

Неорганические материалы, 2021, T. 57, № 7, стр. 796-802
Исследование радиационной и гидролитической устойчивости керамики на основе фосфата Y0.95Gd0.05PO4 со структурой ксенотима
Д. А. Михайлов 1, *, Е. А. Потанина 1, А. И. Орлова 1, А. В. Нохрин 1, М. С. Болдин 1, О. А. Белкин 1, Н. В. Сахаров 1, В. А. Скуратов 2, 3, 4, Н. С. Кирилкин 2, В. Н. Чувильдеев 1
1 Нижегородский государственный университет им. Н.И. Лобачевского
603022 Нижний Новгород, пр. Гагарина, 23, Россия
2 Объединенный институт ядерных исследований
141980 Дубна, ул. Жолио-Кюри, 6, Россия
3 Национальный исследовательский ядерный университет “МИФИ”
115409 Москва, Каширское ш., 31, Россия
4 Государственный университет “Дубна”
141982 Дубна, ул. Университетская, 19, Россия
* E-mail: dmitry.mikhailov@mail.ru
Поступила в редакцию 09.12.2020
После доработки 16.03.2021
Принята к публикации 16.03.2021
Аннотация
Фосфат Y0.95Gd0.05PO4 со структурой ксенотима синтезировали в виде порошка и керамики. Керамику c относительной плотностью ~99% получили методом электроимпульсного плазменного спекания. Максимальная температура спекания составила tmax = 1140°С, общее время процесса спекания – ~18 мин, стадия изотермической выдержки отсутствовала. Исследовали радиационную устойчивость керамики при облучении ионами 132Xe26+ и восстановление образовавшейся метамиктной фазы до кристаллической путем высокотемпературной обработки. Полная аморфизация образцов при примененных флюенсах не достигнута. Расчетное значение критического флюенса составило (9.2 ± 0.1) × 1014 см–2, рассчитанное значение радиуса латентного трека ~2.8 нм. Гидролитические испытания показали, что исследуемый фосфат стабилен в воде в динамических условиях при повышенной температуре. Достигнутые скорости выщелачивания Y и Gd имели значения Ri = = 1.68 × 10–6 и 1.5 × 10–7 г/(см2 сут) соответственно.
ВВЕДЕНИЕ
Минерал ксенотим имеет тетрагональную кристаллическую решетку (пр.гр. I41/amd) и описывается общей формулой RPO4, где R = Ln с к. ч. = 8 [1]. В структуру ксенотима включаются лантаноиды от Tb до Lu, а также редкоземельные элементы Y и Sc [1]. К настоящему времени в литературе описаны соединения, содержащие Pu, Cm, Np [2–4]. Это позволяет рассматривать фосфатные соединения со структурой ксенотима в качестве матрицы для консолидации актиноидов [5, 6].
Природные соединения ксенотима могут содержать Th и U и при этом обладать высокой устойчивостью к радиационным повреждениям, так как доза облучения природного ксенотима может достигать значения (1.4–14) × 1016 α/мг [7]. Известно, что синтетические кристаллы LuPO4, содержащие 1.0 мас. % 244Cm, за 18 лет выдержки накапливали дозу 5 × 1016 α/мг и при этом оставались в высококристаллическом состоянии [7]. Влияние ионной имплантации (Au–, 2 МэВ, дозы: 1 × 1014, 5 × 1014, 1 × 1015 ион/см2) изучали на фосфатах La1 – xYbxPO4 (x = 0–1.0) [8]. Образцы всех составов были подвержены структурным повреждениям, а образцы с x = 0.7 и 1.0 частично рекристаллизовались после облучения максимальной дозой. Образцы при всех х восстанавливались от повреждений после отжига при 300°С.
В работе [9] при исследовании облучения керамики ErPO4 ионами Au и He установлено, что при одновременном облучении ионы He частично препятствовали аморфизации образца.
Керамические образцы с относительной плотностью 96–98% на основе соединений со структурой ксенотима получали прессованием порошков с последующим спеканием [9–11]. Температуры спекания были высокими: ~1300–1600°С, время спекания составляло 1–5 ч.
С точки зрения радиационной безопасности, материал, содержащий компоненты радиоактивных отходов, должен быть скомпактирован до плотности, близкой к теоретической, при пониженной температуре и за минимальное время.
Одним из перспективных методов получения керамик для радиохимических приложений является высокоскоростное электроимпульсное (“искровое”) плазменное спекание (ЭИПС) [12, 13]. Опыт показывает, что технология ЭИПС позволяет получать минералоподобные керамики с высокой относительной плотностью за малые времена, часто – при пониженных температурах [14].
Целью настоящей работы являлось исследование керамики на основе соединения со структурой ксенотима Y0.95Gd0.05PO4, полученной методом ЭИПС, изучение ее радиационной и гидролитической устойчивости. Gd3+ выбрали в качестве имитатора Cm3+ ввиду схожести их электронных конфигураций и ионных радиусов, а следовательно, химических и физических свойств.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Фосфаты Y1 – хGdхPO4 (структурный тип ксенотима) получали золь–гель-методом. В качестве исходных реагентов использовали Y(NO3)3·6H2O, Gd2O3 (растворяли в избытке слабокислого раствора (pH 4–5) HNO3) и дигидрофосфат аммония NH4H2PO4 в качестве осадителя. Добавление 1 М раствора NH4H2PO4 проводили по каплям при постоянном перемешивании. Полученный гель дополнительно интенсивно перемешивали в течение 5 мин до полной гомогенизации и высушивали при 90°С в течение суток. Сухой остаток диспергировали в агатовой ступке, затем поэтапно нагревали при 600, 700, 800 и 900°С в течение 5 ч на каждой стадии без промежуточного диспергирования.
Керамические образцы диаметром 10 и высотой 3–5 мм получали методом ЭИПС с помощью установки Dr. Sinter model SPS-625. Спекание проводили в вакууме в графитовых пресс-формах при давлении p = 70 МПа путем нагрева со скоростью 200°С/мин до 560°С и далее до 1140°С со скоростью 50°С/мин. Выдержка при температуре спекания отсутствовала, общее время процесса спекания составило ~18 мин.
Фазовый состав порошков и керамик, а также долю метамиктной фазы определяли методом рентгенофазового анализа (РФА) на дифрактометре Shimadzu LabX XRD-6000 с применением программного обеспечения Match! и PhasanX v.2.0. Для расчета параметров кристаллической решетки и рентгенографической плотности использовали программное обеспечение KRIST. Микроструктуру образцов исследовали на растровом электронном микроскопе JEOL JSM-6490 с рентгеновским микроанализатором Oxford Instruments INCA-350.
Плотность образцов (ρ) измеряли методом гидростатического взвешивания в дистиллированной воде при помощи весов Sartorius CPA.
Радиационные испытания проводили на циклотроне ИЦ-100 Лаборатории ядерных реакций Объединенного института ядерных исследований (г. Дубна). В ходе экспериментов керамики облучали многозарядными ионами Xe26+ с энергией 167 МэВ до флюенсов в интервале 1 × 1012–3 × × 1013 см–2.
Гидролитические исследования керамических образцов Y0.95Gd0.05PO4 проводили в динамическом режиме, используя аппарат Сокслета в соответствии с ГОСТ Р 52126-2003. Испытания проводили в дистиллированной воде при 90°С в течение 28 дней. Анализ воды на содержание ионов Y3+ и Gd3+ выполняли на атомно-абсорбционном спектрофотометре Shimadzu AA-7000F.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
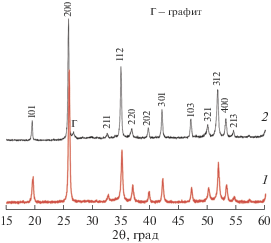
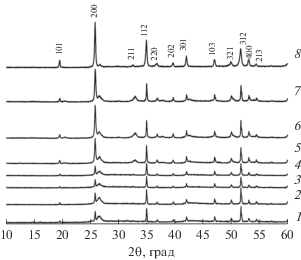
Согласно данным РФА (рис. 1), синтезированный порошок представлял собой однофазный фосфат Y0.95Gd0.05PO4 (ICDD PDF #83-0658, пр. гр. I41/amd, a = b = 6.8905 ± 0.0003 Å, c = = 6.0227 ± 0.0004 Å, α = β = γ = 90°). Рентгенографическая плотность (ρth) исследуемой фазы составила 4.349 ± 0.001 г/см3.
Рис. 1.
Дифрактограммы порошка Y0.95Gd0.05PO4 после синтеза при температуре 900°C (5 ч) (1) и керамики на его основе (2).
Электронные микрофотографии синтезированного порошка Y0.95Gd0.05PO4 представлены на рис. 2. Видно, что частицы порошка крупные (от 10 до 150 мкм), имеют неправильную ограненную форму (рис. 2а) и на их поверхности присутствуют наночастицы размером 100–300 нм.

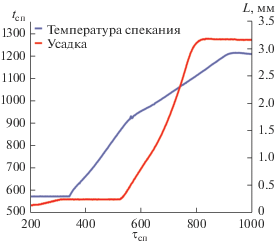
Характерный вид зависимости усадки (L) порошков Y0.95Gd0.05PO4 от времени процесса спекания представлен на рис. 3. Наиболее интенсивная усадка порошков при ЭИПС происходила в интервале 860–1140°C. Относительная плотность спеченных керамических образцов составила 99.9%. Стоит отметить, что температура ЭИПС керамики из порошка со структурой ксенотима была ниже, чем в работах [9–11], а характерное время процесса – намного меньше, чем в [9–11]. Полученный результат свидетельствует о высокой технологичности метода ЭИПС для получения керамики на основе соединения Y0.95Gd0.05PO4 со структурой ксенотима.
Фазовый состав керамических образцов был аналогичен фазовому составу исходных порошков (рис. 1).
Микроструктура изломов керамических образцов приведена на рис 4. Видно, что керамика имеет высокоплотную мелкозернистую структуру. Основная объемная доля зерен керамики имеет размер от 5 до 15 мкм (рис. 4а), при этом встречаются участки мелкозернистой структуры с размером зерна ~1–2 мкм, в которых попадаются единичные поры (рис. 4б). На некоторых участках присутствуют крупные зерна, свидетельствующие об аномальном росте зерен в процессе спекания керамики. Наличие аномально крупных зерен является, по нашему мнению, следствием присутствия крупных частиц и агломератов в составе синтезированных порошков (см. рис. 2).

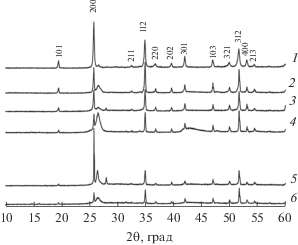
Керамические образцы облучали ионами Xe26+ с флюенсами Ф = 1 × 1012, 3 × 1012, 7 × 1012, 1 × 1013, 3 × 1013 см–2. На рис. 5 представлены результаты РФА облученных образцов. Уменьшение интенсивности дифракционных максимумов, связанное с частичной аморфизацией структуры, наблюдалось уже при флюенсе 1 × 1012 см–2. При флюенсе 1 × 1013 см–2 наблюдается возрастание интенсивности рефлексов по сравнению с флюенсом 7 × × 1012 см–2, что может быть связано с частичной рекристаллизацией образца, обусловленной релаксационным эффектом непосредственно после воздействия тяжелого иона на кристаллическую структуру фосфата [15], но данный эффект в подобном соединении необходимо исследовать подробнее. Полная аморфизация образца Y0.95Gd0.05PO4 при облучении не зафиксирована.
Рис. 5.
Дифрактограмма керамики Y0.95Gd0.05PO4 после облучения ионами Xe26+ с флюенсами: 0 (1), 1 × × 1012 (2), 3 × 1012 (3), 7 × 1012 (4), 1 × 1013 (5), 3 × × 1013 см–2 (6).
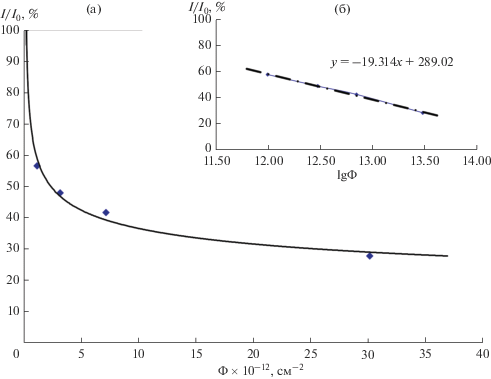
Критический флюенс для исследуемого соединения рассчитывали теоретически, путем анализа зависимости относительной интенсивности (по отношению к интенсивности необлученного образца I0) главного дифракционного максимума соединения 200 от величины флюенса облучения. Результаты расчетов представлены на рис. 6а. Расчетная величина критического флюенса, определенная путем линейной интерполяции зависимости I/I0–Ф в логарифмических координатах (рис. 6б), составила (9.2 ± 0.1) × 1014 см–2.
Рис. 6.
Зависимость относительной интенсивности рефлекса 200 от флюенса в обычных (а) и логарифмических (б) координатах.
Пользуясь рентгенографическими данными облученных образцов, также можно рассчитать оценочный радиус отдельного латентного трека в рамках модели одиночного столкновения применительно к облучению заряженными тяжелыми ионами [16–18]. Идея такой оценки состоит в том, что каждый падающий на поверхность образца тяжелый ион создает отдельный трек, а наблюдаемые на рентгенограммах повреждения (аморфная часть) являются результатом перекрытия множества таких треков [17].
Величина поврежденной (аморфной) части может быть вычислена по данным РФА с помощью следующего уравнения [17, 18]:
Зависимость FD от флюенса выражается уравнением
где Fsat – доля аморфизированной фазы; S – область, окружающая траекторию иона, в которой произошла аморфизация; Ф – флюенс иона. Радиус трека может быть вычислен как R = (S/π)1/2.Расчет FD проводили по рефлексам: 200, 112, 301 и 312. Графическая зависимость FD от флюенса приведена на рис. 7.
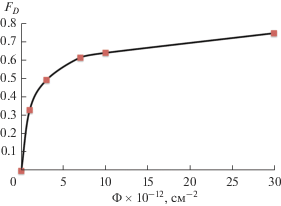
Расчетное значение радиуса латентного трека составило ~2.8 нм.
Восстановление метамиктной фазы образца, облученного максимальным флюенсом 3 × 1013 см–2, проводили при последовательном нагревании от 200 до 700°C в течение 3 ч при каждой из температур и РФА после каждой стадии (рис. 8). Из полученных данных следует, что восстановление кристаллической фазы происходило уже после нагревания при 500°C (общее время нагрева 15 ч). Дальнейший нагрев при 600°C приводил к увеличению интенсивности рефлексов, а после отжига при 700°C (время отжига 18 ч) интенсивность дифракционных максимумов восстановленного образца достигла ~80% от I0.
Рис. 8.
Дифрактограммы керамики непосредственно после облучения (1) и после отжигов при 200 (2), 300 (3), 400 (4), 500 (5), 600 (6) и 700°С (7); дифрактограмма 8 – необлученный образец.
Таким образом, полученные керамики радиационно-устойчивы в изучаемых условиях и способны выдерживать облучение потоками ионов с высокими флюенсами, не претерпевая полной аморфизации, и восстанавливаться до высококристаллического состояния при нагревании.
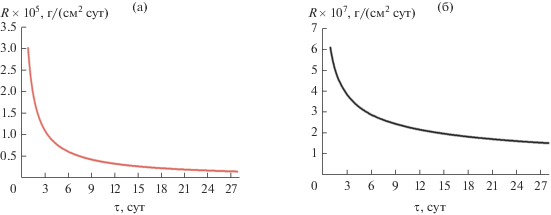
В ходе гидролитических испытаний необлученного керамического образца Y0.95Gd0.05PO4 определяли выщелачиваемость ионов Y3+ и Gd3+ из матрицы. По полученным результатам построили графики скоростей выщелачивания Ri ионов Y и Gd (рис. 9). Обобщение представленных результатов показывает, что с увеличением времени испытания скорость выщелачивания снижается. Минимальные значения скоростей выщелачивания на 28 сут составили 1.68 × 10–6 и 1.51 × × 10–7 г/(см2 сут) для Y и Gd соответственно, что свидетельствует о высокой гидролитической стойкости исследуемого соединения.
ЗАКЛЮЧЕНИЕ
Фосфат Y0.95Gd0.05PO4, где Gd выступал в роли имитатора Cm, в виде порошка был получен золь–гель-методом. Керамические образцы с высокой относительной плотностью (более 99%) получили методом ЭИПС за время ~18 мин.
Образцы керамик демонстрируют высокую стойкость при облучении ионами Xe (E = 167 МэВ). При максимальном флюенсе 3 × 1013 см–2 вещество частично сохранило кристалличность. Расчетное значение флюенса, приводящего к полной аморфизации, составило (9.2 ± 0.1) × 1014 см–2, расчетный радиус латентных треков – 2.8 нм. Восстановление метамиктной фазы происходило после нагревания при 500°С; после нагрева до 700°С степень восстановления достигала ~80%.
Керамика на основе фосфата Y0.95Gd0.05PO4 имела высокую гидролитическую устойчивость в динамической режиме (28 сут, 90°С, испытания в дистиллированной воде). Достигнутые минимальные скорости выщелачивания составили 1.68 × 10–6 и 1.51 × 10–7 г/(см2 сут) для ионов Y и Gd соответственно.
Список литературы
Ni Y., Hughes J.M., Mariano A.N. Crystal Chemistry of the Monazite and Xenotime Structures // Am. Mineral. 1995. V. 80. P. 21–26. https://doi.org/10.2138/am-1995-1-203
Burakov B.E., Ojovan M.I., Lee W.E. Crystalline Materials for Actinide Immobilization // Materials for Engineering. V. 1. L.: Imperial College, 2010. 216 p. https://doi.org/10.1142/p652
Liu G.K., Lia S.T., Beitza J.V., Abraham M. Effects of Self-radiation Damage on Electronic Properties of 244Cm3+ in an Orthophosphate Crystal of YPO4 // J. Alloys Compd. 1998. V. 271–273. P. 872–875. https://doi.org/10.1016/S0925-8388(98)00237-0
Vance E.R., Zhang Y., McLeod T., Davis J. Actinide Valences in Xenotime and Monazite // J. Nucl. Mater. 2011. V. 409. P. 221–224. https://doi.org/10.1016/j.jnucmat.2010.12.241
Structural Chemistry of Inorganic Actinide Compounds / Eds. Krivovichev S.V., Burns P.C., Tananaev I.G. Amsterdam: Elsevier, 2007. 494 p. https://doi.org/10.1016/B978-0-444-52111-8.X5000-5
Emden B., Thornber M.R., Graham J., Lincoln F.J. The Incorporation of Actinides in Monazite and Xenotime from Placer Deposits in Western Australia // Can. Mineral. 1997. V. 35. № 1. P. 95–104.
Lumpkin G.R., Geisler-Wierwille T. Minerals and Natural Analogues // Comprehensive Nuclear Materials / Ed Konings R.J.M. Amsterdam: Elsevier, 2012. V. 5. P. 563–600.
Rafiuddin M.R., Grosvenor A.P. Probing the Effect of Radiation Damage on the Structure of Rare-Earth Phosphates // J. Alloys Compd. 2015. V. 653. P. 279–289. https://doi.org/10.1016/j.jallcom.2015.08.276
Rafiuddin M.R., Seydoux-Guillaume A.-M., Deschanels X. et al. An in-situ Electron Microscopy Study of Dual Ion-beam Irradiated Xenotime-Type ErPO4 // J. Nucl. Mater. 2020. V. 539. P. 152265. https://doi.org/10.1016/j.jnucmat.2020.152265
Hikichi Y., Ota T., Daimon K., Hattori T. Thermal, Mechanical, and Chemical Properties of Sintered Xenotime-Type RPO4 (R = Y, Er, Yb, or Lu) // J. Am. Ceram. Soc. 1998. V. 81. № 8. P. 2216–2218. https://doi.org/10.1111/j.1151-2916.1998.tb02613.x
Cho I.-S., Choi G.K., An J.-S. et al. Sintering, Microstructure and Microwave Dielectric Properties of Rare Earth Orthophosphates, RePO4 (Re = La, Ce, Nd, Sm, Tb, Dy, Y, Yb) // Mater. Res. Bull. 2009. V. 44. P. 173–178. https://doi.org/10.1016/j.materresbull.2008.03.016
Havette J., Iltis X., Palancher H. et al. Spark Plasma Sintering as an Innovative Process for Nuclear Fuel Plate Manufacturing // J. Nucl. Mater. 2021. V. 543. 152541https://doi.org/10.1016/j.jnucmat.2020.152541
Luo F., Tang H., Shu X. et al. Immobilization of Simulated An3+ into Synthetic Gd2Zr2O7 Ceramic by SPS without Occupation or Valence Design // Ceram. Int. 2021. V. 47. № 5. P. 6329–6335. https://doi.org/10.1016/j.ceramint.2020.10.211
Orlova A., Volgutov V., Mikhailov D. et al. Phosphate Ca1/4Sr1/4Zr2(PO4)3 of the NaZr2(PO4)3 Structure Type: Synthesis of a Dense Ceramic Material and its Radiation Testing // J. Nucl. Mater. 2014. V. 446. № 1–3. P. 232–239https://doi.org/10.1016/j.jnucmat.2013.11.025
Rymzhanov R.A., Medvedev N., O’Connell J.H. et al. Recrystallization as the Governing Mechanism of Ion Track Formation // Sci. Rep. 2019. V. 9. Article № 3837. https://doi.org/10.1038/s41598-019-40239-9
Gibbons J.F. Ion Implantation in Semiconductors–Part II: Damage Production and Annealing // Proc. IEEE. 1972. V. 60. № 9. P. 1062–1096. https://doi.org/10.1109/PROC.1972.8854
Moll S., Sattonnay G., Thomé L. et al. Swift Heavy Ion Irradiation of Pyrochlore Oxides: Electronic Energy Loss Threshold for Latent Track Formation // Nucl. Instrum. Methods Phys. Res., Sect. B. 2010. V. 268. № 19. P. 2933–2936.https://doi.org/10.1016/j.nimb.2010.05.012
Janse van Vuuren A., Ibrayeva A., Rymzhanov R.A. et al. Latent Tracks of Swift Bi Ions in Si3N4 // Mater. Res. Express. 2020. V. 7. № 2. Article № 025512. https://doi.org/10.1088/2053-1591/ab72d3
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы