

Неорганические материалы, 2020, T. 56, № 4, стр. 422-428
Формирование структуры цинкзамещенного гидроксиапатита в процессе механохимического синтеза
М. В. Чайкина 1, Н. В. Булина 1, *, И. Ю. Просанов 1, О. Б. Винокурова 1, А. В. Ищенко 2, 3
1 Институт химии твердого тела и механохимии СО Российской академии наук
630128 Новосибирск, ул. Кутателадзе, 18, Россия
2 Институт катализа им. Г.К. Борескова СО Российской академии наук
630090 Новосибирск, пр. Академика Лаврентьева, 5, Россия
3 Новосибирский государственный университет
630090 Новосибирск, ул. Пирогова, 2, Россия
* E-mail: bulina@solid.nsc.ru
Поступила в редакцию 28.05.2019
После доработки 12.09.2019
Принята к публикации 03.10.2019
Аннотация
Механохимическим методом синтезированы образцы цинкзамещенного гидроксиапатита с заданным замещением (Са10 – хZnx(PO4)6(OH)2, x = 0.1–2.0). Показано влияние состава исходных компонентов (Zn(H2PO4)2 ⋅ 2Н2О и ZnО), а также кристаллизационной воды на процесс формирования структуры апатита и степень замещения кальция цинком в процессе механохимического синтеза и отжига. При использовании в качестве допанта ZnО после 5–10 мин обработки реакционной смеси впервые непосредственно в мельнице выявлено образование промежуточного продукта – бета-трикальцийфосфата, который спустя 20 мин исчезает. Конечный продукт синтеза – цинкзамещенный гидроксиапатит – остается однофазным при x ≤ 0.8 в случае Zn(H2PO4)2 ⋅ 2Н2О и при x ≤ 0.25 в случае ZnO. После отжига цинкзамещенный гидроксиапатит однофазный только в случае использования фосфата цинка при x ≤ 0.1.
ВВЕДЕНИЕ
Гидроксиапатит (ГАП) – Са10(РО4)6(ОН)2 – является аналогом минеральной составляющей костных и зубных тканей человека и животных и широко используется в медицине [1–3]. Замещения в структуре апатита изменяют его биологические свойства [4]. Данная работа посвящена синтезу цинкзамещенного ГАП (Zn-ГАП). Цинк является биологически важным элементом, участвующим в различных метаболических процессах роста костной ткани, и бактерицидным элементом, способствующим предотвращению воспалительных процессов [5, 6]. Благодаря этим свойствам Zn-ГАП перспективен для покрытий имплантатов, биосовместимой керамики и композитов, используемых в медицинских целях [7].
Существует ряд методов синтеза Zn-ГАП: путем осаждения из растворов, твердофазный синтез, золь–гель, гидротермальный и другие, подробно описанные в обзоре [8]. В зависимости от метода, условий синтеза и состава исходных веществ изменяются положение ионов цинка в структуре Zn-ГАП, степень замещения и морфология кристаллов.
В работе [9] авторы синтезировали Zn-ГАП методом осаждения из разных исходных веществ в двух вариантах: I – из смеси растворов ацетата кальция и цинка с раствором двухзамещенного фосфата натрия; II – из смеси растворов нитрата кальция и цинка с раствором однозамещенного фосфата аммония. В обоих вариантах концентрация цинка менялась от 0 до 100 ат. %. Выявлено, что присутствие цинка в растворе ингибирует кристаллизацию Zn-ГАП и по мере увеличения его концентрации возрастает содержание аморфной фазы. В продукте, синтезированном по методу I, зарегистрировано ~20 ат. % цинка, а по методу II – ~25 ат. %. При отжиге образцов, однофазный Zn-ГАП-I существует при концентрации Zn ≤ 5 ат. % до 500°С, а Zn-ГАП-II – до 700°С. При более высоких температурах отжига появляется вторая фаза – бета-трикальцийфосфат (β-ТКФ), содержание которого возрастает с увеличением концентрации цинка в растворе и температуры отжига [9].
В работе [10] изучалось влияние цинка и магния на рост грани (0001) кристаллов ГАП. Было обнаружено, что оба катиона ингибируют рост этой грани, причем цинк снижает скорость роста грани в 1000 раз эффективнее, чем магний. Авторы работы [11] предлагают метод получения Zn-ГАП путем гидротермальной обработки аморфного фосфата кальция в нитрате цинка, позволяющий увеличить содержание ионов цинка до 14 мол. % с включением в структуру Zn-ГАП в положение Са2, что почти в 3 раза больше, чем при осаждении его из раствора.
