

Нейрохимия, 2022, T. 39, № 1, стр. 45-58
Гипотетический механизм тремора покоя при болезни Паркинсона
И. Г. Силькис
Федеральное государственное бюджетное учреждение науки Институт высшей нервной деятельности
и нейрофизиологии Российской академии наук
Москва, Россия
Поступила в редакцию 15.08.2021
После доработки 09.09.2021
Принята к публикации 06.10.2021
- EDN: JBDTEJ
- DOI: 10.31857/S1027813322010113
Аннотация
Предложен гипотетический механизм появления тремора покоя при болезни Паркинсона. В основе этого тремора лежит вызванная дефицитом дофамина модификация эффективности возбудительных входов к шипиковым клеткам стриатума, которая приводят к реорганизации активности в нейронной сети, включающей новую кору, таламус, базальные ганглии, мозжечок, субталамическое и педункулопонтийное ядра. В результате этой модификации снижается активность стрионигральных клеток, ведущая к растормаживанию нейронов внутренней части бледного шара. Одновременное увеличивается активность стриопаллидарных клеток, что усиливает ингибирование нейронов наружной части бледного шара и приводит к снижению их активности и уменьшению их ингибирующего действия на нейроны внутренней части бледного шара, а также субталамического ядра и новой коры. В результате активность нейронов этих структур возрастает. Усиление ГАМКергического входа из внутренней части бледного шара к нейронам субталамического ядра, приводящее к гиперполяризации мембраны нейронов, способствует генерации ими пачечных разрядов с частотой тремора. Увеличение активности нейронов субталамического ядра приводит к усилению возбуждения их клеток-мишеней в педункулопонтийном ядре и глубоких ядрах мозжечка. Последующее усиление возбуждающего действия мозжечка на нейроны таламуса передается в новую кору и стриатум. Согласно предлагаемому механизму, степень возбуждения нейронов субталамического ядра определяет амплитуду тремора и позволяет объяснить тот факт, что увеличение активности в мозжечке, таламусе и коре коррелирует с выраженностью тремора. Из предлагаемого механизма следует, что поскольку избирательные агонисты дофаминовых Д2 рецепторов и/или антагонисты аденозиновых А2А рецепторов должны способствовать индукции длительной депрессии на стриопаллидарных клетках, ослаблению их ингибирующего действия на нейроны наружной части бледного шара и последующему усилению торможения субталамического ядра, эти вещества можно использовать для подавления тремора покоя. Эти следствия гипотезы имеют экспериментальное подтверждение.
ВВЕДЕНИЕ
Одним из часто наблюдаемых симптомов болезни Паркинсона (БП) является тремор покоя, характеризующийся частотой 4–6 Гц [1]. На основании клинических данных о том, что дофамин по-разному влияет на тремор покоя и такие симптомы БП, как дискинезия и ригидность, было предположено, что механизмы, лежащие в их основе, отличаются [2]. Брадикинезию и ригидность объясняют снижением уровня дофамина во входной структуре базальных ганглиев (БГ) – стриатуме, тогда как тремор покоя связывают с взаимодействием БГ с цепью мозжечок–таламус–кора (М–Т–К) и увеличением активности нейронов в этой цепи, которое зависит от изменений дофаминергических проекций к этим нейронам [2, 3]. Согласно модели, названной переключатель “диммер”, активность, вызывающая тремор при БП, возникает во внутренней части бледного шара (БШв) – одной из выходных структур БГ и затем распространяется в цепь М‒Т–К, где ритмическая активность тремора поддерживается и усиливается [2, 4, 5]. При этом нейронная активность в новой коре, которую е связывают с амплитудой тремора, влияет на активность всей сети через БГ [5]. Имеются данные и о значительной роли изменений активности нейронов наружной части бледного шара (БШн) в появлении симптомов БП [6]. Поскольку в таламус проецируются преимущественно ГАМКергические клетки выходных ядер БГ – БШв и ретикулярной части черного вещества (ЧВр) [7], увеличение активности нейронов БШв при треморе должно приводить к усилению ингибирования нейронов таламуса. Однако, поскольку при треморе покоя наблюдается увеличение активности в цепи М–Т–К, есть основания полагать, что существует некий механизм, обеспечивающий такое возбуждение нейронов таламуса, которое компенсирует его ингибирование со стороны БГ.
Поскольку у пациентов с БП тремор покоя наблюдается вследствие гибели дофаминергических клеток компактной части черного вещества (ЧВк) и ретро-рубральной области [8, 9] и поскольку плотность чувствительных к дофамину Д1 и Д2 рецепторов самая большая во входной структуре БГ – стриатуме, естественно предположить, что тремор связан с изменением функционирования нейронов стриатума и последующим изменением активности их клеток- мишеней в БШв и БШн (рис. 1). В свою очередь, это приведет к реорганизации активности в структурах, на которые проецируются БШв и БШн. Отмечено, что у пациентов с тремором покоя меняется паттерн разрядов нейронов разных ядер БГ. В них активность соседних нейронов становится более синхронизированной и появляются связанные с тремором пачечные разряды [6, 10]. Такую активность регистрировали в БШ, субталамическом ядре (СТЯ), педункулопонтийном ядре (ППЯ), таламических ядрах [11–16]. При моделировании БП у грызунов и приматов пачечную активность наблюдали и у шипиковых клеток стриатума [17]. Все упомянутые структуры взаимосвязаны, поэтому приведенные экспериментальные данные указывают на распространение осцилляторной активности по всей сети.
Рис. 1.
Упрощенная схема организации взаимодействий в нейронной сети, участвующей в генерации тремора покоя в условиях дефицита дофамина. П – стрионигральные клетки, дающие начало прямому пути через базальные ганглии; Н – стриопаллидарные клетки, дающие начало непрямому пути через базальные ганглии. КЗ – клетки-зерна; КП – клетки Пуркинье; ГЯМ – глубокие ядра мозжечка; МВ – мшистые волокна; ПВ – параллельные волокна; БГ – базальные ганглии; БШн – наружная часть бледного шара; БШв – внутренняя часть бледного шара; ЧВр и ЧВк – ретикулярная и компактная части черного вещества, соответственно; ВПП – вентральное поле покрышки; ЯМ – ядра моста; СТЯ – субталамическое ядро; ППЯ – педункулопонтийное ядро; ПФЯ, ВПЯ, ВЛ – парафасцикулярное, вентральное промежуточное, вентролатеральное ядра таламуса, соответственно; КЯ – красное ядро. Линии, оканчивающиеся белами и черными кружками – возбудительные и тормозные связи, соответственно. Мозжечок и базальные ганглии ограничены пунктирными прямоугольниками. Утолщенные и тонкие линии – потенциированные сильные и депрессированные слабые взаимодействия, соответственно, в условиях дефицита дофамина.
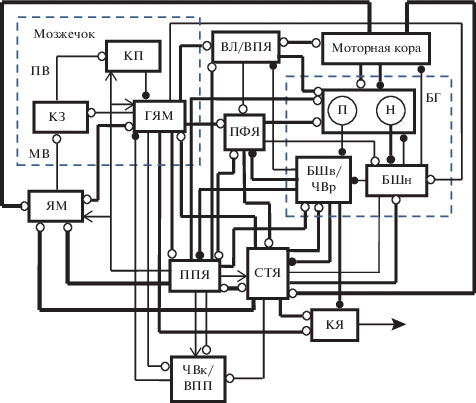
Целью настоящей работы являлся анализ возможных механизмов, лежащих в основе появления пачечной активности, а также увеличения активности нейронов в цепи М–Т–К при тремора покоя, вызванном дефицитом дофамина. При решении поставленной задачи использованы результаты проведенного ранее анализа возможных механизмов влияния дофамина на функционирование нейронной сети, включающей новую кору, БГ, таламус, мозжечок, СТЯ и ППЯ [18].
ГЕНЕРАЦИЯ СИНХРОНИЗИРОВАННОЙ ПАЧЕЧНОЙ АКТИВНОСТИ В РАЗНЫХ СТРУКТУРАХ ПРИ ТРЕМОРЕ ПОКОЯ
Во введении отмечено, что у пациентов с БП при треморе покоя активность в разных структурах возрастает, появляются пачечные разряды с частотой тремора, а также изменяются связи между структурами. В частности, увеличивается активность нейронов в мозжечке и усиливаются связи зубчатого ядра мозжечка с вентральным промежуточным ядром таламуса (ВПЯ), а также связи ВПЯ с первичной моторной корой М1, премоторной корой, дополнительной моторной областью и бледным шаром [19]. При этом сила связи ВПЯ с зубчатым ядром мозжечка и полем М1 положительно коррелировала только с выраженностью тремора, но не акинезии или ригидности [16, 19]. Активность нейронов в некоторых ядрах БГ была когерентна с тремором покоя [20]. У пациентов с тремором выявлено увеличение активности в бледном шаре по сравнению с пациентами, у которых тремора не было [21]. Только при треморе покоя ритмическую активность, которая значимо не отличалась от частоты тремора 4.4 Гц, наблюдали у 19.7% нейронов бледного шара [14]. Активность с частотой тремора наблюдали не только у 15.4% клеток БШв, но и у 46.8% клеток вентрального орального заднего таламического ядра, в которое проецируется БШв, а также у клеток таламического ядра ВПЯ, в которое поступает иннервация из глубоких ядер мозжечка (ГЯМ) [11]. В парафасцикулярном ядре (ПФЯ) и вентролатеральном (ВЛ) ядре таламуса также наблюдали ритмическую пачечную активность, тогда как при произвольных движениях или в ответах на соматосенсорные стимулы этот эффект отсутствовал [14].
При повреждении ЧВк возрастала частота срабатывания нейронов ППЯ и ВЛ, а паттерн срабатывания нейронов ППЯ становился пачечным [13]. При БП пачечные разряды наблюдали примерно у 21% клеток ППЯ [15]. При треморе у пациентов с БП увеличивалась функциональная связь между скорлупой стриатума и мозжечком [22], а также возрастала активность в цепи М–Т–К [23]. Кроме того, была выявлена синхронизация активности между парами нейронов в БШв, ЧВр и БШн, причем осцилляции имели частоту тремора [24]. У приматов с тремором покоя также заметно усиливалась корреляция активности между удаленно расположенными нейронами бледного шара по сравнению со здоровыми приматами [25, 26]. У пациентов с БП только при треморе покоя, но не при брадикинезии или регидности, в активности нейронов СТЯ наблюдали осцилляции с частотой 4–6 Гц [12]. Существенное увеличение числа пачечных разрядов в активности нейронов СТЯ при дефиците дофамина показано и в работе [27]. По сравнению со здоровыми испытуемыми, при БП увеличивается функциональная связь СТЯ с первичной моторной корой М1, а также с первичной сенсорной корой и дополнительной моторной корой [28]. Эти данные позволили предположить, что СТЯ играет важную роль в изменении функционирования цепи, связывающей БГ с моторной корой при БП [28].
ОСОБЕННОСТИ МЕЖНЕЙРОННЫХ ВЗАИМОДЕЙСТВИЙ В СЕТИ, ВКЛЮЧАЮЩЕЙ СТРУКТУРЫ, В КОТОРЫХ ПРИ ТРЕМОРЕ ПОКОЯ НАБЛЮДАЕТСЯ РИТМИЧЕСКАЯ АКТИВНОСТЬ
Упомянутые выше такие структуры как новая кора, таламус, БГ, мозжечок, СТЯ и ППЯ, в которых при БП генерируется активность с частотой тремора покоя, взаимосвязаны и образуют сеть, которая схематически представлена на рис. 1. Особенности функциональной организации нейронной сети кора–базальные ганглии–таламус–кора (К–БГ–Т–К) подробно описаны нами ранее [29, 30]. Кроме возбудительных входов в стриатум из новой коры и таламуса, недавно выявлены прямые входы из новой коры и из зубчатого ядра мозжечка в выходные структуры БГ – БШв и ЧВр. Эта прямая иннервация поступает в основном из сенсомоторных областей коры и мозжечка [31]. Возбуждение из моторной и первичных сенсорных областей коры поступает к входным клеткам-зернам коры мозжечка, а также к нейронам ГЯМ через ядра моста (ЯМ) [32–34]. Возбуждение из ГЯМ возвращается в ЯМ, где оно конвергируют с входами из неокортекса. Сигналы из ГЯМ поступают в неокортекс через таламус [35]. Кроме того, нейроны ГЯМ дисинаптически (через таламус) возбуждают шипиковые клетки стриатума [36]. Также нейроны ГЯМ проецируются в БШн [37]. Проекции из зубчатого ядра мозжечка через таламус в скорлупу стриатума, а также в БШн организованы топографически [37]. Возбуждение к нейронам ЯМ поступает также из СТЯ и ППЯ, находящихся под тормозным контролем со стороны выходных ядер БГ [35], причем такая организация характерна и для человека [38]. Нейроны ГЯМ могут влиять на разные структуры и через ПФЯ таламуса, в которое проецируются нейроны выходных ядер БГ, БШ, а также ВЛ [39] и которое моносинаптически возбуждают нейроны БШн/БШ [40], а также стриатум и СТЯ [41, 42]. Поэтому ГЯМ может влиять на функционирование СТЯ как через БГ, так и через ПФЯ [43]. Нейроны СТЯ обеспечивают основной возбудительный вход к нейронам выходных ядер БГ (ЧВр и БШв/ЭПЯ), а также к нейронам БШн/БШ, причем СТЯ иннервирует те же нейроны ЧВр и БШв/ЭПЯ, которые проецируются в таламус [44, 45]. СТЯ и ППЯ реципрокно возбуждают друг друга [46]. Стимуляция СТЯ и ЧВр вызывала в нейронах ППЯ возбудительные и тормозные ответы, соответственно [47]. Поскольку СТЯ возбуждает нейроны выходных ядер БГ, оно модулирует активность нейронов ППЯ не только за счет прямого возбуждения, но и вследствие ингибирования со стороны БГ [48]. В ППЯ поступает возбуждение также из ПФЯ таламуса [49] и новой коры [50]. Нейроны ППЯ иннервирует выходные ядра БГ и вентральный таламус [51], а также таламо-стриатные клетки [52], моторные зоны ствола мозга и спинного мозга [50].
Следует отметить, что таламо-стриатные входы, так же как входы от дофаминергических клеток. нейронов ППЯ, сенсорных и лимбических областей коры, одинаковы для стрионигральных (С-Н) и стриопаллидарных (С-П) клеток, дающих начало прямому и непрямому пути через БГ [53, 54], в отличие от моторной коры, которая иннервирует преимущественно нейроны непрямого пути [55]. Новая кора возбуждает нейроны СТЯ по гиперпрямому пути, так что при стимуляции коры реакции нейронов СТЯ предшествует возбуждению нейронов бледного шара [56]. Следует отметить, что входы из первичной моторной коры в СТЯ организованы соматотопически [44] и что в сенсомоторной части СТЯ выявлена карта тела [57]. Входы из ПФЯ в СТЯ также топографически организованы [42]. Топографическая организации и замкнутость является важным свойством цепей К–БГ–Т–К и М–Т–К–М [35].
Известно, что ППЯ состоит из нейронов разных типов, содержащих ацетилхолин, ГАМК или глутамат [58]. Холинергические нейроны ППЯ иннервируют практически все таламические ядра [58] и мозжечок, где ацетилхолин действует как на мускариновые, так и никотиновые рецепторы [59]. Поскольку показано, что аппликация антагониста холинорецепторов уменьшала ответы нейронов ЯМ [60], можно полагать, что действие ацетилхолина на ЯМ является потенциирующим. На КП имеются мускариновые М3 рецепторы, а их активация способствует индукции длительной потенциации (ДП) в пути параллельные волокна-КП [61]. С учетом правил модификации для нейронов ГЯМ [18], увеличение активности КП должно способствовать увеличению эффективности возбуждения нейронов ГЯМ. Эти данные позволяют полагать, что холинергическое влияние со стороны ППЯ на ГЯМ также является потенциирующим.
С целью упрощения на рис. 1 не представлены интернейроны, которые имеются в каждой из структур, отображены не все длинноаксонные ГАМКергические клетки, проецирующиеся в другие структуры. К длинноаксонным можно отнести С-Н и С-П шипиковые клетки, иннервирующие соответственно нейроны БШв/ЧВр и БШн; нейроны БШв /ЧВр, проецирующиеся на нейроны таламуса, СТЯ и ППЯ; а также нейроны БШн, ингибирующие СТЯ и БШв/ЧВр (рис. 1). Кроме того, нейроны БШн оказывают тормозное влияние на шипиковые нейроны стриатума [62, 63] и на новую кору, где их аксоны оканчиваются на возбудительных и тормозных клетках [64]. Поскольку БШн находится под тормозным контролем со стороны стриатума [64] (рис. 1), этот вход в кору должен зависеть от функционирования стриатума. В моторной коре также обнаружены длинноаксонные тормозные нейроны, которые моносинаптически иннервируют С-Н и С-П нейроны дорзального стриатума [54]. Поскольку показано, что латентные периоды возбудительных ответов С-Н и С-П клеток на стимуляцию коры одинаковы [65], можно полагать, что кортико-стриатное торможение также поступает на оба типа шипиковых клеток одновременно.
На вовлеченность красного ядра (КЯ) в возникновение тремора указывают результаты работы [66], показавшие, что электрическая стимуляция КЯ может вызвать тремор конечностей. Это ядро получает активацию из ГЯМ и иннервирует мотонейроны, а также премоторные нейроны в стволе мозга и спинном мозге. Те части КЯ, в которые проецируются выходные ядра БГ, также получают иннервацию из ГЯМ – зубчатого ядра и латеральной части ядра интерпозитус [67].
С помощью теоретического моделирования показано, что в нейронной сети с взаимосвязанными тормозными клетками при наличии достаточно сильного возбуждения могут возникать осцилляции, частота которых зависит от силы торможения, а амплитуда (мощность) – от величины возбуждения [68]. Нейронная сеть, представленная на рис. 1, удовлетворяет этим требованиям. В возникновении ритмической активности в сети могут участвовать как длинноаксонные ГАМКергические клетки, так и тормозные интернейроны.
ВЫЗВАННЫЕ ДЕФИЦИТОМ ДОФАМИНА ИЗМЕНЕНИЯ МЕЖНЕЙРОННЫХ СВЯЗЕЙ В СЕТИ, УЧАСТВУЮЩЕЙ В ПОЯВЛЕНИИ ТРЕМОРА ПОКОЯ ПРИ БОЛЕЗНИ ПАРКИНСОНА
В норме в нейронной сети происходит постоянное изменение соотношений между возбуждением и торможением, однако даже небольшой сдвиг в этих соотношениях может привести к чрезмерной синхронизации нейронных разрядов [69]. Очевидно, что БГ должны играть важную роль в таких изменениях, поскольку под действием дофамина или в его отсутствие меняется эффективность тормозного действия со стороны ГАМКергических клеток выходных ядер БГ – ЧВр и БШв (аналогом которого у грызунов является ЭПЯ) на их клетки-мишени в таламусе, СТЯ, ППЯ и ЯМ. Эти ГАМКергические клетки, в свою очередь, находятся под влиянием торможения со стороны С-Н клеток, тогда как С-П клетки контролируют активность нейронов БШн, мишенями которого являются СТЯ, стриатум и новая кора (рис. 1). Активность С-Н и С-П шипиковых клеток существенно зависит от дофамина, так как плотность дофаминовых рецепторов в стриатуме самая высокая в мозге. С учетом данных о том, что на С-Н клетках преимущественно располагаются Д1 рецепторы, а на С-П – Д2 рецепторы, из предложенного нами ранее механизма влияния дофамина на функционирование нейронной сети, включающей кору, БГ и таламус, следует, что поскольку активация Д1 рецепторов на С-Н клетках способствует индукции ДП на их возбудительных входах, при выделении дофамина в стриатуме активность С-Н нейронов должна возрасти, что приведет к усилению торможения их клеток-мишеней в ЧВр и БШв/ЭПЯ [29]. Одновременная активации Д2 рецепторов на С-П клетках, способствующая индукции длительной депрессии (ДД) на их возбудительных входах, должна привести к уменьшению активности С-П клеток и ослаблению их ингибирующего действия на нейроны БШн. В результате усилится тормозное действие со стороны нейронов БШн на их клетки-мишени в БШв/ЭПЯ, ЧВр, а также в СТЯ и новой коре (рис. 1). Последующее снижение активности нейронов СТЯ должно приводить к уменьшению возбуждения нейронов БШв, ЧВр, а также ППЯ и ГЯМ.
В согласии с этими следствиями предложенного механизма показано, что агонист Д2-рецепторов уменьшает выброс ГАМК в БШ (аналог БШн у грызунов) [70], а воздействие на Д1 рецепторы способствовало индукции ДД на входе СТЯ-ЧВр [71], т.е. снижало эффективность возбуждения нейронов ЧВр со стороны СТЯ. Этот эффект мог быть вызван воздействием на Д1 рецепторы на аксонных терминалях С-Н нейронов в ЧВр и увеличением выброса ГАМК.
При наличии тремора покоя у пациентов с БП потеря дофаминергических клеток наибольшая в медиальной части ЧВк (поле А9) и в ретро-рубральной области (поле А8), нейроны которых проецируются в матрикс дорсолатерального стриатума [9]. Поэтому, при БП на С-Н клетках не будет индуцироваться ДП, а на С-П – ДД [29]. Следовательно, активность нейронов БШв/ЧВр должна увеличиться в результате ослабления их ингибирования со стороны С-Н клеток, тогда как активность нейронов БШн должна снизиться, поскольку увеличится их ингибирование со стороны С-П клеток.
Именно такой характер изменений наблюдался в экспериментах в условиях дефицитa дофамина или при использовании антагонистов дофаминовых рецепторов. Так, антагонист Д1-рецепторов уменьшал выброс ГАМК в ЭПЯ и ЧВр [72, 73], что должно способствовать увеличению активности нейронов в этих ядрах. У крыс с полным или почти полным повреждением ЧВк частота разрядов нейронов СТЯ значительно увеличивалась и в их активности появлялось больше пачечных разрядов, чем в норме [74]. В работе [75] также показано, что в условиях дефицита дофамина меняются тормозный вход из БШн в СТЯ и характер активности нейронов СТЯ. То, что частота срабатывания и паттерн активности нейронов СТЯ определяется возбуждением со стороны новой коры и торможением со стороны БШн, отмечено и в работе [76]. Тормозное влияние настолько сильное, что ингибирование нейронов БШн/БШ приводило к увеличению активности нейронов СТЯ на 358% и последующему значительному возбуждению нейронов ЧВр и БШв/ЭПЯ [77]. При повреждении дофаминергических клеток ЧВк увеличивается активность нейронов как в СТЯ, так и в ППЯ, ЧВр и БШв [46, 78]. Полагают, что этот эффект может быть вызван уменьшением тормозного влияния на нейроны СТЯ со стороны БШн [79]. Повреждение СТЯ ослабляло увеличение активности ППЯ и ЧВр, а повреждение ППЯ ослабляло увеличение активности СТЯ и ЧВр [46].
Появление “треморной” активности в БШв связывают с дегенерацией при БП дофаминергических клеток ретро-рубральной области, иннервирующих нейроны БШв [8]. На важную роль этого входа указывает тот факт, что подавление нигро-паллидарного дофаминергического входа (т.е. входа из ЧВк) коррелирует с выраженностью тремора [3]. По мере прогрессирования БП тремор возрастает по амплитуде [80], т.е. выраженность тремора зависит от снижения концентрации дофамина. Дофамин может влиять на функционирование нейронов бледного шара за счет активации Д1 рецепторов на аксонных терминалях С-Н нейронов в ЧВр и БШв/ЭПЯ, а также Д2 рецепторов на аксонных терминалях С-П нейронов в БШн/БШ [81]. Активация Д1 рецепторов должна приводить к увеличению выброса ГАМК, а активация Д2 рецепторов – к уменьшению выброса ГАМК, ослаблению торможения и увеличению активности нейронов БШ/БШн. Действительно показано, что введение в БШ агониста (антагониста) Д2-рецепторов приводит к увеличению (уменьшению) частоты срабатывания нейронов БШ [82].
Как уже указывалось, в БШв и ЧВр имеются и прямые возбудительные входы из новой коры и из ГЯМ [31]. Поскольку в условиях дефицита дофамина С-Н клетки слабее ингибируют нейроны БШв и ЧВр, наличие прямых входов из коры и ГЯМ в дополнение к возбудительному входу из СТЯ может только усилить активность нейронов БШв и ЧВр и увеличить ингибирование их клеток-мишеней в таламусе, СТЯ и ППЯ. Однако экспериментально наблюдалось увеличение частоты разрядов СТЯ и ППЯ [13]. По-видимому, эффект растормаживания СТЯ вследствие ослабления его торможения со стороны БШн превалирует над усилением его торможения со стороны БШн и ЧВр, а усиление возбуждения нейронов ППЯ со стороны СТЯ преобладает над усилением их ингибирования со стороны БШв и ЧВр. Это должно привести к увеличению активности нейронов ГЯМ и таламических ядер (в частности, ВЛ и ПФЯ) и связанных с ними нейронов новой коры. Поскольку при повреждении нейронов ЧВк активность ПФЯ таламуса возрастает [83] и поскольку повреждение ППЯ уменьшало этот эффект, можно полагать, что возбуждение ПФЯ сильно зависит от активности ППЯ. При этом надо учитывать то обстоятельство, что при БП нарушается и холинергический вход из ППЯ в таламус [84].
Естественно предположить, что усиление возбуждения нейронов СТЯ и ППЯ, которое приведет к увеличению возбуждения нейронов ЯМ и ГЯМ, должно усилить связи между СТЯ, ППЯ и мозжечком. Действительно, у пациентов с БП и тремором покоя СТЯ было сильнее связано с мозжечком, чем в норме или в отсутствие тремора, а сила этой связи положительно коррелировала с выраженностью тремора [85]. Как указывалось в наших предшествующих работах [18, 86], активность нейронов ГЯМ, возбуждающих таламус, увеличивается при наличии торможения со стороны клеток Пуркинье (КП). В связи с этим можно ожидать, что КП должны участвовать в появлении тремора. Действительно, на свободно подвижных мышах показано, что возникновение тремора коррелирует с увеличением ритмической активности КП, которая меняет активность нейронов ГЯМ [87].
Показано, что при треморе покоя усиливается активность в мозжечково-таламической цепи и что активность в этой цепи коррелирует с амплитудой тремора [3]. Эти данные указывают на то, что возбуждение нейронов таламических ядер со стороны ГЯМ превалирует над усилением их торможения со стороны БШв и ЧВр. Поскольку амплитуда осцилляций в сети связана с возбуждением, а частота зависит от эффективности торможения [68], можно ожидать, что степень возбуждения нейронов СТЯ и ППЯ будет играть существенную роль в выраженности тремора. Не исключено, что частота тремора зависит от активности в цепи, связывающей СТЯ, БШв и БШн. Это предположение базируется на данных о том, что у пациентов с БП только при треморе покоя в автокоррелограммах нейронов СТЯ появляется начальная пауза и в их наблюдаются осцилляции с частотой 4–6 Гц [12]. Наличие такой паузы указывает на вклад сильного возвратного торможения, которое должно гиперполяризовать мембрану. Если мембранный потенциал становится гиперполяризованным, активность нейронов СТЯ может переключаться с обычной спайковой на пачечную [27]. Цепь БШн–СТЯ не может обеспечить такой паузы, так как в условиях дефицита дофамина торможение нейронов СТЯ со стороны БШн слабее, чем в норме. Поскольку торможение БШв со стороны БШн также слабее, а возбуждение со стороны СТЯ сильнее, активность нейронов БШв должна быть большой, что усилит их тормозящее действие на нейроны СТЯ. Имеется, однако, и точка зрения, что цепь СТЯ–БШн представляет собой пейсмекер, активность в котором модулируется ингибированием нейронов БШн со стороны стриатума, причем при патологии в этой цепи усиливается синхронизированная осцилляторная активность [88].
Кроме того, дофамин может непосредственно влиять на активность нейронов таламуса и СТЯ, поскольку имеются полученные на приматах и человеке данные, что в таламус, включая заднюю часть вентролатерального ядра (ВЛп) и ПФЯ поступают дофаминергические афференты из ретро-рубральной области [39, 89–91], а в СТЯ оканчиваются дофаминергические афференты из ЧВк [92]. Не исключено, что пачечные разряды с частотой 4–6 Гц, возникающие при треморе в активности нейронов СТЯ [12], передаются их клеткам-мишеням в ППЯ и ГМЯ, а от них в таламус и новую кору. Показано, что уровень ГАМК в моторной коре отрицательно коррелирует с выраженностью тремора при БП, а истощение ГАМК может увеличить выраженность моторных симптомов при БП [93]. Как уже указывалось, одним из источников ГАМК в коре является вход из БШн. Поскольку при дефиците дофамина активность ГАМКергических клеток в БШн уменьшена, по сравнению с нормой, это может являться одной из причин повышения активности нейронов моторной коры и увеличения выраженности тремора.
ГИПОТЕТИЧЕСКИЙ МЕХАНИЗМ ВОЗНИКНОВЕНИЯ ТРЕМОРА ПОКОЯ ПРИ БОЛЕЗНИ ПАРКИНСОНА
С учетом вышеизложенного, нами выдвигается следующий гипотетический механизм возникновения тремора покоя при БП. В его основе лежат вызванные дефицитом дофамина изменения эффективности возбудительных входов к ГАМКергическим шипиковым клеткам стриатума, лающим начало прямому и непрямому пути через БГ. Они приводят к снижению активности нейронов БШн, ослаблению его ингибирующего действия на нейроны СТЯ, последующему увеличению активности нейронов СТЯ и усилению возбуждения их клеток-мишеней в ППЯ и ГЯМ. В результате должна увеличиться активность нейронов таламуса, новой коры и КЯ, которые участвуют в проявлениях тремора. Уменьшение тормозного действия со стороны БШн на моторную кору также может увеличить ее активность. От степени возбуждения нейронов СТЯ, ППЯ и новой коры зависит амплитуда тремора. Частота тремора зависит от вызванных дефицитом дофамина изменений эффективности тормозных влияний в цепи, связывающей СТЯ, БШв и БШн, в результате которых в активности нейронов СТЯ появляются пачечные разряды с частотой 4–6 Гц.
Известно, что для цепей, включающих участки стриатума, СТЯ, БШ и ЧВр приматов характерна сомато-топическая организация. Она проявляется в том, что проекции из моторной коры, отображающие различные части тела, проецируются в различные области указанных ядер [92, 94]. При этом нейроны БГ отвечают не только на стимуляцию определенного участка моторной коры, но и на активные и пассивные движения этой части тела. По-видимому, вследствие этого, тремор затрагивает только одну часть тела, которая определяется высокой активностью определенной области коры. Отмечено, что в разных частях тела частота тремора может быть одинаковой, но может и отличаться, а также не быть синхронизированной по фазе. Эти данные указывают на то, что в нейронных цепях, относящихся к функционированию разных частей тела, создаются разные условия для генерации тремора [20].
ЭКСПЕРИМЕНТАЛЬНЫЕ СВИДЕТЕЛЬСТВА В ПОЛЬЗУ ВЫДВИГАЕМОЙ ГИПОТЕЗЫ ВОЗНИКНОВЕНИЯ ТРЕМОРА ПОКОЯ ПРИ БОЛЕЗНИ ПАРКИНСОНА
Если предлагаемый гипотетический механизм тремора покоя при БП верен, то введение в стриатум избирательного агониста Д1 рецепторов должно препятствовать возникновению тремора за счет усиления ингибирования нейронов БШв; а введение в стриатум избирательного агониста Д2 рецепторов должно подавить тремор или уменьшить его амплитуду за счет облегчения индукции ДД на С-П клетках, ослабления ингибирования нейронов БШн и увеличения их активности. С учетом того, что антагонисты А2А рецепторов должны приводить к такому же эффекту, что и агонисты Д2 рецепторов, т.е. облегчать индукцию ДД на С-П клетках [29], их использование также должно уменьшать связанную с тремором активность в разных структурах и подавлять тремор покоя. В пользу этого предсказания предложенной гипотезы свидетельствуют данные о том, что для уменьшения тремора покоя, вызванного дефицитом дофамина, использовали антагонисты А2А рецепторов. В частности, антагонисты А2А рецепторов подавляли тремор лапы, вызванный антагонистом Д2 рецепторов галоперидолом [95, 96]. Использование небольших доз леводопы в комбинации с антагонистом А2А рецепторов KW-6002 ослабляло тремор покоя [97]. Использование KW-6002 полезно еще и потому, что это вещество не вызывает дискинезии, в отличие от больших доз дофаминергических препаратов [98]. У некоторых пациентов с БП дофамин уменьшал связанную с тремором активность в таких структурах, как ВЛп и бледный шар, причем уменьшение активности нейронов ВЛп под действием дофамина коррелировало с клинической выраженностью подавления тремора [99]. Важно отметить, что этот эффект специфически относился к тремору и не был связан с такими симптомами БП, как дискинезия и ригидность [99].
Хотя леводопа может уменьшить вероятность возникновения тремора покоя при БП и его амплитуду [100], в некоторых случаях этот вид тремора был резистентен к дофаминергическим препаратам. Нельзя исключить, что в таких случаях дозы препаратов были недостаточны для воздействия на низко чувствительные Д2 рецепторы. С нашей точки зрения, использование леводопы или неизбирательных антагонистов аденозиновых рецепторов вообще нежелательно, так как активация дофаминовых Д1 рецепторов или инактивация аденозиновых А1 рецепторов должны способствовать индукции ДП на возбудительных входах к нейронам коры, таламуса и СТЯ, что приведет к увеличению их возбуждения [101] и будет препятствовать подавлению тремора.
Если, согласно предлагаемому механизму, СТЯ играет критическую роль в появлении тремора покоя, естественно ожидать, что подавление активности нейронов СТЯ будет препятствовать тремору. Действительно, для лечения тремора при БП используется высокочастотная стимуляция (ВЧС) СТЯ, приводящая к подавлению активности нейронов в этом ядре [102]. Существует точка зрения, что подавление тремора с помощью ВЧС разных структур связано не с возбуждением или ингибированием нейронов стимулируемой структуры, а с прерыванием аномального прохождение информации через эту структуру [103]. Это предположение представляется верным в том смысле, что подавление активности нейронов в одном из звеньев сети может прервать распространение аномальной активности во всей сети. Известные экспериментальные данные указывают именно на то, что тремор можно ослабить или устранить подавлением активности в разных звеньях сети, участвующей в появление тремора. Для подавления тремора, резистентного к дофаминергическим препаратам, используют ВЧС ПФЯ таламуса [104] или ВПЯ таламуса, которое связывает зубчатое ядро мозжечка с первичной моторной областью коры [105]. У пациентов с БП в результате ВЧС ВПЯ тремор уменьшался на 85–95% [106]. Примечательно, что совместная ВЧС СТЯ и ВПЯ таламуса значительно сильнее подавляла тремор, чем стимуляция каждого из ядер в отдельности [107]. Проявления тремора ослабляла и ВЧС ПФЯ таламуса [108]. В частности, она уменьшала тремор рук [109]. Хотя эффект от ВЧС СТЯ был больше, чем от ВЧС ПФЯ, стимуляция последнего не вызывала дискинезии, в отличие от ВЧС СТЯ [108]. Тот факт, что ВЧС СТЯ была эффективнее для подавления тремора, чем ВЧС ПФЯ, указывает на существенный вклад именно СТЯ в появлении тремора.
Как уже указывалось, для сети, участвующей в появлении тремора, характерна сомато-топическая организация. Например, в моторной коре и мозжечке выделены соматотопические области, связанные с тремором руки и головы. Лучшие результаты достигались при ВЧС областей, расположенных в задней субталамической области, заходящей на нижние границы ВПЯ [110]. Вследствие сомато-топической организации увеличение частоты срабатывания и появление пачечных разрядов должно быть характерно только для определенной группы нейронов СТЯ. С учетом данных об увеличении активности в определенной части моторной коры при треморе покоя, интересно было бы проверить в экспериментах на приматах, может ли фиксация на некоторое время той конечности, в которой наблюдается тремор, препятствовать его появлению. Не исключено, что в результате фиксации снизится активность определенной области коры, вследствие чего будут слабо активироваться соответствующие участки БГ, СТЯ и ЯМ и тремор прекратится.
ВОЗМОЖНЫЙ МЕХАНИЗМ УСИЛЕНИЯ ТРЕМОРА ПРИ СТРЕССЕ
Если дефицит дофамина лежит в основе возникновения тремора, то снижение активности дофаминергических клеток должно способствовать его появлению или усилению. Известно, что выраженность тремора даже у одного и того же пациента зависит от его функционального состояния [111]. У многих пациентов с БП тремор покоя существенно усиливался при стрессе [112], а противотреморный эффект леводопы был существенно меньше, чем в спокойном состоянии [113]. Показано, что стресс увеличивал выраженность и частоту тремора покоя [114]. Стресс должен влиять на активность дофаминергических клеток, поскольку выделяемый при стрессе кортизол (кортикостерон) воздействует на расположенные на этих клетках глюкокортикоидные и минералокортикоидные рецепторы. Кроме того, на моделях БП показано, что хронический стресс приводит к гибели дофаминергических клеток ЧВк [115, 116].
У минералокортикоидных рецепторов сродство с кортикостероном значительно больше, чем у глюкокортикоидных рецепторов [117, 118], поэтому влияние кортизола на изменение эффективности возбуждения дофаминергических клеток зависит от его концентрации. На культуре дофаминергической структуры крыс показано, что при низком содержании кортикостерона за счет активации минералокортикоидных рецепторов увеличиваются реакции дофаминергических клеток на деполяризующий стимул, тогда как вовлечение глюкокортикоидных рецепторов при больших уровнях кортикостерона препятствует этому эффекту [119. При сильном стрессе уровень кортизола может быть достаточно высоким для активации глюкокортикоидных рецепторов, что может привести к ДД возбуждения дофаминергических клеток и снижению выделения дофамина. Показано, что стресс может не только усугубить имеющиеся симптомы у пациентов с БП, но и вызвать тремор у здоровых субъектов [120, 121]. Эти данные дополнительно свидетельствуют в пользу важной роли дофаминергической системы в появлении тремора.
СОПОСТАВЛЕНИЕ ВЫДВИГАЕМОГО ГИПОТЕТИЧЕСКОГО МЕХАНИЗМА ВОЗНИКНОВЕНИЯ ТРЕМОРА ПОКОЯ ПРИ БОЛЕЗНИ ПАРКИНСОНА С ИЗВЕСТНЫМИ ГИПОТЕЗАМИ
Ряд положений предлагаемого гипотетического механизма возникновения тремора покоя при БП отличается от гипотез, выдвинутых другими исследователями. Например, авторы работы [122] предположили, что тремор покоя при БП генерируется в нейронной сети, включающей СТЯ и ВЛ. Однако современные исследования не выявили проекций из СТЯ в ВЛ (см., например [123]). Выдвинуто предположение, что в основе тремора, вызванного дефицитом дофамина, лежат такие изменения активности в цепи, включающей кору, таламус, БГ и СТЯ, которые приводят к усилению ингибирования нейронов таламуса со стороны БШв и ЧВр [124]. В таком случае не ясно, как треморная активность передается в новую кору. Тот же аргумент относится к механизму, обсуждаемому в работе [124], авторы которой полагают, что к тремору при БП приводит осцилляторная активность, возникающая в цепи СТЯ–БШн, но вход из ППЯ в БШв подавляет передачу такой активности в кору.
В предлагаемом нами гипотетическом механизме передача “треморной” активности осуществляется благодаря возрастанию активности нейронов ППЯ и ГЯМ, вызванному увеличением активности СТЯ и последующему .усилению возбуждения нейронов таламуса со стороны ППЯ и ГЯМ. Такой механизм позволяет объяснить экспериментально наблюдаемое увеличение активности в цепи М–Т–К при треморе покоя. Хотя в работах [2, 3] отмечено, что СТЯ может влиять на функционирование мозжечка, но не указан механизм этого влияния; не учтено участие ППЯ, не упоминается то обстоятельство, что новая кора моносинаптически возбуждает СТЯ через гиперпрямой путь в БГ. В модели “переключатель диммер”, которая базируется на нейронной сети, состоящей из коры, БГ, таламуса и мозжечка, предполагается, что при БП потеря дофаминергических клеток приводит к нарушению функционирования бледного шара, а через него ВЛп [2, 3, 5, 125, 126]. Было предположено также, что треморная активность возникающая в БШв, затем распространяется в цепь М–Т–К, в которой ритм тремора возрастает и его амплитуда увеличивается, причем на цепь М–Т–К влияет кора, а не мозжечок [127]. При этом опираются на полученные на здоровых испытуемых данные о том, что цепь М–Т–К имеет тенденцию к генерации осцилляторного ритма с частотой 6–9 Гц [127]. Однако эта частота выше частоты тремора покоя. Кроме того, из этой модели также не ясно, почему ГАМКергические клетки БШв способствуют увеличению активности таламуса и новой коры.
Авторы работы [88] предполагают, что к появлению осцилляций в цепи СТЯ–БШн может привести вызванное дефицитом дофамина изменение активности нейронов стриатума, проецирующихся на нейроны БШн. Эти осцилляции с частотой 0.4–1.8 Гц наблюдались в экспериментах in vitro. Однако не известно, как эти осцилляции могут быть связаны с частотой тремора и наблюдаются ли они in vivo. Существует мнение, что тремор покоя появляется, когда высокочастотные (12–15 Гц) осцилляции в БГ трансформируются в осцилляции с частотой (4–6 Гц) в таламическом ядре ВЛ [128]. Трансформация происходит благодаря таламо-паллидарным взаимодействиям, которые определяют частоту тремора во всей сети [128]. Эта предположение базируется на данных, полученных в экспериментах in vitro. Кроме того, частоту осцилляций в БШ с частотой 5 Гц наблюдали и при наличии тремора, и в его отсутствие [129]. Отсутствие специфичности трансформации частоты осцилляций не позволяет рассматривать этот механизм как возможно лежащий в основе тремора покоя.
В работе [130] предложена модель “палец-переключатель диммер”, где тремор покоя при БП запускается патологической активностью БГ (которые играют роль пальца), продолжает генерироваться за счет изменений активности нейронов таламуса (которые играют роль переключателя) и модулируется активностью мозжечка (играет роль диммера). В этой модели ключевой структурой является таламус, в активности нейронов которого могут возникать осцилляции с частотой тремора, если их мембрана гиперполяризована или уменьшен возбудительный драйв [131]. Гиперполяризация мембраны может быть связана с увеличением активности нейронов БШв. Однако, как уже указывалось, уменьшение возбудительного драйва противоречит экспериментальным данным об увеличении активности таламических клеток при треморе. Согласно этой модели, треморная активность в БГ является эфферентной копией корковой или таламической активности [130]. В качестве доказательства приведен тот факт, что повреждение задней части ВЛ или вентрального промежуточного ядра таламуса препятствует тремору [130]. Однако известно, что повреждение и других структур в цепи, в которой распространяется треморная активность, препятствует тремору.
Согласно точке зрения, изложенной в работе [23], влияние дофамина на тремор определяется его воздействием на таламус, а не на мозжечок, на который дофамин не влияет. В нашей модели учтено то обстоятельство, что плотность дофаминовых рецепторов наибольшая в стриатуме, а не в таламусе, поэтому вызванные дефицитом дофамина изменения функционирования должны быть сильнее в БГ, а затем происходить реорганизация активности в структурах, на которые проецируются нейроны БГ. Кроме того, как указывалось выше, на нейронах мозжечка имеются дофаминовые рецепторы, так что дефицит дофамина должен приводить к перестройке активности и в мозжечке.
Предлагаемый нами гипотетический механизм имеет частичное сходство с работами [20, 132], в которых отмечено, что триггером тремора являются БГ, а сеть мозжечка ответственна за его амплитуду, тогда как таламус не является генератором тремора. Кроме того, имеется частичное сходство с механизмом, предложенным в работе [6]. Автор указанной работы опирается на данные о том, что дефицит дофамина в стриатуме приводит к увеличению активности нейронов БШв/ЧВр и снижению активности нейронов БШн. Отмечено, что растормаживание нейронов СТЯ со стороны БШн приводит к увеличению активности нейронов БШв/ЧВр. Отмечена также роль нейронной сети мозжечка, которая через ГЯМ влияет на активность таламуса [6]. Однако автор указанной работы полагает, что механизм участия цепи М–Т–К в появлении тремора покоя остается неизвестным, так как не ясно, каким образом БГ влияют на активность нейронов ГЯМ и как ГЯМ влияет на активность БГ. Возможные механизмы этих влияний предложены в настоящей работе. Также дано объяснение экспериментально наблюдаемого увеличения коррелирующей с тремором активности в мозжечке, таламусе и коре.
ЗАКЛЮЧЕНИЕ
Предложенный гипотетический механизм появления тремора покоя при болезни Паркинсона позволяет объяснить ряд известных экспериментальных данных и лишен ряда неопределенностей, присущих другим гипотезам. Этот механизм базируется на проведенном нами ранее анализе влияния дофамина на функционирование нейронной сети, включающей кору, БГ, таламус, мозжечок, СТЯ и ППЯ. Согласно выдвигаемой гипотезе, в основе появления тремора покоя лежит вызванная дефицитом дофамина модификация эффективности кортико-стриатных и таламо-стриатных входов, ведущая к изменениям функционирования нейронов стриатума и их клеток-мишеней в БШв и БШн. Изменения проявляются в снижении активности нейронов БШн, уменьшении его ингибирующего действия на нейроны новой коры, СТЯ и БШв. В результате увеличивается активность нейронов БШв, новой коры, СТЯ и клеток-мишеней СТЯ в ППЯ, ЯМ и ГЯМ. Последующее усиление возбуждающего действия мозжечка на нейроны таламуса передается в новую кору и стриатум. Согласно предлагаемому механизму, степень возбуждения нейронов субталамического ядра определяет амплитуду тремора. Наблюдающееся при треморе покоя возрастание активности в мозжечково-таламо-кортикальной цепи указывает на то, что коррелирующее с амплитудой тремора увеличение возбуждающего действия нейронов ГЯМ на таламические ядра превалирует над усилением их торможения со стороны БШв. Предположено, что появление в сети пачечной активности с частотой тремора является следствием гиперполяризации мембраны нейронов СТЯ, вызванной усилением торможения со стороны БШв. Основное отличие предлагаемого гипотетического механизма появления тремора покоя при БП от известных к настоящему времени гипотез состоит в предположении, что в появлении пачечной активности с частотой тремора покоя участвует нейронная цепь, включающая СТЯ, БШн и БШв. Кроме того, увеличение активности нейронов в цепи мозжечок–таламус–кора объяснено растормаживанием активности нейронов субталамического ядра. Из предлагаемого механизма следует, что тремор покоя можно подавить или уменьшить его амплитуду не только с помощью ВЧС подкорковых структур, прерывающей “треморную” активность, но и с помощью избирательных агонистов дофаминовых Д2 рецепторов и/или антагонистов аденозиновых А2А рецепторов. Эти вещества должны способствовать индукции ДД на стриопаллидарных клетках, последующему снижению ингибирования нейронов БШн, увеличению их активности и усилению ингибирования нейронов СТЯ и БШв. В литературе имеются экспериментальные свидетельства того, что в ряде случаев тремор удавалось подавить с помощью предлагаемых веществ.
Список литературы
Louis E.D. // Continuum (Minneap. Minn.). 2019. V. 25. № 4. P. 959–975.
Helmich R.C. // Mov. Disord. 2018. V. 33. № 2. P. 219–231.
Helmich R.C., Janssen M.J., Oyen W.J., Bloem B.R., Toni I. // Ann. Neurol. 2011. V. 69. № 2. P. 269–281.
Helmich R.C., Toni I., Deuschl G., Bloem B.R. // Curr. Neurol. Neurosci. Rep. 2013. V. 13. № 9. P. 378.
Dirkx M.F., den Ouden H., Aarts E, Timmer M., Bloem B.R., Toni I., Helmich R.C. // J. Neurosci. 2016. V. 36. № 19. P. 5362–5372.
Wichmann T. // Mov. Disord. 2019. V. 34. № 8. P. 1130–1143.
Wallace M.L., Saunders A., Huang K.W., Philson A.C., Goldman M., Macosko E.Z., McCarroll S.A., Sabatini B.L. // Neuron. 2017. V. 94. № 1. P. 138–152.
Hirsch E.C., Mouatt A., Faucheux B., Bonnet A.M., Javoy-Agid F., Graybiel A.M., Agid Y. // Lancet. 1992. V. 340: P. 125–126.
Jellinger K.A. // J. Neural Transm. Suppl. 2002. V. 62. P. 347–376.
McGregor M.M., Nelson A.B. // Neuron. 2019. V. 101. № 6. P. 1042–1056.
Chen H., Zhuang P., Zhang Y.Q., Li J.Y., Li Y.J. // Chin. Med. J. (Engl.). 2009. V. 122. № 19. P. 2308–2314.
Lintas A., Silkis I.G., Albéri L., Villa A.E.P. // Brain Res. 2012. V. 1434. P. 142–151.
Liu H., Zhang J., Gao D.M. // Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2009. V. 25. № 2. P. 212–216.
Magnin M., Morel A., Jeanmonod D. // Neuroscience. 2000. V. 96. № 3. P. 549–564.
Weinberger M., Hamani C., Hutchison W.D., Moro E., Lozano A.M., Dostrovsky J.O. // Exp. Brain Res. 2008. V. 188. № 2. P. 165–174.
Zhang J.R., Feng T., Hou Y.N., Chan P., Wu T. // CNS Neurosci. Ther. 2016. V. 22. № 5. P. 378–386.
Singh A., Liang L., Kaneoke Y., Cao X., Papa S.M. // J. Neurophysiol. 2015. V. 113. № 5. P. 1533–1544.
Силькис И.Г. // Успехи физиол. наук. 2021. Т. 52. № 1. С. 1–14.
Zhang J., Wei L., Hu X., Xie B., Zhang Y., Wu G.R., Wang J. // Parkinsonism Relat. Disord. 2015. V. 21. № 1. P. 23–30.
Hallett M. // Parkinsonism Relat. Disord. 2012. V. 18. Suppl. 1. P. S85–S86.
Prodoehl J., Planetta P.J., Kurani A.S., Comella C.L., Corcos D.M., Vaillancourt D.E. // JAMA Neurol. 2013. V. 70. № 1. P. 100–106.
Shen B., Pan Y., Jiang X., Wu Z., Zhu J., Dong J., Zhang W., Xu P., Dai Y., Gao Y., Xiao C., Zhang L. // CNS Neurosci, Ther. 2020. V. 26. № 2. P. 207–214.
Madelein van der Stouwe A.M., Nieuwhof F., Helmich R.C. // Curr. Opin. Neurol. 2020. V. 33. № 4. P. 474–481.
Levy R., Hutchison W.D., Lozano A.M., Dostrovsky J.O. // J. Neurosci. 2002. V. 22. № 7. P. 2855–2861.
Bar-Gad I., Heimer G., Ritov Y., Bergman H. // J. Neurosci. 2003. V. 23. P. 4012–4016.
Bergman H., Raz A., Feingold A., Nini A., Nelken I., Hansel D., Ben-Pazi H., Reches A. // Mov. Disord. 1998. V. 13. Suppl. 3. P. 29–34.
Tai C.H., Kuo C.C. // Acta Neurol. Taiwan. 2006. V. 15. № 3. P. 206–216.
Baudrexel S., Witte T., Seifried C., von Wegner F., Beissner F., Klein J.C., Steinmetz H., Deichmann R., Roeper J., Hilker R. // Neuroimage. 2011. V. 55. № 4. P. 1728–1738.
Silkis I. // Biosystems. 2001. V. 59. № 1. P. 7–14.
Силькис И.Г. // Успехи. физиол. наук. 2005. Т. 36. № 2. С. 66–83.
Quartarone A., Cacciola A., Milardi D., Ghilardi M.F., Calamuneri A., Chillemi G., Anastasi G., Rothwell J. // Brain. 2020. V. 143. № 2. P. 396–406.
Glickstein M. // Prog. Brain Res. 1997. V. 114. P. 251–259.
Guell X., D’Mello A.M., Hubbard N.A., Romeo R.R., Gabrieli J.D.E., Whitfield-Gabrieli S., Schmahmann J.D., Anteraper S.A. // Cereb. Cortex. 2020. V. 30. № 4. P. 2401–2417.
Shinoda Y., Sugihara I., Wu H.S., Sugiuchi Y. // Prog. Brain Res. 2000. V. 124. P. 173–186.
Bostan A.C., Strick P.L. // Nat. Rev. Neurosci. 2018. V. 19. № 6. P. 338–350.
Chen C.H., Fremont R., Arteaga-Bracho E.E., Khodakhah K. // Nat. Neurosci. 2014. V. 17. № 12. P. 1767–1775.
Hoshi E., Tremblay L., Feger J., Carras P.L., Strick P.L. // Nat. Neurosci. 2005. V. 8. P. 1491–1493.
Milardi D., Quartarone A., Bramanti A., Anastasi G., Bertino S., Basile G.A., Buonasera P., Pilone G., Celeste G., Rizzo G., Bruschetta D., Cacciola A. // Front. Syst. Neurosci. 2019. V. 13. P. 61.
Royce G.J., Bromley S., Gracco C. // J. Comp. Neurol. 1991. V. 306. № 1. P. 129–155.
Mouroux M., Hassani O.K., Féger J. // Neuroscience. 1997. V. 81. № 2. P. 387–397.
Mandelbaum G., Taranda J., Haynes T.M., Hochbaum D.R., Huang K.W., Hyun M., Umadevi Venkataraju K., Straub C., Wang W., Robertson K., Osten P., Sabatini B.L. // Neuron. 2019. V. 102. № 3. P. 636–652.e7.
Lanciego J.L., Gonzalo N., Castle M., Sanchez-Escobar C., Aymerich M.S., Obeso J.A. // Eur. J. Neurosci. 2004. V. 19. № 5. P. 1267–1277.
Orieux G., Francois C., Féger J., Yelnik J., Vila M., Ruberg M., Agid Y., Hirsch E.C. // Neuroscience. 2000. V. 97. № 1. P. 79–88.
Smith Y., Bevan M.D., Shink E., Bolam J.P. // Neuroscience. 1998. V. 86. № 2. P. 353–387.
Sato F., Parent M., Levesque M., Parent A. // J. Comp. Neurol. 2000. V. 424. № 1. P. 142–152.
Breit S., Lessmann L., Unterbrink D., Popa R.C., Gasser T., Schulz J.B. // Eur. J. Neurosci. 2006. V. 24. № 8. P. 2275–2282.
Takakusaki K., Shiroyama T., Kitai S.T. // Neuroscience. 1997. V.79. № 4. P. 1089–1109.
Florio T., Scarnati E., Confalone G., Minchella D., Galati S., Stanzione P., Stefani A., Mazzone P. // Eur. J. Neurosci. 2007. V. 25. № 4. P. 1174–1186.
Steininger T.L., Rye D.B., Wainer B.H. // J. Comp. Neurol. 1992. V. 321. № 4. P. 515–543.
Benarroch E.E. // Neurology. 2013. V. 80. № 12. P. 1148–1155.
Saper C.B., Loewy A.D. // Brain Res. 1982. V. 252. P. 367–372.
Erro E., Lanciego J.L., Giménez-Amaya J.M. // Exp. Brain Res. 1999. V. 127. № 2. P. 162–170.
Melzer S., Gil M., Koser D.E., Michael M., Huang K.W., Monyer H. // Cell Rep. 2017. V. 19. № 5. P. 1045–1055.
Rock C., Zurita H., Wilson C., Apicella A.J. // Elife. 2016. V. 5. pii: e15890.
Wall N.R., De La Parra M., Callaway E.M., Kreitzer A.C. // Neuron. 2013. V. 79. № 2. P. 347–360.
Nambu A., Tokuno H., Hamada I., Kita H., Imanishi M., Akazawa T., Ikeuch Y., Hasegawa N. // J. Neurophysiol. 2000. V. 84. № 1. P. 289–300.
Romanelli P., Bronte–Stewart H., Heit G., Schaal D.W., Esposito V. // Stereotact. Funct. Neurosurg. 2004. V. 82. № 5–6. P. 222–229.
Lee M.S., Rinne J.O., Marsden C.D. // Yonsei. Med. J. 2000 V. 41. № 2. P. 167–184.
Jaarsma D., Ruigrok T.J., Caffé R., Cozzari C., Levey A.I., Mugnaini E., Voogd J. // Prog. Brain Res. 1997. V. 114. P. 67–96.
Vitale F., Mattei C., Capozzo A., Pietrantoni I., Mazzone P., Scarnati E. // Neuroscience. 2016. V. 317. P. 12–22.
Rinaldo L., Hansel C. // Proc. Natl. Acad. Sci. USA. 2013. V. 110. P. 11181–11186.
Bevan M.D., Booth P.A.C., Eaton S.A., Bolam J.P. // J. Neuroscience. 1998. V. 18. № 22. P. 9438–9452.
Smith A.D., Bolam J.P. // Trends Neurosci. 1990. V. 13. № 7. P. 250–265.
Saunders A., Oldenburg I.A., Berezovskii V.K., Johnson C.A., Kingery N.D., Elliott H.L., Xie T., Gerfen C.R., Sabatini B.L. // Nature. 2015. V. 521. № 7550. P. 85–89.
Kress G.J., Yamawaki N., Wokosin D.L., Wickersham I.R., Shepherd G.M., Surmeier D.J. // Nature Neurosci. 2013. V. 16. P. 665–667.
Gao D.M., Benazzouz A., Piallat B., Bressand K., Ilinsky I.A., Kultas-Ilinsky K., Benabid A.L. // Neuroscience. 1999. V. 88. № 1. P. 201–212.
Pong M., Horn K.M., Gibson A.R. // Brain Res. Rev. 2008. V. 58. № 2. P. 249–264.
Whittington M.A., Traub R.D., Jefferys J.G.R. // Nature. 1995. V. 373. № 6515. P. 612–615.
Magloire V., Mercier M.S., Kullmann D.M., Pavlov I. // Neuroscientist. 2019. V. 25. № 4. P. 344–358.
Floran B., Floran L., Nandi Sierra A., Aceves J. // Neurosci. Lett. 1997. V. 237. № 1. P. 1–4.
Dupuis J.P., Feyder M., Miguelez C., Garcia L., Morin S., Choquet D., Hosy E., Bezard E., Fisone G., Bioulac B.H., Baufreton J. // J. Neurosci. 2013. V. 33. № 36. P. 14331–14341.
Aceves J., Floran B., Sierra A., Mariscal S. // Biol. Psychiatry. 1995. V. 19. № 5. P. 727–739.
Ferre S., O’Connor W.T., Svenningsson P., Bjorklund L., Lindberg J., Tinner B., Stromberg I., Goldstein M., Ogren S.O., Ungerstedt U., Fredholm B.B., Fuxe K. // Eur. J. Neurosci. 1996. V. 8. № 7. P. 1545–1553.
Breit S., Bouali-Benazzouz R., Popa R.C., Gasser T., Benabid A.L., Benazzouz A. // Exp. Neurol. 2007. V. 205. № 1. P. 36–47.
Chu H.Y., Atherton J.F., Wokosin D., Surmeier D.J., Bevan M.D. // Neuron. 2015. V. 85. № 2. P. 364–376.
Bevan M.D., Hallworth N.E., Baufreton J. // Prog. Brain Res. 2007. V. 160. P. 173–188.
Robledo P., Féger J. // Brain Res. 1990. V. 518. № 1–2. P. 47–54.
Bezard E., Boraud T., Bioulac B., Gross C.E. // Eur. J. Neurosci. 1999. V. 11. № 6. P. 2167–2170.
Blandini F., Nappi G., Tassorelli C., Martignoni E. // Prog. Neurobiol. 2000. V. 62. № 1. P. 63–88.
Hughes A.J., Daniel S.E., Blankson S., Lees A.J. // Arch. Neurol. 1993. V. 50. № 2. P. 140–148.
Smith Y., Kieval J.Z. // Trends Neurosci. 2000. V. 23. (10 Suppl.). P. S28–S33.
Querejeta E., Delgado A., Valdiosera R., Erlij D., Aceves J. // Neurosci. Lett. 2001. V. 300. № 2. P. 79–82.
Yan W., Zhang Q.J., Liu J., Wang T., Wang S., Liu X., Chen L., Gui Z.H. // Brain Res. 2008. V. 1240. P. 204–212.
Müller M.L., Albin R.L., Kotagal V., Koeppe R.A., Scott P.J., Frey K.A., Bohnen N.I. // Brain. 2013. V. 136. (Pt. 11). P. 3282–3289.
Wang Z., Chen H., Ma H., Ma L., Wu T., Feng T. // J. Neurol. Sci. 2016. V. 371. P. 137–147.
Silkis I. // Biosystems. 2000. V. 54. № 3. P. 141–149.
Brown A.M., White J.J., van der Heijden M.E., Zhou J., Lin T., Sillitoe R.V. // Elife. 2020. V. 9. e51928.
Plenz D., Kital S.T. // Nature. 1999. V. 400. № 6745. P. 677–682.
García-Cabezas M.A., Martínez-Sánchez P., Sánchez-González M.A., Garzón M., Cavada C. // Cereb. Cortex. 2009. V. 19. № 2. P. 424–434.
García-Cabezas M.A., Rico B., Sánchez-González M.A., Cavada C. // Neuroimage. 2007. V. 34. № 3. P. 965–984.
Sánchez-González M.A., García-Cabezas M.A., Rico B., Cavada C. // J. Neurosci. 2005. V. 25. № 26. P. 6076–6083.
Hamani C., Saint-Cyr J.A., Fraser J., Kaplitt M., Lozano A.M. // Brain. 2004. V. 127. Pt. 1. P. 4-20.
van Nuland A.J.M., den Ouden H.E.M., Zach H., Dirkx M.F.M., van Asten J.J.A., Scheenen T.W.J., Toni I., Cools R., Helmich R.C. // Hum. Brain Mapp. 2020. V. 41. № 4. P. 1017–1029.
Nambu A. // Front. Neuroanat. 2011. V. 5. P. 26.
Betz A.J., Vontell R., Valenta J., Worden L., Sink K.S., Font L., Correa M., Sager T.N., Salamone J.D. // Neuroscience. 2009. V. 163. № 1. P. 97–108.
Correa M., Wisniecki A., Betz A., Dobson D.R., O’Neill M.F., O’Neill M.J., Salamone J.D. // Behav. Brain Res. 2004. V. 148. № 1–2. P. 47–54.
Bara-Jimenez W., Sherzai A., Dimitrova T., Favit A., Bibbiani F., Gillespie M., Morris M.J., Mouradian M.M., Chase T.N. // Neurology. 2003. V. 61. № 3. P. 293–296.
Kanda T., Jackson M.J., Smith L.A., Pearce R.K., Nakamura J., Kase H., Kuwana Y., Jenner P. // Exp. Neurol. 2000. V. 162. № 2. P. 321–327.
Dirkx M.F., den Ouden H.E., Aarts E., Timmer M.H., Bloem B.R., Toni I., Helmich R.C. // Brain. 2017. V. 140. № 3. P. 721–734.
Zach H., Dirkx M., Pasman J.W., Bloem B.R., Helmich R.C. // Parkinsonism Relat. Disord. 2017. V. 35. P. 48–54.
Силькис И.Г. // Нейрохимия. 2014. Т. 31. № 4. С. 287–299.
Diamond A., Shahed J., Jankovic J. // J. Neurol. Sci. 2007. V. 260. № 1–2. P. 199–203.
Chiken S., Nambu A. // Neuroscientist. 2016. V. 22. № 3. P. 313–322.
Sharma V.D., Mewes K., Wichmann T., Buetefisch C., Willie J.T., DeLong M. // Acta Neurochir. (Wien). 2017. V. 159. № 5. P. 789–793.
Akram H., Dayal V., Mahlknecht P., Georgiev D., Hyam J., Foltynie T., Limousin P., De Vita E., Jahanshahi M., Ashburner J., Behrens T., Hariz M., Zrinzo L. // Neuroimage Clin. 2018. V. 18. P. 130–142.
Reinacher P.C., Amtage F., Rijntjes M., Piroth T., Prokop T., Jenkner C., Kätzler J., Coenen V.A. // JMIR Res. Protoc. 2018. V. 7. № 1. P. e36.
Neudorfer C., Hinzke M., Hunsche S., El Majdoub F., Lozano A., Maarouf M. // Neuromodulation. 2019. V. 22. № 4. P. 493–502.
Kerkerian-Le Goff L., Jouve L., Melon C., Salin P. // Parkinsonism Relat. Disord. 2009. V. 15. Suppl. 3. P. S167–S170.
Peppe A., Gasbarra A., Stefani A., Chiavalon C., Pierantozzi M., Fermi E., Stanzione P., Caltagirone C., Mazzone P. // Parkinsonism Relat. Disord. 2008. V. 14. № 6. P. 501–504.
Al-Fatly B., Ewert S., Kübler D., Kroneberg D., Horn A., Kühn A.A. // Brain. 2019. V. 142. № 10. P. 3086–3098.
Zach H., Dirkx M., Bloem B.R., Helmich R.C. // J. Parkinsons Dis. 2015. V. 5. № 3. P. 471–474.
Raethjen J., Austermann K., Witt K., Zeuner K.E., Papengut F., Deuschl G. // Mov. Disord. 2008. V. 23. № 7. P. 1019–1023.
Zach H., Dirkx M.F., Pasman J.W., Bloem B.R., Helmich R.C. // CNS Neurosci. Ther. 2017. V. 23. № 3. P. 209–215.
Lee H.J., Lee W.W., Kim S.K., Park H., Jeon H.S., Kim H.B., Jeon B.S., Park K.S. // J. Neurol. Sci. 2016. V. 362. P. 272–277.
de Pablos R.M., Herrera A.J., Espinosa-Oliva A.M., Sarmiento M., Muñoz M.F., Machado A., Venero J.L. // J. Neuroinflammation. 2014. V. 11. P. 34.
Vyas S., Rodrigues A.J., Silva J.M., Tronche F., Almeida O.F., Sousa N., Sotiropoulos I. // Neural Plast. 2016. V. 2016. P. 6391686.
Pavlides C., Watanabe Y., Magariños A.M., McEwen B.S. // Neuroscience. 1995. V. 68. № 2. P. 387–394.
Payne J.D., Nadel L. // Learn. Mem. 2004. V. 11. № 6. P. 671–678.
Ronken E., Mulder A.H., Schoffelmeer A.N. // Eur. J. Pharmacol. 1994. V. 263. № 1–2. P. 149–156.
Hao Y., Shabanpoor A., Metz G.A. // Neurosci. Lett. 2017. V. 651. P. 79–87.
Metz G.A. // Rev. Neurosci. 2007. V. 18. № 3–4. P. 209–222.
Cagnan H., Little S., Foltynie T., Limousin P., Zrinzo L., Hariz M., Cheeran B., Fitzgerald J., Green A.L., Aziz T., Brown P. // Brain. 2014. V. 137. Pt. 12. P. 3223–3234.
Cavdar S., Özgür M., Çakmak Y.Ö., Kuvvet Y., Kunt S.K., Sağlam G. // Acta Neurobiol. Exp. (Wars.) 2018. V. 78. № 3. P. 251–263.
Hadipour Niktarash A., Shahidi G.A. // J. Comput. Neurosci. 2004. V. 16. № 2. P. 113–127.
Helmich R.C., Hallett M., Deuschl G., Toni I., Bloem B.R. // Brain. 2012. V. 135. Pt. 11. P. 3206–3226.
Kamble N., Pal P.K. // Neurol. India. 2018. V. 66 (Supplement). P. S36–S47.
Gross J., Timmermann L., Kujala J., Dirks M., Schmitz F., Salmelin R., Schnitzler A. // Proc. Natl. Acad. Sci. USA. 2002. V. 99. № 4. P. 2299–2302.
Pare D., Curro’Dossi R., Steriade M. // Neuroscience. 1990. V. 35. P. 217–226.
Rivlin-Etzion M., Marmor O., Saban G., Rosin B., Haber S.N., Vaadia E., Prut Y., Bergman H. // J. Neurosci. 2008. V. 28. P. 633–649.
Duval C., Daneault J.F., Hutchison W.D., Sadikot A.F. // Neurobiol. Dis. 2016. V. 85. P. 49–59.
Llinas R., Urbano F.J., Leznik E., Ramirez R.R., van Marle H.J. // Trends Neurosci. 2005. V. 28. № 6. P. 325–333.
Hallett M. // Parkinsonism Relat. Disord. 2014. V. 20. Suppl. 1. P. S118–S122.
Дополнительные материалы отсутствуют.