаях.
Второй подход основан на использовании современных методов редактирования генома, в т.ч. методов на основе системы CRISPR/Cas9. За последние 8 лет с момента первых публикаций применения на клетках млекопитающих данная система активно развивалась получила широкое распространение [203] и международное признание в виде Нобелевской премии по химии в 2020 году. В настоящий момент несколько десятков генотерапевтических препаратов на основе CRISPR/Cas9 системы проходят клинические испытания [204]. Теоретически, с помощью данного метода можно вернуть последовательность дефектного гена SLC6A1 к природному дикому типу, что с большой вероятностью должно нормализовать работу нервной системы. Само доказательство принципа работы системы CRISPR/Cas9 в нейронах головного мозга было подтверждено несколькими исследованиями [205, 206]. Однако существует несколько проблем, которые ставят под сомнение использование CRISPR/Cas9, равно как и более ранних систем, таких как ZFN или TALEN. Во-первых, низкая эффективность точечного направленного редактирования последовательности ДНК в клетках. Основное действие белка Cas9 – это двухцепочечный разрыв ДНК в необходимом месте гена. Данная система может быть эффективно использована для нокаута генов в клетках, поскольку после подобного разрыва системы репарации ДНК типа NHEJ (non-homology-end-repair) в клетках часто ошибаются и вносят короткие делеции или инсерции, приводящие к сдвигу рамки считывания кодирующей последовательности белка (если гидовая РНК системы CRISPR/Cas9 написана на белок-кодирующую часть гена) [207]. Но для направленного изменения последовательности мутантного гена SLC6A1 необходима система репарации по типу HDR (homology-direct-repair) и одновременно доставка в клетки ДНК-донора – одноцепочечной или двухцепочечной последовательности ДНК, которая послужит матрицей для синтеза “правильной” последовательности гена. Однако в клетках млекопитающих данные системы репарации конкурируют, и эффективность HDR значительно проигрывает по эффективности NHEJ, составляя порядка нескольких единиц процентов [208]. На данный момент актуальной является разработка новых подходов по исправлению точечных мутаций без HDR-рекомбинаций, таких как Prime Editing [209]. Данная система предполагает использование химерной гидовой РНК (pegRNA), содержащей на 3' конце последовательность для “правильной” записи редактируемого участка, и химерного мутантного белка (PE) - nCas9 (никаза, удален один каталитический центр расщепления ДНК) с вирусной обратной транскриптазой, которая синтезирует новую “правильную” цепь ДНК прямо на месте, используя предлагаемую РНК-матрицу. Эффективность данного комплекса может достигать 30–70% в зависимости от редактируемого участка генома. Однако для использования данной системы возникают сложности с ее доставкой, поскольку в клетки необходимо одновременно доставлять как минимум два компонента (pegRNA и PE). Доставка большого количества копий либо плазмидной ДНК либо вирусных векторов в головной мозг по прежнему является нетривиальной задачей, требующей тонких стереотаксических инъекций, для равномерного распределения частиц по достаточно большому объему. После физической доставки векторов кодирующих элементы системы в мозг, необходимо также обеспечить эффективное прохождение векторов через клеточную мембрану, что также является значимым барьером. Так, например, эффективность трансфекции в идеальных условиях чашки Петри, плазмидными векторами с элементами системы CRISPR/Cas9, составляет обычно 10–15% (неопубликованные данные авторов обзора), то есть подавляющее большинство клеток не принимает плазмиды. Проблемой служит размер генетических кассет, поскольку один только ген Cas9 занимает порядка 4,2 т.п.н., а вместе с сопутствующими элементами (ревертаза, промотор, WPRE-сигнал), размеры кассеты Prime Editor – целых 7,2 т.п.н. К сожалению этот факт также служит барьером для доставки Prime Editor в клетки при помощи AAV-векторов, максимальный размер кассет для которых составляет порядка 4.2–4.5 т.п.н [210]. Ну и конечно, традиционной проблемой всех методов с использованием систем редактирования генома является их нецелевая активность (т.н. “off-target” эффекты). Разные исследователи по разному оценивают опасность применения CRISPR/Cas9 систем в плане внесения нежелательных мутаций [211], однако случаи нахождения подобных off-target мутаций являются научным фактом, и с этим неизбежно придется столкнутся при доказательстве безопасности разрабатываемого метода лечения при помощи геномного редактирования.
Таким образом, можно констатировать то, что на пути разработки методов лечения мутаций SLC6A1 при помощи методов редактирования генома в настоящий момент находится множество препятствий, что не позволяет нам надеяться на легкое решение данной проблемы в обозримом будущем. Однако стоит заметить, что вышеуказанные проблемы носят в основном технологический характер, и вполне возможно, что открытие новых эффективных способов доставки и новых эффективных и малоразмерных геномных редакторов помогут сдвинуть процесс с мертвой точки.
ЗАКЛЮЧЕНИЕ
Психические и неврологические полигенные заболевания отличаются сложностью генетической архитектуры. На данный момент ни для одного полигенного психического заболевания нет полноценного генетического портрета. Однако появляется все больше работ, посвященных анализу данных GWAS [98, 212], результатов секвенирования [213, 214] и подходам к их обработке биоинформатическими методами [89, 215]. Расширяются выборки, выявляются новые локусы и гены-кандидаты. Все это приближает к созданию “генетических портфолио” для полигенных заболеваний.
De novo мутации в гене SLC6A1 вызывают дисфункцию ГАМК-транспортера (GAT1), что приводит к нарушению ГАМК-регуляции и значительно повышает риск развития эпилепсии и ряда психических заболеваний, в том числе шизофрении. Изучение мутаций в гене SLC6A1 также важно для составления генетических карт данных заболеваний. Своевременное обнаружение таких мутаций с помощью секвенирования нового поколения и разработка подходов для их исправления могут стать первым шагом к персонализированной терапии полигенных заболеваний.
Список литературы
Sodium- and chloride-dependent GABA transporter 1 [Electronic resource]. URL: https://www.uniprot.org/uniprot/P30531 (accessed: 11.05.2021).
Dalby N.O. // Neuropharmacology. 2000. V. 39. № 12. P. 2399–2407.
Semyanov A., Walker M.C., Kullmann D.M. // Nat. Neurosci. 2003. V. 6. № 5. P. 484–490.
Keros S., Hablitz J.J. // J. Neurophysiol. 2005. V. 94. № 3. P. 2073–2085.
Gong N. et al. // J. Neurosci. 2009. V. 29. № 50. P. 15836–15845.
Andäng M. et al. // Nature. 2008. V. 451. № 7177. P. 460–464.
Ben-Ari Y. // Nat. Rev. Neurosci. 2002. V. 3. № 9. P. 728–739.
Johannesen K.M. et al. // Epilepsia. 2018. V. 59. № 2. P. 389–402.
Goodspeed K. et al. // Brain Commun. 2020. V. 2. № 2. P. fcaa170.
Carvill G.L. et al. // Am. J. Hum. Genet. 2015. V. 96. № 5. P. 808–815.
Feng Y.-C.A. et al. // Am. J. Hum. Genet. 2019. V. 105. № 2. P. 267–282.
Sanders S.J. et al. // Neuron. 2015. V. 87. № 6. P. 1215–1233.
Satterstrom F.K. et al. // Cell. 2020. V. 180. № 3. P. 568–584. e23.
Bolton P.F. et al. // Br. J. Psychiatry. 2011. V. 198. № 4. P. 289–294.
Nelson S.B., Valakh V. // Neuron. 2015. V. 87. № 4. P. 684–698.
Rees E. et al. // Nat. Neurosci. 2020. V. 23. № 2. P. 179–184.
Volk D.W. et al. // Am. J. Psychiatry. 2001. V. 158. № 2. P. 256–265.
Hashimoto T. et al. // Am. J. Psychiatry. 2008. V. 165. № 4. P. 479–489.
Bullock W.M. et al. // Am. J. Psychiatry. 2008. V. 165. № 12. P. 1594–1603.
Hysi P.G. et al. // Nat. Genet. 2020. V. 52. № 4. P. 401–407.
Lawrenson K. et al. // Gynecol. Oncol. 2019. V. 153. № 2. P. 343–355.
Adkins D.E. et al. // Twin Res. Hum. Genet. 2015. V. 18. № 4. P. 335–347.
Thoeringer C.K. et al. // J. Neural Transm. 2009. V. 116. № 6. P. 649–657.
Lasky-Su J. et al. // Am. J. Med. Genet. B Neuropsychiatr. Genet. Wiley, 2008. V. 147B. № 8. P. 1345–1354.
Demontis D. et al. // Nat. Genet. 2019. V. 51. № 1. P. 63–75.
Mattison K.A. et al. // Epilepsia. 2018. V. 59. № 9. P. e135–e141.
Yuan F.-F. et al. // Prog. Neuropsychopharmacol. Biol. Psychiatry. 2017. V. 77. P. 202–208.
Minelli A. et al. // J. Neurosci. 1996. V. 16. № 19. P. 6255–6264.
Melone M., Ciappelloni S., Conti F. // Brain Struct. Funct. 2015. V. 220. № 2. P. 885–897.
Fattorini G., Melone M., Sánchez-Gómez M.V. // Glia. Wiley Online Library, 2017.
Fattorini G. et al. // Glia. 2020. V. 68. № 3. P. 646–655.
Uhlén M. et al. // Science. 2015. V. 347. № 6220. P. 1260419.
Zhang J. et al. // Syst. Biol. Reprod. Med. 2009. V. 55, № 5-6. P. 175–180.
Kanner B.I. // Biochemistry. 1978. V. 17. № 7. P. 1207–1211.
Radian R., Kanner B.I. // J. Biol. Chem. 1985. V. 260. № 21. P. 11859–11865.
Guastella J. et al. // Science. 1990. V. 249. № 4974. P. 1303–1306.
Zafar S., Jabeen I. // Front Chem. 2018. V. 6. P. 397.
Risso S., DeFelice L.J., Blakely R.D. // J. Physiol. 1996. V. 490 (Pt 3). P. 691–702.
Cammack J.N., Schwartz E.A. // Proc. Natl. Acad. Sci. U. S. A. 1996. V. 93, № 2. P. 723–727.
Stark E. et al. // Neuron. 2013. V. 80. № 5. P. 1263–1276.
Pouille F., Scanziani M. // Science. 2001. V. 293. № 5532. P. 1159–1163.
Hájos N. et al. // J. Neurosci. 2004. V. 24. № 41. P. 9127–9137.
Akam T., Kullmann D.M. // Neuron. 2010. V. 67. № 2. P. 308–320.
Brown D.A. // Neuropharmacology. 2018. V. 136. Pt A. P. 3–9.
Borden L.A. // Neurochem. Int. 1996. V. 29. № 4. P. 335–356.
Iversen L.L., Kelly J.S. // Biochem. Pharmacol. 1975. V. 24. № 9. P. 933–938.
Dingledine R., Korn S.J. // J. Physiol. 1985. V. 366. P. 387–409.
Isaacson J.S., Solís J.M., Nicoll R.A. // Neuron. 1993. V. 10. № 2. P. 165–175.
Overstreet L.S., Westbrook G.L. // J. Neurosci. 2003. V. 23. № 7. P. 2618–2626.
Liu G.-X. et al. // J. Neurosci. Res. 2007. V. 85. № 3. P. 649–655.
Liu G.-X. et al. // Neuropsychopharmacology. 2007. V. 32. № 7. P. 1531–1539.
Xu Y.F. et al. // J. Neurosci. Res. 2008. V. 86. № 2. P. 465–470.
Cai Y.-Q. et al. // J. Neurosci. Res. 2006. V. 84. № 2. P. 255–267.
Chiu C.-S. et al. // J. Neurosci. 2002. V. 22. № 23. P. 10251–10266.
Chiu C.-S. et al. // J. Neurosci. 2005. V. 25. № 12. P. 3234–3245.
Quandt G., Höfner G., Wanner K.T. // Bioorg. Med. Chem. 2013. V. 21. № 11. P. 3363–3378.
Kristensen A.S. et al. // Pharmacol. Rev. 2011. V. 63. № 3. P. 585–640.
Zhou Y., Danbolt N.C. // Front. Endocrinol. 2013. V. 4. P. 165.
Scimemi A. // Front. Cell. Neurosci. 2014. V. 8. P. 161.
Kavanaugh M.P. et al. // J. Biol. Chem. 1992. V. 267. № 31. P. 22007–22009.
Ikegaki N. et al. // Brain Res. Mol. Brain Res. 1994. V. 26. № 1–2. P. 47–54.
Conti F., Minelli A., Melone M. // Brain Res. Brain Res. Rev. 2004. V. 45. № 3. P. 196–212.
Zhou Y. et al. // Am. J. Physiol. Renal Physiol. 2012. V. 302. № 3. P. F316–F328.
GeneCards Human Gene Database. SLC6A1 Gene - GeneCards [Electronic resource]. URL: https://www.genecards.org/cgi-bin/carddisp.pl?id= P30531 (accessed: 12.05.2021).
Braestrup C. et al. // J. Neurochem. 1990. V. 54. № 2. P. 639–647.
P30531 [Electronic resource]. URL: https://swissmodel.expasy.org/repository/uniprot/P30531?csm= 094DB39C1D3B75D2 (accessed: 15.11.2020).
Dichter M.A., Ayala G.F. // Science. 1987. V. 237. № 4811. P. 157–164.
Markram H. et al. // Nat. Rev. Neurosci. 2004. V. 5. № 10. P. 793–807.
Markram H. et al. // Cell. 2015. V. 163. № 2. P. 456–492.
Rudy B. et al. // Dev. Neurobiol. 2011. V. 71. № 1. P. 45–61.
Petilla Interneuron Nomenclature Group et al. // Nat. Rev. Neurosci. 2008. V. 9. № 7. P. 557–568.
Kawaguchi Y., Kubota Y. // Cereb. Cortex. 1997. V. 7. № 6. P. 476–486.
Buzsáki G., Draguhn A. // Science. 2004. V. 304. № 5679. P. 1926–1929.
Gaetz W. et al. // Neuroimage. 2011. V. 55. № 2. P. 616–621.
Lundqvist M. et al. // Neuron. 2016. V. 90. № 1. P. 152–164.
Rojas D.C., Wilson L.B. // Biomark. Med. 2014. V. 8. № 3. P. 353–368.
McNally J.M., McCarley R.W. // Curr. Opin. Psychiatry. 2016. V. 29. № 3. P. 202–210.
Le Roux N. et al. // Eur. J. Neurosci. 2008. V. 27. № 12. P. 3244–3256.
den Boon F.S. et al. // Pflugers Arch. 2015. V. 467. № 7. P. 1551–1564.
Gao R., Penzes P. // Curr. Mol. Med. 2015. V. 15. № 2. P. 146–167.
Turrigiano G.G., Nelson S.B. // Nat. Rev. Neurosci. 2004. V. 5. № 2. P. 97–107.
Kang E. et al. // Cell Rep. 2019. V. 28. № 6. P. 1419–1428.e3.
Arslan A. // Int. J. Mol. Sci. 2018. V. 19. № 1.
Akbarian S. et al. // Arch. Gen. Psychiatry. 1995. V. 52. № 4. P. 258–266.
Volk D.W. et al. // Arch. Gen. Psychiatry. 2000. V. 57. № 3. P. 237–245.
Guidotti A. et al. // Arch. Gen. Psychiatry. 2000. V. 57. № 11. P. 1061–1069.
Hashimoto T. et al. // J. Neurosci. 2003. V. 23. № 15. P. 6315–6326.
Williams H.J. et al. // Am. J. Psychiatry. 2005. V. 162. № 9. P. 1736–1738.
Arslan A. // Prog. Neuropsychopharmacol. Biol. Psychiatry. Elsevier Inc., 2018. V. 80. P. 155–165.
Bormann J. // Trends Pharmacol. Sci. 2000. V. 21. № 1. P. 16–19.
Zhang D. et al. // Trends Pharmacol. Sci. 2001. V. 22. № 3. P. 121–132.
Chebib M. // Clin. Exp. Pharmacol. Physiol. 2004. V. 31. № 11. P. 800–804.
Mehta A.K., Ticku M.K. // Brain Res. Brain Res. Rev. 1999. V. 29. № 2–3. P. 196–217.
Costa E. // Annu. Rev. Pharmacol. Toxicol. 1998. V. 38. P. 321–350.
Bianchi M.T., Haas K.F., Macdonald R.L. // J. Neurosci. 2001. V. 21 № 4. P. 1127–1136.
Lee Y.H., Kim J.-H., Song G.G. // Gene. 2013. V. 525. № 1. P. 107–115.
Lencz T. et al. // Nat. Commun. 2013. V. 4. P. 2739.
Ripke S. et al. // Nat. Genet. 2013. V. 45. № 10. P. 1150–1159.
Bettler B., Tiao J.Y.-H. // Pharmacol. Ther. 2006. V. 110, № 3. P. 533–543.
Jackson M.F., Esplin B., Capek R. // Epilepsy Res. 1999. V. 37. № 1. P. 25–36.
Brooks-Kayal A.R. et al. // Nat. Med. 1998. V. 4. № 10. P. 1166–1172.
Bethmann K. et al. // Neurobiol. Dis. 2008. V. 31. № 2. P. 169–187.
Jones K.A. et al. // Nature. 1998. V. 396. № 6712. P. 674–679.
Couve A., Moss S.J., Pangalos M.N. // Mol. Cell. Neurosci. 2000. V. 16. № 4. P. 296–312.
Mott D.D., Lewis D.V. // Int. Rev. Neurobiol. 1994. V. 36. P. 97–223.
Scanziani M. // Neuron. 2000. V. 25. № 3. P. 673–681.
Misgeld U., Bijak M., Jarolimek W. // Prog. Neurobiol. 1995. V. 46. № 4. P. 423–462.
Andrade R., Malenka R.C., Nicoll R.A. // Science. 1986. V. 234. № 4781. P. 1261–1265.
Freund T.F., Buzsáki G. // Hippocampus. 1996. V. 6. № 4. P. 347–470.
Miles R. et al. // Neuron. 1996. V. 16. № 4. P. 815–823.
Gaspary H.L., Wang W., Richerson G.B. // J. Neurophysiol. 1998. V. 80, № 1. P. 270–281.
Liu Q.Y. et al. // J. Neurophysiol. 2000. V. 84. № 3. P. 1392–1403.
Overstreet L.S., Westbrook G.L. // J. Neurophysiol. 2001. V. 86. № 2. P. 596–603.
Brickley S.G., Cull-Candy S.G., Farrant M. // J. Physiol. 1996. V. 497 (Pt 3). P. 753–759.
Rossi D.J., Hamann M. // Neuron. 1998. V. 20, № 4. P. 783–795.
Rivera C. et al. // Nature. 1999. V. 397, № 6716. P. 251–255.
Ganguly K. et al. // Cell. 2001. V. 105, № 4. P. 521–532.
Ben-Ari Y. et al. // Prog. Brain Res. 1994. V. 102. P. 261–273.
Jensen K. et al. // J. Neurophysiol. 2003. V. 90. № 4. P. 2690–2701.
Bragina L. et al. // J. Neurochem. 2008. V. 105. № 5. P. 1781–1793.
Conti F. et al. // Front. Cell. Neurosci. 2011. V. 5. P. 2.
Owens D.F., Kriegstein A.R. // Nat. Rev. Neurosci. 2002. V. 3. № 9. P. 715–727.
Cellot G., Cherubini E. // Front. Neural Circuits. 2013. V. 7. P. 136.
Manent J.-B. et al. // J. Neurosci. 2005. V. 25. № 19. P. 4755–4765.
Behar T.N. et al. // Cereb. Cortex. 2001. V. 11. № 8. P. 744–753.
Behar T.N. et al. // Cereb. Cortex. 2000. V. 10. № 9. P. 899–909.
Aronne M.P. et al. // Exp. Neurol. 2011. V. 229, № 2. P. 364–371.
Cuzon V.C. et al. // J. Neurosci. 2008. V. 28, № 8. P. 1854–1864.
Haas M. et al. // Eur. J. Neurosci. 2013. V. 37. № 10. P. 1584–1593.
Manent J.-B. et al. // Epilepsia. 2007. V. 48. № 4. P. 684–693.
Thompson B.L., Levitt P., Stanwood G.D. // Nat. Rev. Neurosci. 2009. V. 10. № 4. P. 303–312.
Wu X. et al. // J. Neurosci. 2012. V. 32. № 1. P. 331–343.
Trinchero M.F., Giacomini D., Schinder A.F. // Curr. Opin. Neurobiol. 2021. V. 69. P. 124–130.
Huang E.J., Reichardt L.F. // Annu. Rev. Neurosci. 2001. V. 24. P. 677–736.
Marín O., Rubenstein J.L. // Nat. Rev. Neurosci. 2001. V. 2. № 11. P. 780–790.
Park H., Poo M.-M. // Nat. Rev. Neurosci. 2013. V. 14, № 1. P. 7–23.
Knüsel B. et al. // J. Neurosci. 1994. V. 14. № 3. Pt 2. P. 1542–1554.
Polleux F. et al. // Development. 2002. V. 129. № 13. P. 3147–3160.
Woo N.H., Lu B. // Neuroscientist. 2006. V. 12. № 1. P. 43–56.
Gorski J.A. et al. // J. Neurosci. 2003. V. 23. № 17. P. 6856–6865.
Vicario-Abejón C. et al. // Nat. Rev. Neurosci. 2002. V. 3. № 12. P. 965–974.
Paus T., Keshavan M., Giedd J.N. // Nat. Rev. Neurosci. 2008. V. 9. № 12. P. 947–957.
Farhan H., Freissmuth M., Sitte H.H. // Handb. Exp. Pharmacol. 2006. № 175. P. 233–249.
Berninger B. et al. // Development. 1995. V. 121. № 8. P. 2327–2335.
Marty S. et al. // J. Neurosci. 1996. V. 16. № 2. P. 675–687.
Arion D., Lewis D.A. // Arch. Gen. Psychiatry. 2011. V. 68. № 1. P. 21–31.
Hyde T.M. et al. // J. Neurosci. 2011. V. 31. № 30. P. 11088–11095.
Ben-Ari Y. et al. // J. Physiol. 1989. V. 416. P. 303–325.
Blume W.T. et al. // Epilepsia. 2001. V. 42. № 9. P. 1212–1218.
Chen L. et al. // Acta Neuropsychiatr. 2015. V. 27. № 6. P. 368–374.
Cope D.W. et al. // Nat. Med. 2009. V. 15. № 12. P. 1392–1398.
Han H.A., Cortez M.A., Snead O.C. III. GABAB Receptor and Absence Epilepsy // Jasper’s Basic Mechanisms of the Epilepsies / Ed. Noebels J.L. et al. Bethesda (MD): National Center for Biotechnology Information (US), 2012.
Khosravani H., Zamponi G.W. // Physiol. Rev. 2006. V. 86. № 3. P. 941–966.
Snead O.C. 3rd. // Eur. J. Pharmacol. 1992. V. 213. № 3. P. 343–349.
Zhang L. et al. // Neuropsychopharmacology. 2016. V. 41. № 6. P. 1467–1476.
Macdonald R.L., Kang J.-Q., Gallagher M.J. // J. Physiol. 2010. V. 588. Pt 11. P. 1861–1869.
Yu Z. et al. // PLoS One. 2013. V. 8. № 7.
Cai K. et al. // Exp. Neurol. Elsevier, 2019. V. 320. January. P. 112973.
Parikshak N.N. et al. // Cell. 2013. V. 155. № 5. P. 1008–1021.
Iakoucheva L.M., Muotri A.R., Sebat J. // Cell. 2019. V. 178. № 6. P. 1287–1298.
Grove J. et al. // Nat. Genet. Nature America, Inc., 2019. V. 51. № 3. P. 431–444.
Di J. et al. // Int. J. Dev. Neurosci. 2020. V. 80. № 2. P. 73–85.
Delahanty R.J. et al. // Mol. Psychiatry. 2011. V. 16. № 1. P. 86–96.
Han S. et al. // Nature. 2012. V. 489. № 7416. P. 385–390.
Kang J.-Q., Barnes G. // J. Autism Dev. Disord. 2013. V. 43. № 1. P. 68–79.
Wang J. et al. // Mol. Brain. 2020. Vol. 13, № 1. P. 76.
Devries S. et al. // Cold Spring Harb Mol Case Stud. 2020.
Posar A., Visconti P. // J. Pediatr. Neurosci. 2019. V. 14. № 2. P. 100–102.
Ferguson B.R., Gao W.-J. // Front. Neural Circuits. 2018. V. 12. P. 37.
Singh T. et al. // Nat. Genet. Nature Publishing Group, 2017. V. 49. № 8. P. 1167–1173.
Fromer M. et al. // Nature. Nature Publishing Group, 2014. V. 506. № 7487. P. 179–184.
Lee J., Park S. // J. Abnorm. Psychol. 2005. V. 114. № 4. P. 599–611.
Insel T.R. // Nature. 2010. V. 468. № 7321. P. 187–193.
Tse M.T., Piantadosi P.T., Floresco S.B. // Biol. Psychiatry. Elsevier, 2015. V. 77. № 11. P. 929–939.
Hashimoto T. et al. // Mol. Psychiatry. 2008. V. 13. № 2. P. 147–161.
Wray N.R., Visscher P.M. // Schizophr. Bull. 2010. V. 36. № 1. P. 14–23.
Gejman P.V., Sanders A.R., Duan J. // Psychiatr. Clin. North Am. 2010. V. 33. № 1. P. 35–66.
Martin A.R. et al. // Biol. Psychiatry. 2019. V. 86. № 2. P. 97–109.
Ma C. et al. // Transl. Psychiatry. 2018. V. 8. № 1.
Pardiñas A.F. et al. // Nat. Genet. 2018. V. 50. № 3. P. 381–389.
Tansey K.E. et al. // Mol. Psychiatry. 2016. V. 21. № 8. P. 1085–1089.
Pocklington A.J. et al. // Neuron. 2015. V. 86. № 5. P. 1203–1214.
Ripke S. et al. // Nature. 2014. V. 511. № 7510. P. 421–427.
Wray N.R. et al. // J. Child Psychol. Psychiatry. 2014. V. 55. № 10. P. 1068–1087.
Bush W.S., Moore J.H. // PLoS Comput. Biol. 2012. V. 8. № 12. P. e1002822.
Sullivan P.F., Daly M.J., O’Donovan M. // Nat. Rev. Genet. Nature Publishing Group. 2012. V. 13. № 8. P. 537–551.
St Clair D. // Schizophr. Bull. 2009. V. 35, № 1. P. 9–12.
Cook E.H., Jr., Scherer S.W. // Nature. 2008. V. 455. № 7215. P. 919–923.
Gulsuner S. et al. // Cell. 2013. V. 154. № 3. P. 518–529.
Singh T. et al. // Eur. Neuropsychopharmacol. 2019. V. 29. P. S813–S814.
Lewis D.A., Hashimoto T., Volk D.W. // Nat. Rev. Neurosci. 2005. V. 6. № 4. P. 312–324.
Opris I., Bruce C.J. // Brain Res. Brain Res. Rev. 2005. V. 48. № 3. P. 509–526.
Семьянов А.В. // Нейрофизиология. researchgate.net, 2002.
Somogyi P., Klausberger T. // J. Physiol. 2005. V. 562. Pt 1. P. 9–26.
Lewis D.A. et al. // Trends Neurosci. 2012. V. 35. № 1. P. 57–67.
Lisman J. // Curr. Opin. Neurobiol. 2012. V. 22. № 3. P. 537–544.
Basar-Eroglu C. et al. // Int. J. Psychophysiol. 2007. V. 64, № 1. P. 39–45.
Wang D., Tai P.W.L., Gao G. // Nat. Rev. Drug Discov. 2019. V. 18. № 5. P. 358–378.
Foust K.D. et al. // Nat. Biotechnol. 2010. V. 28. № 3. P. 271–274.
Fu H. et al. // Mol. Ther. 2011. V. 19. № 6. P. 1025–1033.
Kaplitt M.G. et al. // Lancet. 2007. V. 369. № 9579. P. 2097–2105.
Bolt M.W. et al. // J. Toxicol. Sci. 2021. V. 46. № 2. P. 57–68.
Shivram H. et al. // Nat. Chem. Biol. 2021. V. 17. № 1. P. 10–19.
Weiss W.F. // Ther Innov Regul Sci. 2021.
Nishiyama J., Mikuni T., Yasuda R. // Neuron. 2017. V. 96. № 4. P. 755–768.e5.
Matsuda T., Oinuma I. // Sci. Rep. 2019. V. 9. № 1. P. 11309.
Xue C., Greene E.C. // Trends Genet. 2021.
Fu Y.-W. et al. // Nucleic Acids Res. 2021. V. 49. № 2. P. 969–985.
Anzalone A.V. et al. // Nature. 2019. V. 576. № 7785. P. 149–157.
Ridley R.B., Walsh E.M., Ildefonso C.J. // Methods Mol. Biol. 2021. V. 2225. P. 77–92.
Sledzinski P. et al. // Biotechnol. Adv. 2021. V. 49. P. 107737.
Visscher P.M. et al. // Am. J. Hum. Genet. 2012. V. 90. № 1. P. 7–24.
Malhotra D., Sebat J. // Cell. 2012. V. 148. № 6. P. 1223–1241.
Yuan H. et al. // BMC Med. Genet. 2020. V. 21. № 1. P. 93.
Golov A.K. et al. // Cells. 2020. V. 9. № 1. P. 246.
Дополнительные материалы отсутствуют.