В ряде работ исследуется механизм замещения и стабильность ионов цинка в структуре апатита. В работе [12] рассчитаны электронные состояния при замещении цинком ионов кальция в двух позициях. Выявлено, что цинк при замещении вакансий кальция второй позиции (Zn2) оказывается “более ковалентным”, с более короткими связями и энергетически в более выгодной тетраэдрической координации, чем цинк в первой позиции (Zn1), находящийся в октаэдрической координации. Подобные данные получены также авторами работы [13], согласно которым ион цинка сдвигается с позиции Са2 в направлении гидроксильной группы, что сопровождается уменьшением параметра a элементарной ячейки апатита с 9.488 до 9.319 Å.
В статье [14] предложен механизм синтеза цинкзамещенного дефицитного по кальцию апатита (ДКА) из растворов, при котором цинк занимает вакансии ионов Са2 в дефектных комплексах на стадии формирования структуры ДКА. Замещение Са2 цинком в этом случае ограничено несколькими мол. %.
Согласно приведенным выше результатам, при синтезе Zn-ГАП ионы цинка замещают Са2 в количестве от 5 до 25 ат. %. Однако появились работы, где в синтезированных золь–гель-методом отожженных образцах цинк встраивается в позиции не катионов кальция, а ОН-групп, вдоль оси с структуры апатита, образуя цепочки О–Zn–О [15]. Изучена термостабильность Zn-ГАП с такой локализацией ионов цинка [16]. Максимальное содержание цинка в структуре Zn-ГАП получено при отжиге образцов в интервале температур 900–1100°С с образованием продукта состава Ca10Zn0.25(PO4)6(ОН)1.5О0.5. Необычную локализацию цинка в структуре Zn-ГАП авторы этих работ объясняют электронной спецификой 3d-элементов, приводя в качестве примера подобную локализацию ионов меди в положении ОН-групп на оси с структуры ГАП [17].
Ведутся исследования бактерицидных и токсических свойств Zn-ГАП [18, 19].
Синтез ГАП методом осаждения нередко осложняется образованием примеси других ортофосфатов [4]. В последние десятилетия для получения различных материалов широко используется механохимический синтез (МХС) [20]. Преимущество МХС в его простоте. Весьма перспективным является “мягкий” МХС [21], когда в качестве исходных компонентов используют оксиды и кислые соли, реакция между которыми при совместной активации в мельницах аналогична кислотно-основным взаимодействиям компонентов в жидкой фазе. В результате такого взаимодействия образуется заданный готовый продукт и вода. В большинстве работ механическая активация компонентов длится десятки часов вследствие использования мельниц с низкой мощностью [22]. Нами используется мощная планетарная мельница [23], позволяющая за десятки минут получить готовый продукт в нанокристаллическом состоянии [24].
Целью данной работы является исследование процесса формирования структуры цинкзамещенного ГАП при взаимодействии компонентов реакционной смеси с разным составом допанта для определения оптимальных условий МХС и получения однофазного продукта.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходными компонентами для синтеза были: монетит CaHPO4 (квалификации “ч.”, Вектон), свежепрокаленный CaO (квалификации “ч. д. а.”, Вектон). В качестве допантов использовались: Zn(H2PO4)2 ⋅ 2H2O (квалификации “ч. д. а.”) и ZnO (квалификации “ч.”). Синтез образцов Zn-ГАП цинка проводился по реакциям (1) и (2), которые отличаются количеством выделяющейся воды:
(1)
$\begin{gathered} 6{\text{СaHP}}{{{\text{O}}}_{4}} + {\text{(}}4 - x{\text{)CaО}} + x{\text{ZnO}}\xrightarrow{{{\text{MXC}}}} \\ \to {\text{С}}{{{\text{a}}}_{{10 - x}}}{\text{Z}}{{{\text{n}}}_{x}}{{({\text{P}}{{{\text{O}}}_{4}})}_{6}}{{({\text{OH}})}_{2}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O,}} \\ \end{gathered} $(2)
$\begin{gathered} (6 - 2x){\text{CaHP}}{{{\text{O}}}_{4}} + (4 + x){\text{CaO}} + \\ + \,\,x{\text{Zn(}}{{{\text{H}}}_{2}}{\text{P}}{{{\text{O}}}_{4}}{{{\text{)}}}_{2}} \cdot 2{{{\text{Н}}}_{2}}{\text{О}}\xrightarrow{{{\text{MXC}}}} \\ \to {\text{С}}{{{\text{a}}}_{{10 - x}}}{\text{Z}}{{{\text{n}}}_{x}}{{({\text{P}}{{{\text{O}}}_{4}})}_{6}}{{({\text{OH}})}_{2}} + {\text{ }}n{{{\text{Н}}}_{2}}{\text{О,}} \\ \end{gathered} $МХС образцов проводился в планетарной мельнице АГО-2 [23] в двух охлаждаемых водой стальных барабанах объемом 150 мл стальными шарами массой 200 г со скоростью вращения барабанов 1800 об./мин. Соотношение навески реакционной смеси и массы шаров составляло 1 : 20. Во избежание загрязнения продукта намолом железа перед синтезом проводили “футеровку” рабочей зоны мельницы реакционной смесью того же состава. Отжиг образцов проводился при температуре 1000°С в течение 5 ч в электрической печи марки ПВК 1.4-8 со скоростью 10°C/мин.
Образцы после МХС и после отжига исследовались методами рентгенографического анализа, ИК-спектроскопии и просвечивающей электронной микроскопии высокого разрешения (ВРЭМ). Рентгенограммы регистрировались на порошковом дифрактометре Bruker D8 Advance в геометрии Брегга–Брентано с CuKα-излучением, никелевым Kβ-фильтром и сверхбыстрым позиционно-чувствительным одномерным детектором Lynx-Eye (угол захвата 3°). Рентгенофазовый анализ соединений проводился с использованием базы данных порошковых рентгенограмм ICDD PDF-4 (2011 г.). Уточнение параметров элементарной ячейки, размера кристаллитов, а также расчет концентраций фаз проводили по методу Ритвельда в программе Topas 4.2 (Bruker, Германия). ИК-спектры снимали на спектрометре “Инфралюм-801”, таблетки образцов получали по стандартной методике прессованием с KBr. Исследование образцов методом ВРЭМ проводили на электронном микроскопе JEM-2010 (JEOL, Япония). Локальный анализ элементного состава образцов осуществляли с использованием энергодисперсионного спектрометра QUANTAX 200-TEM (Bruker, Германия) с XFLASH-детектором.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Взаимодействие исходных компонентов при МХС. На рис. 1 представлены ИК спектры реакционных смесей, полученных после первой минуты взаимодействия компонентов по реакциям (1) и (2) – образцы 1 и 2 соответственно. Полосы поглощения в области волновых чисел 500–1100 см–1 практически идентичны для обоих образцов и соответствуют полосам поглощения деформационных и валентных колебаний связей Р–О в структуре СаНРО4 [25]. В области более высоких значений волнового числа спектры разные. В образце 2 наблюдается интенсивная полоса поглощения при 3640 см–1, относящаяся к валентным колебаниям групп ОН в Са(ОН)2, образовавшегося при взаимодействии оксида кальция с кристаллизационной водой, выделяющейся в ходе реакции кислотно-основного взаимодействия оксида кальция с однозамещенным фосфатом цинка по реакции (2). При взаимодействии компонентов по реакции (1) в первую минуту воды выделилось мало, поэтому в спектре образца 1 полоса 3640 см–1 едва заметна. ИК-спектр продукта реакции (1) почти не изменился и идентичен спектру исходного СаНРО4 (рис. 1, спектр 3). Малое количество выделившейся воды сказывается на скорости взаимодействия соединений кальция с ${\text{НРО}}_{4}^{{2 - }}$-группами исходного СаНРО4. В ИК-спектре образца 2 видны также изменения в области волновых чисел 1130–1400 см–1, относящихся к колебаниям связей кислых групп ${\text{НРО}}_{4}^{{2 - }}$ и плоским колебаниям ОН-групп в структуре СаНРО4 [25] вследствие их взаимодействия с Са(ОН)2, что характерно для процессов “мягкой механохимии” [21]. При увеличении времени активации структура СаНРО4 разрушается, что проявляется в постепенном вырождении валентных и деформационных колебания связей Р–О фосфатных групп в ИК-спектрах (рис. 2, спектры 2–4). Через семь минут синтеза в ИК-спектре образца появляется слабая полоса поглощения при частоте 602 см–1 (рис. 2, отмечено стрелкой), относящаяся к деформационному колебанию связей О–Р–О в апатите, что свидетельствует о начале формирования структуры апатита.
Рис. 1.
ИК-спектры образцов 1 и 2 c х = 0.5, активированных в течение 1 мин, в сравнении с ИК-спектрами исходных реагентов: 3 – СаНРО4, 4 – Zn(H2PO4)2 · 2Н2О, 5 – ZnO.
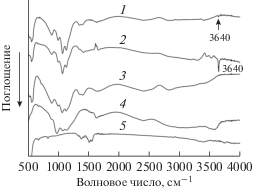
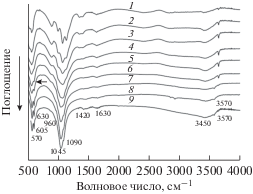
Рис. 2.
ИК-спектры образца 1 активированного в течение 1 (1), 3 (2), 4 (3), 6 (4), 7 (5), 10 (6), 20 (7), 30 (8).
По данным ВРЭМ (рис. 3а), реакционная смесь к этому моменту состоит из агрегатов микронных частиц оксида кальция, покрытых нанометровыми (30–90 нм) частицами фосфата кальция. На рис. 3б представлена частица этого же образца с межплоскостными расстояниями, близкими к кристаллической решетке β-ТКФ [26] и атомным отношением (Ca + Zn)/P ~ 1.5 (вставка на рис. 3б).
Рис. 3.
Электронные микрофотографии и данные рентгенодисперсионного микроанализа образца 1, активированного 7 мин: общий вид (а); протяженная решетка с межплоскостными расстояниями, близкими к таковым для β-ТКФ (б).

Через 30 мин МХС, по данным ИК-спектроскопии, образуется структура апатита с полосами поглощения колебания связей Р–О (рис. 2, спектр 8), характерными и для стехиометрического незамещенного апатита (рис. 2, спектр 9). В ИК-спектре МХС-ГАП присутствуют полосы поглощения деформационных колебаний связей О–Р–О ν4 570 и 605 см–1, три полосы валентных колебаний связей О–Р ν1 960 см–1, ν3 1045 и 1090 см–1, полоса поглощения либрационных 630 см–1 и валентных 3570 см–1 колебаний ОН-группы в структуре ГАП. Синтез проходил на воздухе, поэтому имело место взаимодействие компонентов реакционной смеси с СО2 воздуха с образованием ионов ${\text{CО}}_{3}^{{2 - }},$ входящих в структуру ГАП. В спектре 9 незамещенного ГАП присутствуют слабая полоса валентного колебания связей С–О при 1420 см–1 и едва заметное плечо при 1460 см–1, указывающие на образование карбонатапатита типа II (рис. 2). Полосы поглощения с волновыми числами 1630, 3450 см–1 принадлежат молекулам воды, выделившейся в процессе реакции (1) и сорбированной на поверхности частиц Zn-ГАП. Следует отметить, что полосы поглощения валентных колебаний ОН-групп в Zn-ГАП и незамещенном ГАП имеют одну и ту же частоту – 3570 см–1. Следовательно, можно полагать, что при МХС Zn-ГАП ионы цинка в канале ОН-групп отсутствуют.
ИК-спектры образцов, допированных фосфатом цинка, после 5 и более минут МХС аналогичны спектрам образцов с оксидом цинка.
На рис. 4 приведены рентгенограммы образца 1 с x = 0.5 в зависимости от времени механической активации. Установлено, что на рентгенограмме образца после 1 мин активации рефлексы Са(ОН)2 отсутствуют и появляются только после 3 мин взаимодействия. Это повлияло на формирование промежуточных продуктов реакции в процессе “мягкого механохимического” взаимодействия. После 5–10 мин обработки реакционной смеси 1 наблюдается образование промежуточного продукта с рефлексами при 27.77° и 31.01°, характерными для β-ТКФ (отмечены стрелками на рис. 4). В пользу образования β-ТКФ свидетельствуют данные ВРЭМ (рис. 3б). Известно, что β-ТКФ обычно образуется при отжиге образцов при 600–1000°С [4]. Это позволяет полагать, что уровень подводимой механической энергии при МХС образцов в мощных мельницах эквивалентен этим температурам. В работе [27] показано, что при активации порошков в мельнице АГО-2 возможно локальное повышение температуры до 600°С.
Рис. 4.
Рентгенограммы образцa 1 с x = 0.5, активированного в течение 1 (1), 3 (2), 4 (3), 5 (4), 6 (5), 7 (6), 10 (7), 20 мин (8).
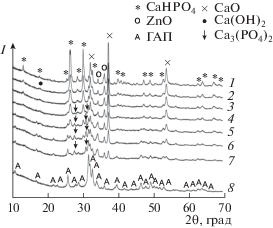
При увеличении времени синтеза до 20 мин происходит дальнейшее взаимодействие компонентов смеси 1 с образованием структуры Zn-ГАП, а β-ТКФ уже не обнаруживается (рис. 4). Следует отметить, что при синтезе Zn-ГАП по реакции (2) образование β-ТКФ не наблюдалось.
Влияние концентрации вводимых соединений цинка. Был проведен МХС Сa10 – xZnx(PO4)6(OH)2, где х = 0.1, 0.25, 0.5, 0.8, 1.0, 1.5, 2.0 при введении двух типов допантов цинка в соответствии с реакциями (1) и (2). Обнаружено, что образцы, синтезированные по реакции (1) в течение 30 мин, остаются однофазными до x = 0.25 (табл. 1). При x ≥ 0.5 обнаруживается небольшая примесь непрореагировавшего ZnО. При синтезе по реакции (2) однофазный Zn-ГАП образуется при x ≤ 0.8 (табл. 1). При введении x > 0.8 в образце появляется аморфная фаза неидентифицированного состава (табл. 1).
Таблица 1.
Концентрации фаз, образующихся в результате МХС Ca10 –xZnx(PO4)6(OH)2 по реакциям (1) и (2)
| x | Концентрация, мас. % | Rwp, % | xрасч | Концентрация, мас. % | Rwp, % | xрасч | ||
|---|---|---|---|---|---|---|---|---|
| Zn-ГAП | ZnO | Zn-ГАП | аморфная фаза | |||||
| (1) | (2) | |||||||
| 0.1 | 100 | – | 7.0 | 0.1 | 100 | – | 6.8 | 0.1 |
| 0.25 | 100 | – | 6.8 | 0.25 | 100 | – | 6.8 | 0.25 |
| 0.5 | 99.7(1) | 0.3(1) | 6.5 | 0.46 | 100 | – | 6.8 | 0.5 |
| 0.8 | 99.3(1) | 0.7(1) | 6.7 | 0.71 | 100 | – | 7.4 | 0.8 |
| 1.0 | 99.1(1) | 0.9(1) | 6.7 | 0.89 | 100 | + | 7.6 | Не определяли |
| 1.5 | 98.7(1) | 1.3(1) | 7.2 | 1.28 | 100 | ++ | 8.6 | Не определяли |
| 2.0 | 98.5(1) | 1.5(1) | 7.4 | 1.81 | 100 | +++ | 10.2 | Не определяли |
Определение параметров элементарных ячеек МХС-образцов показало, что при введении разных концентраций ZnO и Zn(H2PO4)2 ⋅ 2Н2О параметр с апатитовой фазы ведет себя одинаково (рис. 5). Это позволяет предположить, что при синтезе по реакциям (1) и (2) в структуру апатита входит равное количество цинка. Снижение параметра с с ростом концентрации цинка обусловлено замещением цинком ионов кальция, так как радиус иона Zn2+ (0.74 Å) значительно меньше радиуса Ca2+ (0.99 Å). Поведение же параметра а при введении ZnO и Zn(H2PO4)2 ⋅ 2Н2О немного отличается, что, вероятно, связано с отличающимся ближним окружением ионов цинка. Стоит отметить, что с увеличением концентрации вводимого Zn снижается размер кристаллитов Zn-ГАП, что свидетельствует об ингибировании процесса кристаллизации при введении обоих допантов (рис. 5г).
Рис. 5.
Изменение параметров (а, б, д, е), объемa элементарной ячейки (в, ж), а также размера кристаллитов (г, з) в образцах Zn-ГАП до (а–г) и после отжига (д–з) при введении разных концентраций ZnO и Zn(H2PO4)2 ⋅ 2Н2О.
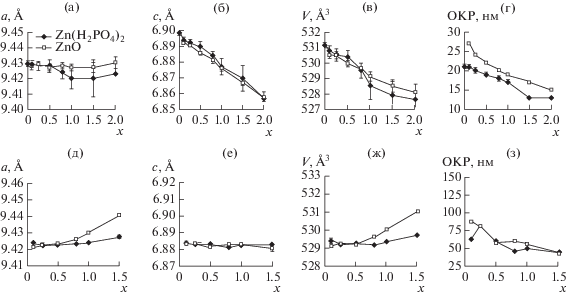
В отожженных образцах апатита наблюдается более заметное изменение параметра а при введении оксида цинка (рис. 5д), причем в сторону увеличения, и практически постоянное значение параметра с для обоих допантов (рис. 5е). Это можно объяснить возможным изменением положения иона-заместителя в структуре апатита в процессе отжига. Локализация заместителя в положении ионов Са2+ либо ОН-групп может влиять на параметры решетки синтезированного апатита.
Методом рентгенофазового анализа установлено, что при x = 0.1 (~0.65 мас. % Zn2+) в случае Zn(H2PO4)2 ⋅ 2Н2О отожженные образцы (1000°С, 5 ч) остаются однофазными (табл. 2). При x ≥ 0.25 начинает выделяться оксид цинка, а при x ≥ 1 и выше образец становится трехфазным, т.к. добавляется еще фаза β-ТКФ (табл. 2). Подобные данные были получены и при введении ZnО, но β-ТКФ в этом случае появлялся уже при x = 0.8. Это объясняется тем, что расчет проводили для замещения цинком заданного количества ионов кальция (см. реакции (1) и (2)), но т.к. цинка вошло меньше, продукт становился дефицитным по катиону – ДКА и при отжиге переходил в β-ТКФ.
Таблица 2.
Концентрации фаз в отожженных образцах, полученных при МХС Ca10 –xZnx(PO4)6(OH)2 по реакциям (1) и (2)
| x | Концентрация, мас. % | Rwp, % | xрасч | Концентрация фаз, мас. % | Rwp, % | xрасч | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Zn-ГAП | ZnO | CaO | ТКФ | Zn-ГAП | ZnO | ТКФ | |||||
| (1) | (2) | ||||||||||
| 0.1 | 99.4(1) | 0.3(1) | 0.3(1) | – | 8.2 | 0.08 | 100 | – | – | 7.8 | 0.10 |
| 0.25 | 99.5(1) | 0.5(1) | – | – | 8.3 | 0.19 | 99.6(1) | 0.4(1) | – | 8.3 | 0.20 |
| 0.5 | 99.0(1) | 1.0(1) | – | – | 7.7 | 0.38 | 99.0(1) | 1.0(1) | – | 7.7 | 0.38 |
| 0.8 | 86.3(2) | 1.3(2) | – | 12.4(3) | 7.5 | 0.60 | 98.7(1) | 1.3(1) | – | 7.4 | 0.60 |
| 1.0 | 70.5(2) | 1.7(2) | – | 27.8(3) | 7.4 | 0.79 | 86.5(2) | 1.6(2) | 11.9(3) | 7.1 | 0.80 |
| 1.5 | 39.6(3) | 2.4(3) | – | 58.0(2) | 8.7 | 1.20 | 51.0(3) | 2.5(3) | 46.5(4) | 7.8 | 1.20 |
| 2.0 | 5(1) | 3(1) | – | 92(1) | 11.9 | – | 13.3(3) | 3.5(3) | 83.2(5) | 9.5 | 1.78 |
Согласно расчетным данным, в структуру апатита при МХС с введением ZnO без отжига может войти до x = 1.81 (табл. 1), а после отжига – до x = = 0.38. При использовании в качестве допанта цинка Zn(H2PO4)2 ⋅ 2Н2О в структуру Zn-ГАП при МХС входит до x = 0.8, после отжига – до x = 0.6 моль. При бóльших концентрациях вводимого допанта, в той или иной форме, в МХС-образцах будет примесь непрореагировавшего источника цинка, а после отжига также β-ТКФ.
ЗАКЛЮЧЕНИЕ
“Мягким” механохимическим методом синтезированы образцы Zn-ГАП (Ca10 – xZnx(PO4)6(OH)2) с использованием ZnО и Zn(H2PO4)2 ⋅ 2Н2О в качестве допантов. Установлено, что при введении ZnО, когда в процессе взаимодействия компонентов выделяется незначительное количествo воды, в качестве промежуточного продукта образуется β-ТКФ. При введении Zn(H2PO4)2 ⋅ 2Н2О промежуточным продуктом является аморфный фосфат. Выявлена более высокая степень замещения при использовании в качестве допанта Zn(H2PO4)2 ⋅ 2Н2О по сравнению с ZnО.
Установлено, что при использовании в качестве допанта Zn(H2PO4)2 ⋅ 2Н2О при x ≤ 0.8 в процессе МХС образуется однофазный Zn-ГАП. При x ≥ 1 кроме Zn-ГАП образуется аморфная фаза. При МХС с использованием ZnО однофазный Zn-ГАП наблюдается при x ≤ 0.25. При более высоких концентрациях присутствует непрореагировавший ZnО.
Отжиг образцов, синтезированных с разной степенью замещения, приводит к изменению динамики параметров решетки, что, вероятно, связано с изменением положения ионов цинка в структуре Zn-ГАП.
Список литературы
Pokhrel S. Hydroxyapatite: Preparation, Properties and Its Biomedical Applications // Adv. Chem. Eng. Sci. 2018. V. 8. P. 225–240 https://doi.org/10.4236/aces.2018.84016
Баринов С.М., Комлев В.С. Биокерамика на основе фосфатов кальция. 2-е изд. М.: Наука, 2014. 201 с.
Захаров Н.А., Полунина И.А., Полунин К.Е., Ракитина Н.М., Кочеткова Е.И., Соколова Н.П., Калинников В.Т. Гидроксиапатит кальция как основа для создания новых лекарственных форм // Неорган. материалы. 2004. Т. 40. № 6. С. 735–743.
Elliott J.C. Structure and Chemistry of Apatite and Other Calcium Orthophosphates. Amsterdam: Elsevier, 1994. 389 p.
Begam H., Kumar N.S., Chanda A., Kundu B. Effect of Bone Morphogenetic Protein on Zn-HAp and Zn-HAp/ Collagen Composite: a Systematic in Vivo Study // Res. Veterinary Sci. 2017. V. 115. P. 1–9. https://doi.org/10.1016/j.rvsc.2017.01.012
Thian E.S., Konishi T., Kawanobe Y., Lim P.N., Choong C., Ho B., Aizawa. M. Zinc-Substituted Hydroxyapatite: a Biomaterial with Enhanced Bioactivity and Antibacterial Properties // J. Mater Sci: Mater. Med. 2013. V. 24. P. 437–445. https://doi.org/10.1007/s10856-012-4817-x
Šupová M. Substituted Hydroxyapatites for Biomedical Applications: a Review // Ceram. Int. 2015. V. 41. P. 9203–9231. https://doi.org/10.1016/j.ceramint.2015.03.316
Ptáček P. Synthetic Phase with the Structure of Apatite. // Apatites and Their Synthetic Analogues – Synthesis, Structure, Properties and Applications. INTECH. 2016. P. 177–244.
Bigi A., Foresti E., Gandolfi M., GazzanoM., Roveri N. Inhibiting Effect of Zinc on Hydroxylapatite Crystallization // J. Inorg. Biochem. 1995. V. 58. P. 49–58. https://doi.org/10.1016/0162-0134(94)00036-A
Kanzaki N., Onuma K., Treboux G., Tsutsumi S., Ito A. Inhibitory Effect of Magnesium and Zinc on Crystallization Kinetics of Hydroxyapatite (0001) Face // J. Phys. Chem. B. 2000. V. 104. P. 4189–4194. https://doi.org/10.1021/jp9939726
Gross K.A., Komarovska L., Viksna A. Efficient Zinc Incorporation into Hydroxyapatite through Crystallization of an Amorphous Phase Could Extend the Properties of Zinc Apatites // J. Australian Ceram. Soc. 2013. V. 49. № 2. P. 129–135.
Matos M., Terra J., Ellis Donald E. Mechanism of Zn Stabilization in Hydroxyapatite and Hydrated (001) Hurfaces of Hydroxyapatite // J. Phys. Condens. Matter. 2010. V. 22. № 14. Article N145502. https://doi.org/10.1088/0953-8984/22/14/145502
Tang Y., Chappell H.F., Dove M.T., Reeder R.J., Lee Y. Zinc Incorporation into Hydroxylapatite // Biomater. 2009. V. 30. P. 2864–2872. https://doi.org/10.1016/j.biomaterials.2009.01.043
Matsunaga K., Murata H., Mizoguchi T., Nakahira A. Mechanism of Incorporation of Zinc into Hydroxyapatite // Acta Biomaterialia. 2010. V. 6. P. 2289–2293. https://doi.org/10.1016/j.actbio.2009.11.029
Gomes S., Nedelec J.M., Jallot E., Sheptyakov D., Renaudin G. Unexpected Mechanism of Zn2+ Insertion in Calcium Phosphate Bioceramics // Chem. Mater. 2011. V. 23. P. 3072–3085. https://doi.org/10.1021/cm200537v
Gomes S., Nedelec J.M., Renaudin G. On the Effect of Temperature on the Insertion of Zinc into Hydroxyapatite // Acta Biomaterialia. 2012. V. 8. P. 1180–1189. https://doi.org/10.1016/j.actbio.2011.12.003
Karpov A.S., Nuss J., Jansen M., Kazin P. E., Tretyakov Y.D. Synthesis, Crystal Ctructure and Properties of Calcium and Barium Hydroxyapatites Containing Copper Ions in Hexagonal Channels // Solid State Sci. 2003. V. 5. P. 1277–1283. https://doi.org/10.1016/S1293-2558(03)00152-3
Фадеева И.В., Бакунова Н.В., Комлев В.С., Медвецкий Л., Фомин А.С., Гурин А.Н., Баринов С.М. Цинк- и серебросодержащие гидроксиапатиты: синтез и свойства // Доклады академии наук. 2012. Т. 442. № 6. С. 780–783.
Ito A., Ojima K., Naito H., Ichinose N., Tateishi T. Preparation, Solubility, and Cytocompatibility of Zinc-Releasing Calcium Phosphate Ceramics // J. Biomed. Mater. Res. 2000. V. 50. P. 178–183.
Nasiri-Tabrizi B., Fahami A., Ebrahimi-Kahrizsangi R., Ebrahimi F. New Frontiers in Mechanosynthesis: Hydroxyapatite- and Fluorapatite-Based Nanocomposite Powders // Nanocomposites – New Trends and Developments. INTECH. 2012. P. 259–297.
Avvakumov E., Senna M., Kosova N. Soft Mechanochemical Synthesis: A Basis for New Chemical Technologies. Kluwer: Academic Publishers, 2001. 207 p.
Adzila S., Azmil K., Radde R., Hassan M. F., Arifin A.M.T., Yunos M.Z., Rahman M.N.A., Haq R.H.A. Effect of Zinc Doped Calcium Phosphate Trough Mechanochemical Synthesis // ARPN J. Eng. Appl. Sci. 2015. V. 10. № 15. P. 6246–6249.
Аввакумов Е.Г., Поткин Ф.З., Самарин О.И. Планетарная мельница: Пат. 975068 РФ. В 02 С 17/08. / БИ № 43, 1982.
Булина Н.В., Чайкина М.В., Просанов И.Ю. Механохимический синтез Sr-замещенного гидроксиапатита // Неорган. материалы. 2018. № 8. С. 866–872.
Petrov I., Šoptrajanov B., Fuson N., Lawson J.R. Infra-Red Investigation of Dicalcium Phosphates // Spectrochim. Acta. A. 1967. V. 23. № 10. P. 2637–2646. https://doi.org/10.1016/0584-8539(67)80155-7
Calvo C., Gopal R. The Crystal Structure of Whitlockite from the Palermo Quarry // Am. Mineral. 1975. V. 60. P. 120–133.
Kwon Y.-S., Gerasimov K.B., Yoon S.-K. Ball Temperatures During Mechanical Alloying in Planetary Mills // J. Alloys Compd. 2002. V. 346. P. 276–281. https://doi.org/10.1016/S0925-8388(02)00512-1
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы