

Нейрохимия, 2022, T. 39, № 2, стр. 131-143
Тормозный контроль кратковременной пластичности при парной стимуляции зависит от полимеризации актина
И. В. Кудряшова
Федеральное государственное бюджетное учреждение науки Институт высшей нервной деятельности
и нейрофизиологии Российской академии наук
Москва, Россия
Поступила в редакцию 29.10.2021
После доработки 31.10.2021
Принята к публикации 02.11.2021
- EDN: YFXYDV
- DOI: 10.31857/S1027813322020108
Аннотация
Исследование влияния джасплакинолида на кратковременную пластичность при парной стимуляции в переживающих срезах гиппокампа крыс выявило значимые различия при тестировании парными стимулами с межстимульными интервалами 15 и 70 мс. Это предполагает, что помимо общих механизмов, определяющих величину фасилитации при парной стимуляции (PPF), от полимеризации актина зависит степень участия дополнительных подавляющих PPF сигналов, включая суммацию с бисинаптическим ТПСП. Применение методов, контролирующих общие для разных межстимульных интервалов модификации, показало, что характер изменения специфичных для 15 мс интервала механизмов подавления PPF зависел от их исходных характеристик до аппликации джасплакинолида. Если в начале эксперимента эффект подавления PPF15 относительно PPF70 отсутствовал или был минимальным, джасплакинолид приводил к активации исходно слабого торможения. В противоположность этому, на более зависимые от тормозного компонента реакции джасплакинолид оказывал растормаживающее влияние. Предполагается, что дифференцированное влияние джасплакинолида на девиантные реакции содействует восстановлению нормального уровня активации и может иметь отношение к реполимеризации актина в ходе гомеостатической регуляции баланса возбуждения и торможения.
Кратковременная синаптическая пластичность вносит существенный вклад в формирование пространственно-временного паттерна активации, создавая условия для последовательного включения нейронов в реализацию приспособительного поведения [1]. При повторной активации индивидуальные синапсы настроены преимущественно на фасилитацию или депрессию, демонстрируя при этом большое разнообразие свойств кратковременной пластичности даже в одной и той же популяции синапсов, в частности оканчивающихся на пирамидных нейронах CA3-CA1 синапсов.
Исследования кратковременной синаптической пластичности используют целый ряд методических подходов, одним из которых является метод парной стимуляции моносинаптических входов с разными межстимульными интервалами. Считается, что наблюдаемая при такой стимуляции фасилитация синаптических реакций (PPF) имеет преимущественно пресинаптическую природу [2] и связана со следовыми эффектами, возникающими в пресинапсе после прохождения потенциала действия. Прирост амплитуды ответа экспоненциально снижается при увеличении межстимульного интервала (20–200 мс), что находится в соответствии (хотя и не всегда совпадает) с кинетикой изменения остаточного кальция. Парная стимуляция коллатералей Шаффера чаще всего сопровождается фасилитацией синаптических реакций пирамидных нейронов.
Вместе с тем, имеется множество синапсов, в которых парная фасилитация проявляется в заметно ослабленной форме или не проявляется вовсе. Более того, некоторые синапсы реагирует на парную стимуляцию депрессией второго ответа (PPD). Считается, что величина PPF пропорциональна вероятности выброса медиатора [2]. Наибольшая PPF наблюдается в синапсах с исходно низкой вероятностью выброса медиатора, тогда как максимально высокая вероятность выброса не может еще больше увеличиться даже в присутствие остаточного кальция. PPD таких высокоэффективных синапсов объясняют недостаточно быстрым восстановлением запасов медиатора [3]. Однако маловероятно, чтобы одиночный импульс мог приводить к существенному истощению запасов медиатора низкоэффективных синапсов, учитывая достаточно быструю кинетику его восстановления. Еще одним фактором, контролирующим кратковременную пластичность, является состояние ассоциированных с секреторными везикулами белков, участвующих в регуляции восприимчивости секреторного аппарата к ионам кальция. Остаточный кальций может приводить к активации этих Ca2+ сенсоров, которые влияют на заякоривание, проводимость и инактивацию кальциевых каналов, а также на восстановление доступного пула секреторных везикул [2, 4].
Несмотря на максимальное содержание остаточного кальция, многие синапсы демонстрируют подавление парной фасилитации, вплоть до депрессии только при сокращении межстимульного интервала (менее 20–40 мс). Механизмы такой “ранней” депрессии, по всей вероятности, отличаются от тех, которые лежат в основе депрессии высокоэффективных синапсов. Рефрактерный период сразу после спайкового разряда пресинаптического окончания связывают с инактивацией потенциал-зависимых натриевых и кальциевых каналов [2], хотя по некоторым данным PPF может быть обнаружена как минимум через 5 мс после первого стимула. В некоторых случаях снижение амплитуды ответа при повторной активации может объясняться десенситизацией постсинаптического рецептора [5]. Учитывая достаточно быстрый обмен между синаптическими и экстрасинаптическими рецепторами, не исключена также латеральная диффузия постсинаптических рецепторов [6]. Все эти механизмы в равной степени влияют на кратковременную пластичность при парной стимуляции с межстимульными интервалами 15 и 70 мс. Вместе с тем, даже совпадение всех этих свойств не гарантирует одну и ту же величину PPR и реакция постсинаптического нейрона на повторную активацию может быть ограничена из-за суммации с бисинаптическим ТПСП [7].
В пирамидных нейронах поля СА1 в ответ на стимуляцию коллатералей Шаффера регистрируется сложный постсинаптический потенциал, который состоит из первоначального ВПСП, на фоне которого развивается более поздний ТПСП. ТПСП генерируется тормозными интернейронами поля СА1, которые, также как и пирамидные нейроны иннервируются коллатералями Шаффера [7, 8]. На пирамидных нейронах поля СА1 аксоны тормозных интернейронов оканчиваются преимущественно на дендритах, образуя множественные синапсы, которые шунтируют проведение возбуждения, по некоторым данным, тормозные нейроны, могут иннервировать отдельные шипики [9]. Такая структурно-функциональная организация предполагает возможность локальных изменений возбудимости мембраны в каждом отдельном компартменте. Если при парной стимуляции (в том числе и с межстимульным интервалом 15 мс) бисинаптический ТПСП по времени совпадает с потенциалом действия, генерируемым в ответ на второй стимул, прирост ВПСП вследствие пресинаптической фасилитации частично или полностью компенсируется гиперполяризационным потенциалом (рис. 1а). Более мощный ТПСП может иногда провоцировать специфичную для соответствующих межстимульных интервалов депрессию [7, 8].
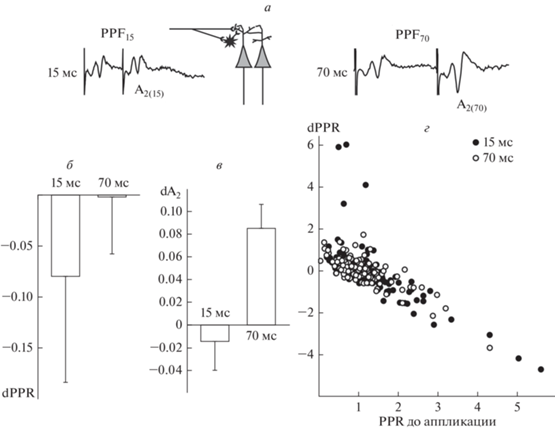
Рис. 1.
Комплексное влияние джасплакинолида на общие и специфичные для коротких межстимульных интервалов механизмы кратковременнрй пластичносии при парнрй стимуляции. а: примеры реакций на парную стимуляцию с межстимульными интервалами 15 и 70 мс. Измеряемые величины обозначены используемыми в тексте символами. Схема объясняет возможность подавления PPF15 за счет бисинаптического тпсп. б: изменение PPR после аппликации джасплакинолида (dPPR = PPRпосле – PPRдо) при разных межстимульных интервалах. Здесь и на других рисунках представлены средние значения ± ошибка среднего по всей выборке в целом. Для выявления значимых изменений потребовалось учитывать комплекс действующих факторов, включающих величину исходной PPR. в: изменение амплитуды второго в паре ответа (dA2 = А2(после) – А2(до)) при тех же межстимульных интервалах. г: зависимость dPPR (ордината) от исходной величины PPR70 (абсцисса) при парной стимуляции с межстимульным интервалом 15 мс (темные символы) или 70 мс (светлые символы).
Таким образом, по соотношению кратковременных модификаций при парной стимуляции зависимых и независимых от бисинаптического ТПСП реакций даже при экстраклеточной регистрации фокальных потенциалов гиппокампа можно в одном и том же эксперименте судить о функциональном состоянии не только возбуждающих, но и тормозных синапсов, что подтверждается также изменением этого соотношения на фоне блокатора ГАМКА рецепторов [10]. Систематические исследования показывают, что для идентификации локуса кратковременной пластичности достаточно применить дискриминантный анализ, способный изолировать вклад всех других сопутствующих факторов [2]. При этом влияние ТПСП до настоящего времени в расчет не принималось. Проблема заключается в том, что любое изменение вероятности выброса медиатора и других, независимых от ТПСП участников (включая различия в амплитуде ответа по причинам не физиологического характера, например расположение электродов при сравнении результатов разных экспериментов) по-разному влияют на PPF15 и PPF70, тем самым изменяя их баланс. Это снижает надежность определения зависимых от ТПСП модификаций, и необходимо подобрать метод, позволяющий исключить действие общих для разных межстимульных интервалов факторов.
Молекулярные основы фасилитации и депрессии сосуществуют в каждом из синапсов. От их баланса зависит динамика кратковременных модификаций, как отдельных синапсов, так и популяции в целом [2, 7]. Молекулярное обеспечение механизмов кратковременной пластичности включает, в том числе и зависимые от полимеризации актина реакции [11]. Участие актина в пресинаптической кратковременной пластичности дополняется его влиянием на скорость латеральной диффузии постсинаптических глутаматных [12, 13] и ГАМКА рецепторов [14, 15]. Существенно, что ГАМКА рецепторы менее устойчивы к латеральной диффузии по сравнению с расположенными на том же шипике глутаматными рецепторами [16]. Вместе с тем, гефириновые кластеры содержат много неполимеризованного актина [17], обеспечивая дополнительные возможности для зависимой от полимеризации актина пластичности.
В электрофизиологических экспериментах процедуры, влияющие на полимеризацию и деполимеризацию актина, чаще использовались при тестировании свойств возбуждающих синапсов, тогда как данные об изменении амлитуды и частоты спонтанных ТПСП представлены отдельными работами на культуре нейронов и переживающих срезах гиппокампа [18, 19]. Для блокады деполимеризации и индукции полимеризации актина применяют проникающий через клеточную мембрану джасплакинолид [20] и в зоне его влияния находятся все синаптические и внесинаптические компартменты. Вот почему желательно в одном эксперименте хотя бы косвенно контролировать изменения в разных компартментах, предположительно участвующих в исследуемых модификациях.
Целью данной работы было исследование влияния джасплакинолида на соотношение модификаций зависимых и независимых от бисинаптического ТПСП реакций в поле СА1 переживающих срезов гиппокампа крыс на парную стимуляцию коллатералей Шаффера с межстимульными интервалами 15 и 70 мс. С целью дальнейшей оптимизации количественного определения величины подавляющего фасилитацию тормозного компонента предполагалось сравнить результаты, полученные при использовании разных способов измерения.
МЕТОДИКА
В работе использованы 43 крысы линии Вистар 1–1.5 месячного возраста. Животные получены из питомника “Столбовая” РАМН (Московская обл.). До начала эксперимента животные содержались в виварии и имели неограниченный доступ к пище и воде, световая фаза суточного цикла продолжалась с 8.00 до 20.00. Эксперименты с животными выполнены в соответствии с требованиями международного и национального законодательства в области защиты и использования животных в экспериментальных исследованиях. Протокол эксперимента утвержден Этической комиссией ИВНД и НФ РАН.
Электрофизиологические эксперименты проводили на переживающих срезах гиппокампа. Состав перфузионной среды (мМ): NaCl 124; KCl 5; MgSO4 · 7H2O 1.3; CaCl2 2.5; NaH2PO4 1; NaHCO3 26; D-глюкоза – 10; карбоген – 95% O2 и 5% CO2; pН 7.3–7.4, температура 32°С. Для регистрации фокальных потенциалов в пирамидном слое поля CА1 использовали стеклянные микроэлектроды, заполненные 0.33 M раствором хлористого натрия. Раздражающие биполярные электроды устанавливали в радиальном слое на коллатерали Шаффера. Для того, чтобы гарантировать тестирование разных синаптических входов в срезах гиппокампа одного и того же животного, использовалась разная интенсивность раздражения. Перфузия раствором, содержащим джасплакинолид (0.2 мкМ), применяемый in vitro для индукции полимеризации актина [20], производилась для каждого среза индивидуально. Тестирование каждого из синаптических входов в режиме парной стимуляции производили каждые 30 с до и на фоне джасплакинолида, чередуя межстимульные интервалы 70 и 15 мс.
Измеряли амплитуду первого (A1) и второго (A2) в паре ответа (рис. 1а), а также их соотношение (paired pulse ratio, PPR). Для количественного сравнения величины прироста при разных межстимульных интервалах определяли разницу между PPF70 и PPF15 (PPF15 – PPF70) и амплитудами второго ответа (А2(15 мс) – А2(70 мс)), а также соотношение амплитуд второго ответа (А2(15 мс)/А2(70 мс)). Для определения статистической значимости при попарном сравнении показателей до и после аппликации джасплакинолида был использован t критерий Стьюдента для зависимых переменных. Чтобы подтвердить зависимость эффектов джасплакинолида от длительности межстимульного интервала и исходных свойств кратковременной пластичности был использован регрессионный анализ и трехфакторный ANOVA или ANOVA Repeated measures.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЙ
С целью определения возможного влияния джасплакиналида на вклад тормозных потенциалов в кратковременную пластичность при парной стимуляции в поле СА1 переживающих срезов гиппокампа крыс было использовано 14 животных. Перфузия раствором, содержащим джасплакинолид, производилась для каждого среза индивидуально. В контрольных экспериментах было использовано 32 среза от 29 животных. Чтобы оценить возможный вклад тормозных потенциалов в кратковременную пластичность при парной стимуляции в гиппокампе крыс и, в частности, в поле СА1, в переживающих срезах тестирование каждого из синаптических входов (n = 10 для каждого животного) производилось в режиме парной стимуляции с межстимульными интервалами 15 и 70 мс. Контрольные срезы подвергались тем же процедурам без добавления джасплакинолида.
Влияние джасплакинолида проявлялось в тесном взаимодействии с величиной межстимульного интервала и свойствами кратковременной пластичности до начала его применения (F(1,548) = 5.21, p = = 0.02). Чтобы ограничить вариабельность исходных условий, данные были сгруппированы в две отдельные выборки в зависимости от исходной величины PPF. В поле СА1 наиболее типичной реакцией на парную стимуляцию является фасилитация, и первоначально предполагалось не включать в статистический анализ данные нестандартных экспериментов, в которых исходная PPF70 <1. Основным эффектом, наблюдаемым на фоне перфузии срезов с джасплакинолидом, было снижение PPF (F(1,144) = 5.61, p = 0.01), что может быть связано с увеличением вероятности выброса медиатора и свидетельствовать о пресинаптических механизмах наблюдаемого на фоне джасплакинолида увеличения амплитуды ответа (t = 6.14, p = 0.000000) и их зависимости от полимеризации актина. Однако при очевидном влиянии на PPF70 (t = 3.45, p = 0.0009) снижение PPF15 было лишь незначительным, а инспекция остальных данных (исходная PPF70 <1) показала значимую зависимость эффектов джасплакинолида от межстимульного интервала (взаимодействие факторов F(1,130) = 5.16, p = 0.02). И хотя это становится заметным только в тех условиях, когда PPF70 изначально отсутствовала и, соответственно, можно было исключить ее снижение за счет увеличения вероятности выброса медиатора, не исключено, что джасплакинолид по-разному влияет на PPF15 и PPF70 во всех экспериментах. Величина dPPF = PPFпосле – PPFдо показывает насколько изменилась PPF после аппликации джасплакинолида и этот способ измерения, если не учитывать исходные характеристики, также не выявил значимых различий при разных межстимульных интервала (t = 0.61, p = 0.54, рис. 1б). В то же время, изменения амплитуды второго в паре ответа (А2(после) – А2(до)) значимо различались (t = = 3.36, p = 0.001) и при 15-миллисекундном интервале было заметно усиление тормозного влияния (рис. 1в). По всей вероятности, вмешательство разных механизмов не позволяет составить четкого представления о степени их участия.
Сравнение выборочных средних, в частности до и после аппликации джасплакинолида, в качестве единственного метода исследования существенно обедняет возможности интерпретации полученных результатов. Поэтому далее представлен анализ модификаций индивидуальных реакций в зависимости от исходных свойств кратковременной пластичности. В соответствии с экспоненциальным затуханием генерируемого в пресинапсе Са2+ сигнала можно ожидать, что сокращение межстимульного интервала больше способствует увеличению секреции на второй стимул. Тем не менее, это не гарантирует соответствующего увеличения PPF из-за действия некоторых дополнительных факторов, включая суммацию с бисинаптическим ТПСП. Действительно, связь между PPF70 и PPF15 плохо выявлялась, что особенно заметно на фоне джасплакинолида (соответственно r = 0.17, p = 0.04, n = 139 до и r = 0.15, p = 0.06, n = 139 после аппликации). Хотя при визуальной инспекции регрессии было видно, что некоторые данные все же в той или иной степени соответствуют теоретически ожидаемому росту фасилитации при сокращении межстимульного интервала, что может наблюдаться, в том числе и в отсутствие явно выраженного торможения. Все вместе означает, что при слишком коротких межстимульных интервалах на величину PPF в той или иной степени оказывают влияние сопутствующие факторы, маскирующие связь с вероятностью выброса медиатора, тем более, что их модификации вполне вероятно могут в той или иной степени компенсировать стремление к снижению PPF. А главное, не было никакого соответствия между изменениями PPF15 и PPF70 (r = –0.19, p = = 0.02, n = 139). Значимая корреляция, скорее всего, отражает обратную зависимость этих модификаций от исходных свойств PPF. Вероятно, по этой причине потенциация на фоне джасплакинолида развивалась независимо от изменения PPF15 (r = –0.07, p = 0.93, n = 139), в отличие от значимой корреляции с изменениями PPF70 (r = –0.53, p = 0.000, n = 139).
Несомненно, изменение вероятности выброса медиатора вносит одинаковый вклад в снижение PPF15 и PPF70, о чем свидетельствует зависимость модификаций от исходной величины PPF (рис. 1г). При исходно низкой PPF70 (а, следовательно, высокой вероятности выброса медиатора) эффекты джасплакинолида заметно ослабевали, что может объясняться меньшим диапазоном возможных модификаций. В полном соответствии с результатами, полученными при тестировании парными стимулами с межстимульным интервалом 70 мс (r = = –0.78, p = 0.000, n = 139), чем больше была величина PPF15 до аппликации джасплакинолида, тем заметнее было ее снижение (r = –0.7, p = 0.000, n = 139, рис. 1г). С точки зрения основной задачи исследования для выявления специфики влияния джасплакинолида на независимые от вероятности выброса медиатора компоненты кратковременной пластичности важно было применить метод, позволяющий исключить из анализа действие этого фактора. В частности при межстимульном интервале 15 мс, по соотношению модификаций зависимых и независимых от бисинаптического ТПСП реакций на фоне джасплакинолида можно судить о функциональном состоянии не только возбуждающих, но и тормозных синапсов и их зависимости от полимеризации актина.
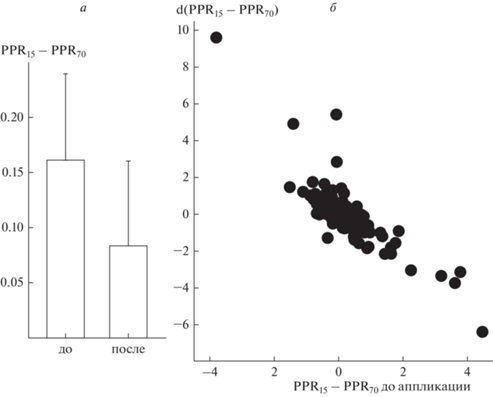
Влияние бисинаптического ТПСП на кратковременную пластичность, в отличие от многих других подавляющих PPF механизмов, ограничено коротким периодом времени и не распространяется на PPF70. Поэтому любые модификации тормозного потенциала могут изменять соотношение зависимых и независимых от ТПСП реакций, и для дальнейшего анализа необходимо было подобрать подходящие показатели, способные количественно определить меру расхождения в свойствах кратковременной пластичности при разных межстимульных интервалах. Анализ различий между PPF15 и PPF70 (PPF15 – PPF70) не выявил значимого изменения этого показателя после аппликации джасплакинолида (t = 0.61, p = 0.54, рис. 2а). Необходимо отметить, что видимость стабильного поддержания относительно постоянного уровня различий между PPF15 и PPF70 при сравнении средних величин по всей выборке в целом сама по себе не означает отсутствие эффекта. Это еще один пример того, что сравнение средних величин может быть недостаточным для выявления возможных модификаций, поскольку инспекция индивидуальных реакций на аппликацию показала, что этот эффект джасплакинолида, также как и изменение вероятности выброса медиатора, зависел от исходных характеристик. В каждом отдельном эксперименте после аппликации джасплакинолида степень подавления PPF15 относительно PPF70 либо усиливалась, либо ослаблялась в зависимости от исходных свойств кратковременной пластичности. В целом, изменение величины (PPF15 – PPF70) положительно коррелировало с исходной PPF70 (r = 0.47, p = 0.000, n = 139) и отрицательно с исходной PPF15 (r = –0.59, p = = 0.000, n = 139). Причем частный коэффициент корреляции, исключающий вклад PPF70 как общего, зависимого от вероятности выброса медиатора фактора, дополнительно подтверждает обратную зависимость эффекта от действия подавляющих PPF сигналов (rчастн = –0.77, p = 0.000, n = 139). В соответствии с этим меняется также и разница между PPF15 и PPF70 (r = –0.82, p = 0.000, n = 139, рис. 2б), величина независимая или, по крайней мере, менее зависимая от вероятности выброса медиатора. Все вместе означает, что джасплакинолид усиливал тормозный компонент реакции на парную стимуляцию при высокой вероятности выброса медиатора (по данным PPF теста) и/или в отсутствие подавляющих PPF сигналов, но ослаблял при исходно сильном торможении.
Рис. 2.
Баланс PPR15 и PPR70 определяет его разнонаправленные изменения в ответ на аппликацию джасплакинолида. а: разница между PPR15 и PPR70 (PPR15 – PPR70) значимо не различается до и после аппликации джасплакинолида. б: Изменение (PPR15 – PPR70) (ордината) коррелирует с его исходной величиной (абсцисса).
Допуская возможность участия ТПСП в подавлении PPF15, изменение амплитуды второго в паре ответа является более подходящим для количественной оценки и интерпретации результатов показателем. Существенно, что при парной стимуляции первый и второй потенциалы по-разному реагировали на подведение джасплакинолида. При тестировании с межстимульным интервалом 15 мс после подведения джасплакинолида амплитуда второго в паре ответа то увеличивалась, то снижалась, хотя в среднем изменения относительно контроля (до аппликации) не достигали порога достоверности, что естественно, учитывая высокую вариабельность эффектов в зависимости от исходных характеристик. Обращает на себя внимание обратная зависимость модификаций второго в паре ответа от исходной величины PPF при любом способе тестирования (r = –0.35, p = = 0.000, n = 139 при тестировании с межстимульным интервалом 15 мс и r = –0.23, p = 0.006, n = 139 при тестировании с межстимульным интервалом 70 мс). Снижение PPF более склонных к фасилитации реакций не сопровождалось заметным изменением А2–70 (t = 0.33, p = 0.74 для зависимых выборок при сравнении амплитуд до и после аппликации). Однако при 15 мс интервале те же реакции демонстрировали снижение амплитуды второго ответа. С другой стороны, реакции с исходно низкой PPF или даже PPD после подведения джасплакинолида увеличивали амплитуду второго в паре ответа, причем не только А2(15), но и А2(70), что свидетельствует о снижении влияния факторов, способствующих кратковременной депрессии. Означает ли это увеличение ТПСП, или задействованы другие зависимые от полимеризации актина механизмы подавления, можно определить, сопоставив модификации при разных межстимульных интервалах.
Приведенный выше анализ изменения амплитуды второго в паре ответа не контролирует возможность совместного участия ТПСП и неспецифичных для межстимульного интервала 15 мс механизмов. В частности, влияние джасплакинолида может распространяться на постсинаптические рецепторы. Значимость участия общих механизмов подтверждает скоррелированность модификаций амплитуды вторых ответов при тестировании парными стимулами с межстимульными интервалами 15 и 70 мс (r = 0.18, p = 0.03, n = 139), которая определяется их общей зависимостью от исходной величины PPF. Однако судя по величине коэффициента корреляции, основной вклад в эти модификации вносят независимые от базовой PPF компоненты кратковременной пластичности и, вероятно, именно поэтому влияние джасплакинолида на амплитуду второго ответа зависело от межстимульного интервала (взаимодействие факторов F(1,276) = 9.16, p = 0.002). Исходя из этих соображений, целесообразнее обратить внимание не на абсолютные величины, а на изменение баланса амплитуд вторых ответов при разных межстимульных интервалах.
Очевидно, что разница между амплитудами вторых ответов (А2(15 мс) – А2(70 мс)) максимальна в отсутствие тормозного потенциала, а высокоамплитудный ТПСП снижает, или даже инвертирует разницу, делая ее отрицательной. Направленность изменений этой величины, в среднем положительной до, и отрицательной после аппликации джасплакинолида (t = 3.36, p = 0.001, рис. 3а) свидетельствует в пользу гипотезы о преимущественном увеличении ТПСП. Наиболее существенное смещение разницы между А2(15 мс) и А2(70 мс) в сторону отрицательных значений происходило при исходно слабом торможении. Однако при наличии хорошо выраженной депрессии А2(15 мс) относительно А2(70 мс) еще до подведения джасплакинолида, реакции чаще всего демонстрировали снижение тормозного компонента. Достаточно четкую зависимость изменения величины (А2(15 мс) – А2(70 мс)) от ее исходных значений можно видеть на рис. 3б (r = = –0.59, p = 0.000, n = 139). Складывается впечатление, что наблюдаемые модификации могут иметь отношение к гомеостатической регуляции и оптимизации тормозного контроля.
Рис. 3.
Джасплакинолид усиливает депрессию второго ответа при коротких межстимульных интервалах. а: разница амплитуд (А2(15) – А2(70)) до и после аплликации джасплакинолида. Значимость различий p = 0.001. б: Изменение (А2(15) – А2(70))(d(А2(15) – А2(70)), ордината) коррелирует с его исходной величиной (абсцисса). в: соотношение амплитуд А2(15)/А2(70) до и после аплликации джасплакинолида. Значимость различий p = 0.02. г: Изменение А2(15)/А2(70) (ордината) коррелирует с его исходной величиной (абсцисса).

Чтобы окончательно убедиться в правильности этого вывода, необходимо было исключить влияние всех других факторов. В этом отношении недостатком показателя (А2(15 мс) – А2(70 мс)) является его зависимость не только от степени подавления фасилитации при парной стимуляции с межстимульным интервало 15 мс, но и от индивидуальных различий по амплитуде, в том числе по нефизиологическим причинам, зависящим от условий регистрации. И действительно, чем больше была амплитуда второго ответа, тем больше она различалась при разных межстимульных интервалах (r = 0.31, p = 0.000, n = 139), хотя изменения этой разницы на фоне джасплакинолида в основном не зависели от исходной активности, за исключением отдельных зависимых от тормозного компонента показателей. Тем не менее, предпочтительнее использовать не абсолютные, а относительные показатели различий. В отличие от всех других показателей, соотношение амплитуд А2(15 мс)/А2(70 мс) не зависит от амплитудных характеристик, что подтверждается полным отсутствие корреляции между величинами А2(15 мс)/А2(70 мс) и А2(70 мс) (r = –0.003, p = 0.74, n = 139). До аппликации джасплакинолида амплитуда второго ответа А2(15 мс) была почти в полтора раза больше, чем А2(70 мс) (в среднем в 1.38 ± 0.16 раза). После аппликации величина А2(15 мс)/А2(70 мс) достоверно снижалась (t = 2.2, p = 0.02, рис. 3в), и их величина в среднем практически совпадала (в среднем 1.0 ± 0.04), что также свидетельствует в пользу увеличения ТПСП. Тем не менее, в тех экспериментах, когда соотношение (А2(15 мс)/А2(7 0мс)) было меньше 1, реже меньше 2, реакция на подведение джасплакинолида была нетипичной для всей группы в целом. Тот факт, что соотношение (А2(15 мс)/А2(70 мс)) в этих экспериментах увеличивалось, может, по-видимому, объясняться растормаживающим действием джасплакинолида. Характер изменения этой независимой от амплитудных характеристик величины на фоне джасплакинолида, также как и других показателей, еще раз подтверждал активацию слабого и подавление сильного торможения (r = –0.96, p = 0.000, n = 139 и r = –0.83, p = 0.000, n = 138 после удаления из выборки слишком далеко отстоящей от основного массива точки, рис. 3г). Судя по всему, активация полимеризации сама по себе не определяет направленность изменений, а, по всей вероятности, стабилизирует те модификации, которые происходили до аппликации.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В работе обнаружено, что блокада деполимеризации и активация полимеризации актина влияет на краковременную пластичность при парной стимуляции коллатералей Шаффера в переживающих срезах гиппокампа крыс. Результаты работы в основном подтверждают полученные ранее нами и другими авторами данные об участии актина в пресинаптических механизмах кратковременной пластичности [11]. Вместе с тем, сравнение изменений при использовании парной стимуляции с разными межстимульными интервалами показало, что наряду с общими закономерностями влияние джасплакинолида распространяется на механизмы, подавляющие кратковременную фасилитацию при парной стимуляции с межстимульным интервалом 15 мс. С большой долей вероятности это может быть связано с влиянием джасплакинолида на амплитуду бисинаптического ТПСП. Если торможение действительно не участвует в наблюдаемых на фоне джасплакинолида модификациях, появляется необходимость привлекать неизвестные механизмы к объяснению дифференцированного влияния на PPF15 и PPF70.
Для количественного определения тормозного компонента реакции на парную стимуляцию был использован эффект подавляющего влияния бисинаптической активации тормозных интернейронов на PPF15. Этот межстимульный интервал выбран, чтобы по возможности избежать более ранних связанных с рефрактерным периодом модификаций. Очевидно, что любые изменения амплитуды тормозного потенциала вносят рассогласование между модификациями PPR15 и PPR70. Для определения особенностей влияния джасплакинолида при разных межстимульных интервалах было использовано несколько методов, каждый из которых давал возможность, так или иначе, учитывать разные аспекты не свзанных с ТПСП модификаций. Все эти методы выявили, что полимеризация актина чаще всего способствует потенциации как возбуждающих, так и тормозных синапсов. Такое заключение в целом соответствует тем данным, которые были получены на культуре нейронов и переживающих срезах гиппокампа [18].
Активирующее влияние джасплакинолида на эффективность тормозных синапсов может быть связано с дополнительной полимеризацией актина пресинаптических и постсинаптических компартментов. Учитывая общие принципы регуляции пресинаптических функций, полимеризация актина может облегчать секрецию медиатора не только возбуждающих [11], но и тормозных синапсов [21]. Заякоривание транспортеров содействует росту запасов медиатора [22]. К тому же, увеличение выброса глутамата может оказывать активирующее влияние на синтез ГАМК [23, 24] и даже образование новых синапсов [25, 26]. И хотя простое тестирование в условиях переживающих срезов вряд ли может обеспечить индукцию структурных модификаций, не искючено, что это возможно при отклонении от нормального уровня синаптической активации.
Кроме того, механизмы увеличения эффективности тормозных синапсов могут быть связаны и с полимеризацией постсинаптического актина [15]. Тот факт, что джасплакинолид предотвращает слипание шипиков [27], предполагает его влияние на функциональные характеристики расположенных на них рецепторов, в том числе менее стабильных в этом отношении ГАМКА рецепторов [16]. Активирующее влияние джасплакинолида на тормозную передачу может быть связано с ограничением двигательной активности постсинаптических рецепторов. В частности, полимеризация актина может приостанавливать их латеральную диффузию [28]. Кратковременная пластичность чаще всего сопровождается усилением латеральной диффузии, в том числе обнаружено увеличение коэффициента диффузии экстрасинаптических и синаптических ГАМКА рецепторов [14, 29, 30]. Актиновые филаменты участвуют в заякоривании рецепторов [14, 15, 17], а также могут служить естественным барьером, ограничивающим латеральную диффузию [6, 14, 31].
Постсинаптические механизмы регуляции эффективности тормозных синапсов зависят от баланса процессов эндоцитоза и экзоцитоза ГАМКА рецепторов [14, 15, 29]. Транслокация рецепторов из эндоплазматического ретикулума регулируется комплексом белков и ферментов, в том числе ассоциированных с цитоскелетом [15, 32]. Экзоцитоз и эндоцитоз ГАМКА рецепторов происходит в экстрасинаптической зоне, и пластичность тормозных синапсов связана с их диффузией и стабилизацией в зоне синаптического контакта [33]. Экстрасинаптические ГАМКА рецепторы в целом более мобильны [29], однако их латеральную диффузию ограничивает цитоскелетный белок радиксин, который, связывает актиновый цитоскелет с плазматической мембраной [31]. При его фосфорилировании экстрасинаптические рецепторы начинают свободно диффундировать, в том числе и в зону постсинапса [31]. Высокое содержание таких включающих альфа5-субъединицу рецепторов характерно для гиппокампа. Полимеризация актина может способствовать мобилизации и встраиванию дополнительных ГАМКА рецепторов [29, 32]. В том числе, гомеостатическая регуляция баланса возбуждения и торможения включает пополнение синаптического пула за счет экстрасинаптических ГАМКА рецепторов при более интенсивной активации нейронов. Кроме того, активация глутаматергических синапсов при полимеризации актина может быть сигналом для снижения интернализации ГАМКА рецепторов [34].
Вместе с тем необходимо отметить, что увеличение вероятности выброса медиатора, которое, судя по общему снижению PPF происходит в возбуждающих синапсах, расположенных на пирамидных нейронах поля СА1, предполагает возможность такого же активирующего влияния джасплакинолида и на другие пресинаптические окончания, в том числе и на тормозных интернейронах [35]. Подключение дополнительных интернейронов само по себе в состоянии обеспечить рост суммарного ТПСП даже без изменения эффективности каждого из синапсов, тем более что один интернейрон иннервирует большое число пирамидных нейронов. Хотя по другим данным, при потенциации расположенных на пирамидных нейронах глутаматергических синапсов, синапсы на интернейронах реагируют долговременной депрессией [36]. В результате часть интернейронов не достигает порога генерации потенциала действия и, исключительно за счет этого, может снижаться число вовлекаемых в реакцию тормозных синапсов.
Полимеризация, как известно, не определяет ход модификаций, и, в частности, джасплакинолид может усиливать или ослаблять синаптическую активность [11, 37]. В наших экспериментах часть данных не соответствует тем выводам, которые были сделаны на основании сравнения средних величин. Судя по изменению показателей характеризующих степень подавления PPF при коротких межстимульных интервалах, прирост ТПСП на фоне джасплакинолида был максимальным при исходно слабом торможении. В противоположность этому, при более или менее заметной PPD до подведения джасплакинолида наблюдалось увеличение амплитуды второго в паре ответа и, соответственно, активация PPF. Поскольку, снижение эффективности тормозных влияний увеличивает вероятность генерации потенциала действия, прирост амплитуды второго в паре ответа при межстимульном интервале 15 мс может быть связан с растормаживающим действием джасплакинолида. В целом тип реакции на подведение джасплакинолида зависел от исходных характеристик.
Такое заключение возможно еще и потому, что зависимость синаптической пластичности от интенсивности торможения широко известный факт [38, 39]. Гиперполяризация постсинаптического нейрона снижает вероятность открытия зависимых от деполяризации NMDA каналов, тогда как растормаживание способствует более активному входу кальция [40, 41]. В соответствии с этим меняется пластичность не только возбуждающих, но и тормозных синапсов [15, 16, 42, 43], включая пресинаптические модификации [23]. Зависимость от исходных свойств торможения может быть связана с разной степенью активации ГАМКВ рецепторов, модулирующих связь ГАМКА рецепторов с гефирином [44], хлорный градиент [45] и секреторный аппарат пресинапса [45, 46].
Вероятность латеральной диффузии синаптических ГАМКА рецепторов увеличивается при исходно сильном торможении [29, 30, 47]. Эффект растормаживания тесно связан с активацией NMDA рецепторов, расположенных в синапсах тормозных интернейронов поля СА1 [29]. Обнаружено, что наблюдаемое при этом дефосфорилирование ГАМКА рецепторов активизирует их двигательную активность и, соответственно, декластеризацию, что стимулирует латеральную диффузию рецепторов за пределы зоны синаптического контакта [29, 47]. Вместе с тем, активация возбуждающих и тормозных синапсов по-разному влияет на заякоривание ГАМКА рецепторов [31]. Соответственно, ГАМК стимулирует, а глутамат снижает стабилизацию экстрасинаптических рецепторов и их взаимодействие с актиновым цитоскелетом. В соответствии с этим меняется вероятность их транслокации к зоне синаптического контакта [31]. Вследствие этого соотношение синаптических и экстрасинаптических ГАМКА рецепторов может меняться при одном и том же режиме стимуляции в зависимости от исходных характеристик и, в частности, баланса возбуждения и торможения. К тому же ГАМКА рецепторы более подвержены десенситизации при высокой концентрации ГАМК. От контекста и от общего уровня активации зависит динамическая реорганизация образованных гефирином кластеров, как известно, стабилизирующих постсинаптические ГАМКА рецепторы [29, 32, 48, 49] и тесно связанных с актиновый цитоскелетом [50]. Причем учитывая активирующее влияние джасплакинолида на глутаматергические синапсы, можно думать, что это происходит достаточно быстро, по некоторым данным в течение нескольких минут [51].
Отличительной особенностью тормозных синапсов является так называемая “ионная пластичность” [45, 52]. Величина тормозного потенциала при активации ГАМКА рецепторов определяется проводимостью каналов и потенциалом реверсии EGABA. Сдвиг потенциала инверсии может усиливать, ослаблять, или даже инвертировать токи, индуцируемые активацией ГАМКА рецепторов. Увеличение объема шипиков при полимеризации актина меняет расположение K–Cl транспортеров [52]. Данные о том, что в ходе модификаций может происходить как деполяризационный, так и гиперполяризационный сдвиг потенциала реверсии [52–55], позволяют рассматривать ионную пластичность в качестве общего механизма, объединяющего все наблюдаемые в данной работе модификации. Деполяризационный сдвиг потенциала реверсии может быть одним из механизмов растормаживающего действия джасплакинолида [53, 56]. При исходно сильном торможении деполяризационный сдвиг и, соответственно, снижение амплитуды ТПСП могут быть связаны с активацией ГАМКВ рецепторов [45]. К тому же, при парной стимуляции с межстимульным интервалом 15 мс этому способствует сочетание тормозного потенциала с генерацией потенциалов действия в постсинаптическом нейроне [53, 57].
Необходимо отметить, что активирующее влияние полимеризации актина на пресинаптические механизмы PPF [11, 58], а также зависимый от актина вклад постсинаптических [6, 19], а то и структурных модификаций [9, 25, 51], не исключает возможность увеличения амплитуды второго в паре ответа и без снижения ТПСП. Тем более что этот эффект джасплакинолида не был специфичным относительно межстимульного интервала. Однако с этих позиций не очень понятными становятся закономерности изменений баланса между амлитудами А2(15 мс) и А2(70 мс), и прежде всего, их зависимость от исходной величины. Возможно, снижение секреции ГАМК вместе с более надежным заякориванием транспортеров [22] сокращает длительность тормозного влияния. Транслокация рецепторов в зону синаптического контакта может сопровождаться изменением композиционного состава ГАМКА рецепторов и кинетики тормозного потенциала [19, 31, 59], тем самым влияя на динамические характеристики реакций на парную стимуляцию.
В целом, эффект джасплакинолида, по всей вероятности, является комплексным. С одной стороны, полимеризация актина способствует восстановлению функций кратковременной пластичности, которые могут быть в той или иной степени ослаблены вследствие подготовительных манипуляций. С другой стороны, зависимость эффектов от тех свойств кратковременной пластичности, которые тесно связаны с эффективностью глутаматергических и тормозных синапсов, могут быть следствием активации метаботропных глутаматных и ГАМКВ рецепторов [36, 42]. Вероятным объяснением разнонаправленных эффектов джасплакинолида представляется “консолидация” тех модификаций, которые связаны с поддержанием оптимального баланса активности возбуждающих и тормозных входов [16, 39, 57, 60]. Гомеостатическая регуляция пресинаптических и постсинаптических функций является одной из причин изменения характера кратковременной пластичности [48, 57, 61].
ЗАКЛЮЧЕНИЕ
Таким образом, исследование влияния джасплакинолида на кратковременную пластичность выявило значимые различия при тестировании парными стимулами с межстимульными интервалами 15 и 70 мс. Дифференцированное влияние при разных межстимульных интервалах свидетельствует о том, что помимо общих механизмов, определяющих величину PPF, могут быть задействованы и другие модулирующие факторы и, в частности, изменение амплитуды тормозных потенциалов, вовлеченных в реакцию при коротких межстимульных интервалах. Полученные результаты дают основание полагать, что полимеризация актина имеет отношение к модуляции активности не только возбуждающих, но и тормозных синапсов. Судя по всему, активация полимеризации сама по себе не определяет направленность изменений, а, по всей вероятности, стабилизирует те модификации, которые происходили до аппликации. Предполагается, что все эффекты джасплакинолида в данных экспериментах, включая растормаживание, активацию исходно слабого торможения и обнаруженное ранее увеличение вероятности выброса медиатора, могу быть связаны с гомеостатической пластичностью.
Список литературы
Кудряшова И.В. // Нейрохимия. 2014. Т. 31. № 1. С. 5–15.
Jackman S.L., Regehr W.G. // Neuron. 2017. V. 94. P. 447–464.
Schneggenburger R., Sakaba T., Neher E. // Trend. Neurosci. 2002. V. 25. № 4. P. 206–212.
Mochida S. // Neurosci. Res. 2011. V. 70. № 1. P. 16–23.
Jones M.V., Westbrook G.L. // Trend. Neurosci. 1996. V. 19. № 3. P. 96–101.
Renner M., Schweizer C., Bannai H., Triller A., Lévi S. // PLoS ONE. 2012. V. 7. P. e43032.
Bartley A.F., Dobrunz L.E. // Eur. J. Neurosci. 2015. V. 41. P. 1402–1415.
Willadt S., Nenniger M., Vogt K.E. // PLoS One. 2013. V. 8. № 11. P. e80984.
Villa K.L., Berry K.P., Subramanian J., Cha J.W., Oh W.C., Kwon H.B., Kubota Y., So P.T., Nedivi E. // Neuron. 2016. V. 89. P. 756–769.
Кудряшова И.В. // Журн. высш. нервн. деят. 2015. Т. 65. № 2. С. 131–138.
Кудряшова И.В. // Нейрохимия. 2021. Т. 38. № 3. С. 195–204.
Gu J., Lee C.W., Fan Y., Komlos D., Tang X., Sun C., Yu K., Hartzell H.C., Chen G., Bamburg J.R., Zheng J.Q. // Nat. Neurosci. 2010. V. 13. № 10. P. 1208–1215.
Rust M.B., Gurniak C.B., Renner M., Vara H., Morando L., Görlich A., Sassoè-Pognetto M., Banchaabouchi M.A., Giustetto M., Triller A., Choquet D., Witke W. // EMBO J. 2010. V. 29. № 11. P. 1889–18902.
Maynard S.A., Triller A. // Front. Mol. Neurosci. 2019. V. 12. P. 313.
Mele M., Costa R.O., Duarte C.B. // Front. Cell Neurosci. 2019. V. 13. P. 77.
Chiu C.Q., Barberis A., Higley M.J. // Nat. Rev. Neurosci. 2019. V. 20. P. 272–281.
Giesemann T., Schwarz G., Nawrotzki R. Berhörster K., Rothkegel M., Schlüter K., Schrader N., Schindelin H., Mendel R.R, Kirsch J., Jockusch B.M. // J. Neurosci. 2003. V. 23. № 23. P. 8330–8339.
Wei J., Zhang M., Zhu Y., Wang J.-H. // Neuroscience. 2004. V. 127. № 3. P. 637–647.
Ivanov A., Esclapez M., Pellegrino Ch., Shirao T., Ferhat L. // J. Cell Sci. 2009. V. 122. P. 524–534.
Bubb M.R., Spector I., Beyer B.B., Fosen K.M. // J. Biol. Chem. 2000. V. 275. P. 5163–5170.
Yan Zh., Kim E., Datta D., Lewis D.A., Soderling S.H. // J. Neurosci. 2016. V. 36. № 45. P. 11411–11417.
Imoukhuede P.I., Moss F.J., Michael D.J., Chow R.H., Lester H.A. // Biophys. J. 2009. V. 96. P. 2949–2960.
Peng Y.-R., Zeng S.-Y., Song H.-L., Li M.-Y., Yamada M.K., Yu X. // J. Neurosci. 2010. V. 30. P. 16220–16231.
Lau C.G., Murthy V.N. // J. Neurosci. 2012. V. 32. № 25. P. 8521–8531.
Bourne J.N., Harris K.M. // Hippocampus. 2011. V. 21. P. 354–373.
Dobie F.A., Craig A.M. // J. Neurosci. 2011. V. 31. P. 10481–10493.
Jaworski J., Kapitein L.C., Gouveia S.M. Dortland B.R., Wulf P.S., Grigoriev I., Camera P., Spangler S.A., Di Stefano P., Demmers J., Krugers H., Defilippi P., Akhmanova A., Hoogenraad C.C. // Neuron. 2009. V. 61. № 1. P. 85–100.
Hanus C., Ehrensperger M.-V., Triller A. // J. Neurosci. 2006. V. 26. P. 4586–4595.
Bannai H., Lévi S., Schweizer C., Inoue T., Launey T., Racine V., Sibarita J.B., Mikoshiba K., Triller A. // Neuron. 2009. V. 62. P. 670–682.
de Luca E., Ravasenga T., Petrini E.M., Polenghi A., Nieus T., Guazzi S., Barberis A. // Neuron. 2017. V. 95. № 1. P. 63–69.e5.
Hausrat T.J., Muhia M., Gerrow K., Thomas P., Hirdes W., Tsukita S., Heisler F.F., Herich L., Dubroqua S., Breiden P., Feldon J., Schwarz J.R, Yee B.K., Smart T.G., Triller A., Kneussel M. // Nat. Commun. 2015. V. 6. P. 6872.
Luscher B., Fuchs T., Kilpatrick C.L. // Neuron. 2011. V. 70. № 3. P. 385–409.
Bogdanov Y., Michels G., Armstrong-Gold C., Haydon P., Lindstrom J., Pangalos M., Moss S. // EMBO J. 2006. V. 25. P. 4381–4389.
Rannals M.D., Kapur J. // J. Neurosci. 2011. V. 31. № 48. P. 17701–17712.
Harney S.C., Anwyl R. // J. Physiol. 2012. V. 590. № 16. P. 3771–3786.
McBain C.J. Kaue J.A. // Curr. Opin. Neurobiol. 2009. V. 19. № 3. P. 254–262.
Bleckert A., Photowala H., Alford S. // J. Neurophysiol. 2012. V. 107. № 12. P. 3479–3492.
Кудряшова И.В. // Нейрохимия. 2015. Т. 32. № 3. С. 181–191.
Gandolfi D., Bigiani A., Porro C.A., Mapelli J. // Int. J. Mol. Sci. 2020. V. 21. № 5. P. 1805.
Bar-Ilan L., Gidon A., Segev I. // Front. Neural. Circuits. 2013. V. 6. P. 118.
Hayama T., Noguchi J., Watanabe S., Takahashi N., Hayashi-Takagi A., Ellis-Davies G.C., Matsuzaki M., Kasai H. // Nat. Neurosci. 2013. V. 16. № 10. P. 1409–1416.
Bannai H., Niwa F., Sherwood M.W., Shrivastava A.N., Arizono M., Miyamoto A., Sugiura K., Lévi S., Triller A., Mikoshiba K. // Cell Rep. 2015. V. 13. P. 2768–2780.
Mapelli J., Gandolfi D., Vilella A., Zoli M., Bigiani A. // Proc. Natl. Acad. Sci. USA. 2016. V. 113. P. 9898–9903.
Gerrow K., Triller A. // Mol. Cell. Neurosci. 2014. V. 60. P. 97–107.
Wright R., Newey S.E., Ilie A., Wefelmeyer W., Raimondo J.V., Ginham R.,R.A. Mcllhinney J., Akerman C.J. // J. Neurosci. 2017. V. 37. № 22. P. 5447–5462.
Fu Y., Wu X., Lu J., Huang Z.J. // Front. Cell Neurosci. 2012. V. 6. P. 57.
Muir J., Arancibia-Carcamo I.L., MacAskill A.F., Smith K.R., Griffin L.D., Kittler J.T. // Proc. Natl Acad. Sci. USA. 2010. V. 107. № 38. P. 16679–16684.
Vlachos A., Reddy-Alla S., Papadopoulos T., Deller T., Betz H. // Cereb. Cortex. 2013. V. 23. P. 2700–2711.
Fritschy J., Panzanelli P. // Eur. J. Neurosci. 2014. V. 39. P. 1845–1865.
Pizzarelli R., Griguoli M., Zacchi P., Petrini E.M., Barberis A., Cattaneo A., Cherubini E. // Neuroscience. 2020. V. 439. P. 125–136.
Lushnikova I., Skibo G., Muller D., Nikonenko I. // Neuropharmacology. 2011. V. 60. P. 757–764.
Chamma I., Heubl M., Chevy Q., Renner M., Moutkine I., Eugène E., Poncer J. Ch., Lévi S. // J. Neurosci. 2013. V. 33. № 39. P. 15488–15503.
Woodin M.A., Ganguly K., Poo M. // Neuron. 2003. V. 39. P. 807–820.
Jedlicka P., Deller T., Gutkin B.S., Backus K.H. // Hippocampus. 2011. V. 21. P. 885–898.
Lee H.H., Deeb T.Z., Walker J.A., Davies P.A., Moss S.J. // Nat. Neurosci. 2011. V. 14. P. 736–743.
Viitanen T., Ruusuvuori E., Kaila K., Voipio J. // J. Physiol. 2010. V. 588. P. 1527–1540.
Hennequin G., Agnes E.J., Vogels T.P. // Ann. Rev. Neurosci. 2017. V. 25. P. 557–579.
Guzman G.A., Guzman R.E., Jordan N., Hidalgo P. // Front. Cell Neurosci. 2019. V. 13. P. 125.
Vargas-Caballero M., Martin L.J., Salter M.W., Orser B.A. Paulsen O. // Neuropharmacology. 2009. V. 58. P. 668–675.
Sprekeler H. // Curr. Opin. Neurobiol. 2017. V. 43. P. 198–203.
Ortega J.M, Genç Ö., Davis G.W. // eLife. 2018. V. 7. P. e40385.
Дополнительные материалы отсутствуют.