

Неорганические материалы, 2022, T. 58, № 10, стр. 1118-1125
Особенности стеклообразования и кристаллизация стекол в системе СdO–В2O3–SiO2
А. Ю. Колобов 1, Е. А. Семенова 2, Г. А. Сычева 2, *
1 ОАО “ДИНУР”,
623103 Свердловская обл., Первоуральск, ул. Ильича, 1, Россия
2 Институт химии силикатов им. И.В. Гребенщикова Российской академии
наук
199034 Санкт-Петербург, наб. Макарова, 2, Россия
* E-mail: sycheva_galina@mail.ru
Поступила в редакцию 28.03.2022
После доработки 30.05.2022
Принята к публикации 17.06.2022
- EDN: WYQYRZ
- DOI: 10.31857/S0002337X22100086
Аннотация
Исследовано стеклообразование и кристаллизация стекол в системе СdO–В2O3–SiO2. Стекла получены в диапазоне составов с содержанием CdO от 21.12 до 87.00 мол. %. Для системы СdO–В2O3–SiO2 фазовая диаграмма отсутствует, поэтому режимы синтеза выбирали на основе существующих фазовых диаграмм тройных щелочноземельных боросиликатных систем, а также подбирали эмпирическим путем. Установлена область стеклообразования в системе СdO–В2O3–SiO2 и идентифицированы кристаллические фазы, образующиеся при выработке стекол.
ВВЕДЕНИЕ
Боратные стекла привлекают к себе повышенное внимание, во-первых, в связи с обнаружением нелинейно-оптических свойств у многих кристаллов, полученных на их основе [1]. Кадмиевоборатные составы используются для получения “лантановой” оптики, создавая уникальность оптических постоянных [2]. Во-вторых, они представляют большой интерес для практики, так как являются основой для синтеза стекол, керамики и огнеупорных материалов. Изучение областей стеклообразования в этих системах актуально также в связи с проблемами захоронения радиоактивных отходов в боросиликатные матрицы. Одно из первых упоминаний о системе СdO–В2O3 приведено в работе [3, C. 55]. Именно там приведена таблица (рис. 1), в которой среди прочих указана информация о СdO–В2O3. Рядом мелким шрифтом в скобках указана цифра 156, смысл которой раскрывается на стр. 76 в разделе “Литература”: Mazzetti and de Carli // Gazzetta chimica italiana. 1926. T. 56 II. P. 19–29. Таким образом, одними из первых исследователей, обративших внимание на кадмиевоборатную систему, были итальянские ученые Мазетти и де Карли. Затем после долгого перерыва в 1956 г. выходит работа Субарро и Хаммеля [4], посвященная изучению системы СdO–В2O3; в 1962 г. – Хэнда и Крог–Му [5]: новые данные о кадмиевоборатной системе; а в 1964 г. – Хаяши, Накаяма и др. [6]: фазовые превращения в кадмиевом оливине. Систематический анализ накопленного к настоящему времени экспериментального материала о стеклообразовании в системе СdO–В2O3–SiO2 представляет собой достаточно трудную задачу, что можно проиллюстрировать на некоторых примерах. В литературе представлены физико-химические свойства отдельных составов стекол этой системы. О стеклообразовании в двойных системах можно составить представление по данным электронного информационного справочника [7] из разделов, посвященных определению температур стеклования и плотностей стекол СdO–В2O3 и СdO–SiO2, существующих в достаточном объеме. Отдельные составы системы СdO–В2O3–SiO2 приведены в [8]. Однако содержание СdO, В2O3, SiO2 в них не анализировалось, составы приводятся по синтезу. Из экспериментальных работ авторов по синтезу стекол в боратных системах следует, что в процессе их синтеза происходит улетучивание боратной составляющей, что ведет к изменению содержания СdO и SiO2 в синтезированных стеклах.
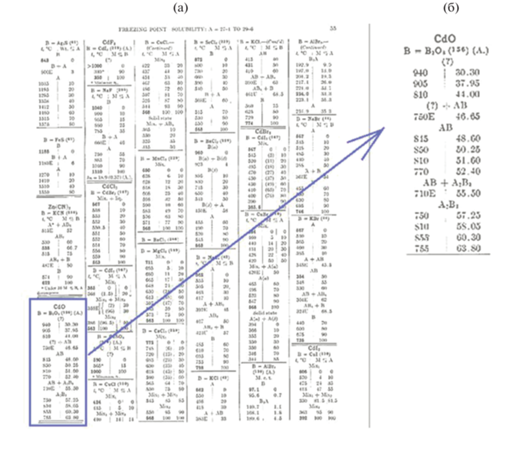
Целью настоящего исследования было проведение синтеза кадмиевоборосиликатных стекол, их химический анализ, установление области стеклообразования в системе CdO–B2O3–SiO2 и идентификация кристаллических фаз, образующихся при выработке стекол.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методы исследования и аппаратура. Химический анализ проводился по классическим методикам “мокрой химии”. Рентгенофазовый анализ (РФА) выполняли на установках ДРОН-7 и “Дифрей-401”. Оптическая микроскопия в отраженном свете осуществлялась на микроскопе Neophot 32, в проходящем – на микроскопе Jenaval (оба производства фирмы Carl Zeiss, Jena, Германия).
Для синтеза стекол использовали реактивы квалификаций “ос. ч.” и “х. ч.”: карбонат кадмия (СdCO3), борную кислоту (H3ВO3) и аморфный диоксид кремния (SiO2). Количество исходного материала для приготовления шихты рассчитывали на 75 г готового продукта. При синтезе щелочноборатных стекол рекомендуется температуру синтезу выбирать выше температуры ликвидуса на фазовой диаграмме в среднем на 50°С, однако фазовая диаграмма для системы CdO–В2O3–SiO2 не найдена. Варку стекол проводили в платиновом тигле в силитовой печи при температурах синтеза 850–1250°С в течение 1–2 ч в зависимости от заданного состава. Выработку стекол выполняли методом “молота и наковальни”: быстрая закалка на металлическую плиту с последующим прижатием расплава молотом. Сразу после выработки свежеприготовленные стекла из-за их гигроскопичности помещали в эксикатор, в который добавляли пентаоксид фосфора и гидроксид калия для поглощения углекислоты и влаги воздуха.
На рис. 2 приведен внешний вид синтезированных стекол. Видно, что образцы 1–6 – прозрачные с легким желтоватым оттенком. В образцах 7 и 8 наблюдается частичная кристаллизация, образцы 9 и 10 полностью закристаллизованы, образец 10 разрушается в процессе выработки.

Определение содержания компонентов в исследуемых стеклах. Для определения содержания компонентов в исследуемых стеклах использовали классические методы “мокрой химии”: гравиметрический (кремний) [9], комплексонометрический (кадмий) [10] и потенциометрический (бор) [11] методы анализа.
Определение содержания кремния. Кремний определяли в виде хинолята кремнемолибденовой кислоты после сплавления навески образца с содой и борной кислотой и последующего растворения плава в HCl [9]. Погрешность определения кремния составила ±0.4 отн. %.
Определение содержания кадмия. В литературе предлагается много разнообразных методов комплексонометрического титрования кадмия [10, 11]. Выбор того или иного способа зависит в первую очередь от состава анализируемого материала. Отсутствие в образцах мешающих элементов, вступающих в реакцию с комплексоном III, позволяет успешно проводить титрование кадмия при pH 5 как прямым титрованием комплексоном III, так и обратным титрованием его избытка. Предварительные испытания показали, что второй способ предпочтительнее, так как в этом случае переход окраски индикатора более четкий, что позволяет получить точные результаты.
Содержание кадмия определяли в том же растворе, что и кремния. Кадмий образует достаточно прочный комплекс с комплексоном III в широком диапазоне pH. Поэтому определение кадмия проводили следующим образом: к аликвотной порции раствора добавляли избыток раствора комплексона III, буферный раствор с рН 5 и индикатор – ксиленоловый оранжевый. Титрование проводили раствором сернокислого цинка до перехода окраски из желтой в персиковую. Погрешность составляла ±1 отн. %.
Определение содержания бора. Для определения бора отдельную навеску стекла сплавляли с содой и растворяли в соляной кислоте. Для устранения влияния мешающих ионов авторы [12] предлагали добавлять комплексон III при растворении плава в незначительном избытке, так как большой его избыток завышает результаты. Наши предварительные испытания показали, что предпочтительнее добавлять комплексон III непосредственно в титруемый раствор. Было установлено, что его избыток не должен превышать 0.5 мл, так как в противном случае скорость титрования замедляется и возможно искажение результатов.
Определение бора проводили следующим образом: к аликвотной порции раствора добавляли рассчитанный по отношению к кадмию в анализируемом растворе объем комплексона III с небольшим превышением необходимого количества, но не более 0.5 мл. Нейтрализовали раствором гидроксида натрия примерно до рН 5 и кипятили в течение 2–3 мин для удаления углекислого газа. Охлажденный раствор нейтрализовали раствором гидроксида натрия до рН 6.9. Затем добавляли маннит и образовавшуюся борноманнитовую кислоту титровали раствором гидроксида натрия до рН 6.9. Величину рН устанавливали с помощью рН-метра (рН-метр–миливольтметр рН-673.М). Погрешность составляет ±0.4 отн. %.
Составы полученных стекол по анализу приведены в табл. 1.
Таблица 1.
Результаты химического анализа стекол
| Образец | С, мол. % | ||
|---|---|---|---|
| CdO | B2O3 | SiO2 | |
| 1 | 21.12 | 11.53 | 67.35 |
| 2 | 27.81 | 6.61 | 65.58 |
| 3 | 34.47 | 23.46 | 42.07 |
| 4 | 37.45 | 9.53 | 53.01 |
| 5 | 40.28 | 10.48 | 49.24 |
| 6 | 47.50 | 29.40 | 23.09 |
| 7 | 48.44 | 14.71 | 36.86 |
| 8 | 48.92 | 10.45 | 40.64 |
| 9 | 85.44 | 14.56 | – |
| 10 | 87.00 | 13.00 | – |
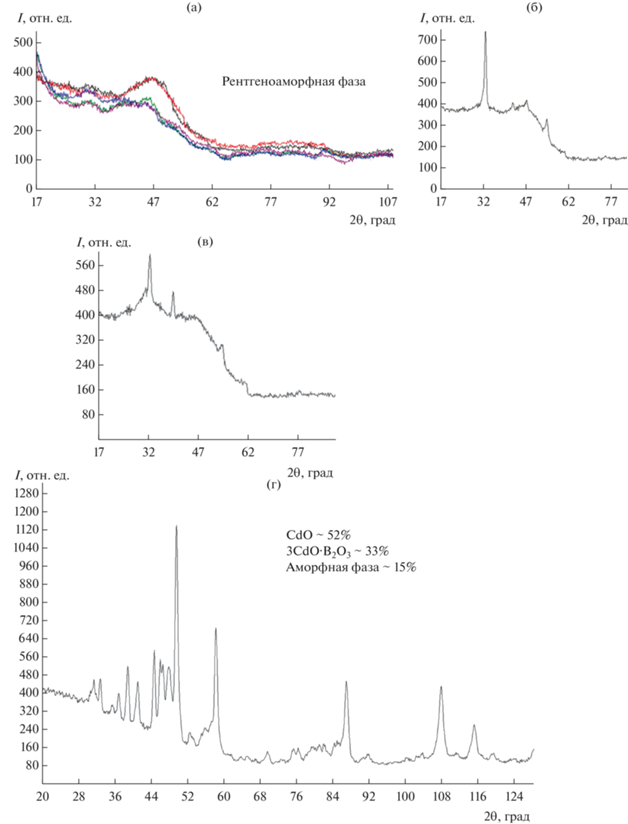
РФА синтезированных стекол. РФА показал, что образцы 1–6 рентгеноаморфны. В образцах стекол 7, 8 наблюдается некоторое количество кварца и (или) кристобалита. На рентгенограммах образцов 9 и 10 присутствовали четкие кристаллические пики кварца и (или) кристобалита (рис. 3). Кроме того, образцы синтезированных стекол просматривали в оптических микроскопах NEOPHOT-32 (в отражающем свете) и JENAVAL (в проходящем свете). Рентгеноаморфные образцы 1–6 были оптически прозрачны, в образцах 7, 8 наблюдались единичные кристаллы кварца и кристобалита.
Измерения массовой доли кварца и (или) кристобалита в исследуемых пробах выполняли с помощью РФА методом внешнего стандарта.
Метод количественного определения кристобалита и кварца, использованный в работе, основан на измерении интенсивности дифракционного отражения в зависимости от содержания остаточного кварца и (или) кристобалита. В общем виде задача количественного определения остаточного кварца (кристобалита) представляет собой несложную задачу РФА.
Подготовка проб для РФА. Полученные пробы измельчали до полного прохождения через сетку с номером 0063 по ГОСТ 6613 [13] в агатовых ступках, исключая загрязнение пробы. Пробы материала перед проведением анализа высушивали в сушильном шкафу при 105–110°С (точность поддержания температуры ±5°С). После высушивания пробы охлаждали в эксикаторе до комнатной температуры. После истирания в агатовой ступке пробу материала засыпали в лунку кюветы, набивали, припрессовывали и смачивали спиртом. При этом достигается значительная разориентация, т. к. быстрое улетучивание спирта препятствует возникновению преимущественной ориентации кристалликов образца. Измерения и обработку результатов проводили в соответствии с руководствами по эксплуатации [14, 15]. Съемку рентгенограммы проводили в диапазоне углов 2θ от 10° до 80°, качественный анализ исследуемых образцов выполняли, используя базу порошковых дифракционных данных PDF-2.
Определение количества кварца и кристобалита в исследуемых образцах. Для дифрактометра ДРОН-7 устанавливали следующий режим работы рентгеновской трубки: U = 40 кВ, I = 30 мА. Воспользовавшись программой модифицированного сбора, задавали следующие параметры съемки дифрактограмм для определения содержания кварца и кристобалита на дифрактометре ДРОН-7 (элемент анода Cu, элемент фильтра Ni): угол сканирования 2θ для кристобалита 20.6°–22.6°, для кварца 26.0°–27.2°; шаг сканирования 0.02°; время экспозиции в одной точке 10 с.
Для дифрактометра “Дифрей-401” (элемент анода Cr) угол рентгеновской трубки 2θ выставляли на 15°, угол детектора – на 20°. Напряжение на рентгеновской трубке U = 20–25 кВ, ток I = 5–10 мА выставляли в зависимости от конкретных задач съемки, содержания кварца, кристобалита и аморфной фазы в исследуемом образце после приблизительной их оценки.
На одной пробе проводили съемку двух дифрактограмм в идентичных условиях. После завершения измерений при помощи программы предварительной обработки вычитали фон и определяли интегральную интенсивность пика.
Определение массовой доли кварца и (или) кристобалита в исследуемых пробах проводили методом внешнего стандарта при помощи программы количественного анализа в следующей последовательности: загружали в программу Data Collection для ДРОН-7 файл полученных первичных данных; вводили параметры геометрии съемки; запускали процесс обработки данных; записывали полученные результаты измерений фазового состава и содержания (мас. доли) остаточного кварца (кристобалита) в образце.
Содержание кварца и (или) кристобалита в исследуемых пробах рассчитывали по формуле
где Wэт – содержание фазы в эталоне, %; Iпр, Iэт – интегральные интенсивности фазы в исследуемой пробе и эталоне соответственно, отн. ед.В расчетах использовали следующие эталоны: для кварца – кварц молотый пылевидный с массовой долей оксида кремния не менее 99.0% по ГОСТ 9077 [16]; для кристобалита – кварцевое стекло, обожженное при температуре 1600°С с выдержкой 4 ч. Для количественной оценки содержания кристобалита в кварцевом стекле был выбран наиболее интенсивный кристобалитовый рефлекс 101 (d = 4.04 Å). Для получения точных результатов интегральную интенсивность эталона кварца или кристобалита измеряли 1 раз в неделю. Это позволило с высокой точностью определить содержание кварца и кристобалита в образцах 7 (48.44CdO⋅14.71В2O3⋅36.86SiO2) и 8 (48.92CdO⋅10.45В2O3⋅40.64SiO2). В образце 7 содержание кристобалита составило 4 мас. %, в образце 8 кристобалита – 2 мас. % и кварца – 1 мас. %.
Расчет содержания аморфной фазы. Хотя рентгеновская дифрактометрия не предназначена для определения содержания аморфной фазы, без учета последней любое заключение по РФА будет неинформативно [17]. Можно сформулировать по крайней мере три непрямых подхода для расчета содержания аморфной фазы.
Аморфную фазу можно рассчитать, во-первых, как разность 100% и суммы кристаллических фаз в образце:
(2)
$\begin{gathered} \sum {\left( {{\text{аморфных фаз}}} \right)} = \\ = \,\,100\% {\text{ }}--\sum {\left( {{\text{кристаллических фаз}}} \right)} . \\ \end{gathered} $Каждую из кристаллических фаз в отдельности, если есть возможность, можно определить с использованием метода внешнего стандарта, в нашем случае для образцов 7 и 8, – это эталонный образец кварца или кристобалита со 100%-ным содержанием чистой фазы.
Во-вторых, можно воспользоваться отношением интенсивности нелинейного фона исследуемого образца к интенсивности нелинейного фона известной рентгеноаморфной фазы. Расчет возможен с использованием специального программного обеспечения. Данный подход также использовался в нашей работе.
В-третьих, с использованием данных химического анализа, если известна структура аморфной фазы и процентное содержание в ней конкретного оксида. Зная содержание данного оксида (за вычетом его содержания в имеющихся кристаллических фазах), можно без труда посчитать содержание аморфной фазы в исследуемом образце.
Определение массовой доли кристаллических фаз было проведено в образце 9 85.44CdO⋅14.56В2O3. Сначала с помощью автоматизированного программного комплекса с возможностью ручной корректировки и вводом необходимых параметров качественно (по трем совпавшим линиям) идентифицировали кристаллические фазы. О наличии стеклофазы в образце можно судить по гало (рис. 3г). Расчет аморфной фазы проводили с учетом интенсивности нелинейного фона образца 9 и анализа содержания аморфной фазы в образцах 1–8. Следует отметить, что метод Ритвельда [18] и другие количественные и полуколичественные бесстандартные методы не позволяют определить содержание аморфной фазы. После оценки содержания аморфной фазы сумму кристаллических фаз в образце 9 вычислили по формуле
(3)
$\begin{gathered} \sum {\left( {{\text{кристаллических фаз}}} \right)} = 100{\text{ }}\% -- \\ - \,\,\sum {\left( {{\text{аморфных фаз}}} \right)} = 100--15 = 85\% . \\ \end{gathered} $Затем идентифицированные и рассчитанные с помощью встроенного программного комплекса дифрактометра кристаллические фазы были отнормированы на коэффициент 0.85 для учета вклада аморфной фазы. В результате расчетов содержания кристаллических и аморфных фаз в образце 9 получены следующие значения (мас. %): СdO – 52, 3СdO⋅В2O3 – 33, аморфная фаза – 15. Определение кристаллических фаз в образце 9 проводили по картотеке PDF-2 (Card 11049 и 90130 соответственно).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
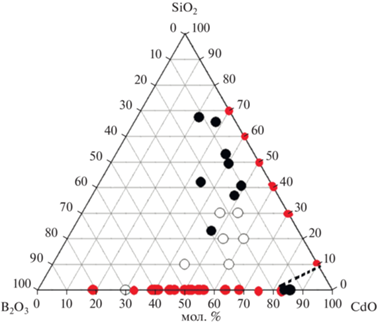
На рис. 4 приведен треугольник составов СdO–В2O3–SiO2. На нем нанесены экспериментальные результаты, полученные в настоящей работе, и данные [7, 8], совокупность которых позволяет определить область стеклообразования в системе.
Известно, что стеклообразующая способность оксидов зависит от размеров ионов и увеличивается с ростом ионного радиуса. Попытка прогнозирования верхних границ стеклообразования была предпринята в работе [19]. В ней приведена формула для определения предельной концентрации модификатора в двухкомпонентных стеклах
(4)
$S = \left( {{{{\text{e}}}^{{ - \frac{1}{{{{N}_{m}}{{R}_{m}}}}}}} - \frac{1}{{{{N}_{m}}}}} \right)\frac{{\frac{{2{{M}_{m}}}}{{{{n}_{m}}}} \times 100}}{{\frac{{2{{M}_{m}}}}{{{{n}_{m}}}} + {{M}_{g}}}},$Верхние границы стеклообразования, определенные по этой формуле, дают заниженные значения по сравнению с результатами, полученными в настоящей работе. Нами показано, что в бинарной системе CdO–В2O3 нельзя получить стекла, содержащие более 85.44 мол. % CdO. Образцы кристаллизуются в процессе выработки стекла. Также кристаллизовались стекла 7 (48.44CdO⋅14.71В2O3⋅36.86SiO2) и 8 (48.92CdO⋅10.45В2O3⋅40.64SiO2) в тройной системе CdO–В2O3–SiO2. В них обнаружены кварц и кристобалит.
Структура стекол системы CdO–В2O3–SiO2, определенная по положению аморфных пиков на дифрактограммах. Как было показано ранее, можно делать определенные выводы о структуре стекол системы CdO–В2O3–SiO2 на основании положения аморфных пиков на дифрактограммах.
В работе [8] выполнен расчет межатомных расстояний по приближенной формуле
где ${{\lambda }}$ = 1.54 Å – длина волны рентгеновского излучения [20].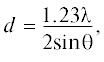
При исследовании дифрактограмм кадмиевоборатных и кадмиевоборосиликатных стекол были измерены интенсивности рассеяния (рис. 5а). Для сравнения на рис. 5б приведены дифрактограммы близких по составу стекол из работы [8]. На дифрактограммах наблюдаются три дифракционных максимума. По данным [8], они соответствуют характерным межатомным расстояниям Cd–Cd (максимум I ~ 17°), Cd–О второй координационной сферы (максимум II ~ 30°), Cd–О (максимум III ~ 47°–50°). Борокислородная сетка в кадмиевоборатных стеклах обеспечивает упорядоченное распределение ионов-модификаторов. Из рис. 5а видно (красная и черная кривые), что добавки SiO2 (система CdO–В2O3–SiO2) приводят к уменьшению и практически исчезновению максимума при углах около 17°, т.е. к исчезновению упорядоченности в распределении атомов кадмия.
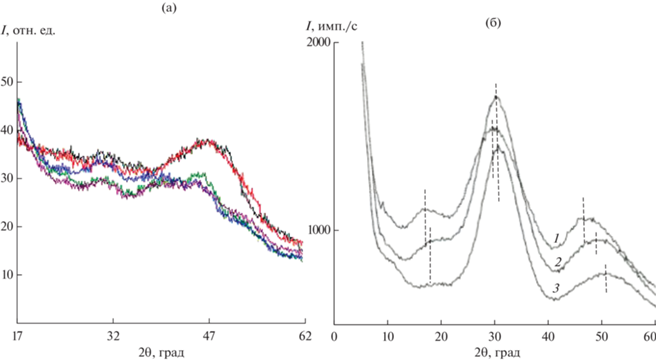
Интенсивность дифракционных максимумов существенно различается из-за различия в химических составах стекол, полученных в настоящей работе и исследованных в [8]. Три ярко выраженных дифракционных максимума на дифрактограммах обусловлены наличием повторяющихся межатомных расстояний в структуре ближнего и среднего порядков. Это связано с образованием структурных группировок с участием ионов-модификаторов или с упорядоченностью в борокислородном окружении иона-модификатора. Максимум при угле ~17° наблюдается при высоком содержании оксида бора. Он соответствует межатомному расстоянию Cd–Cd. Максимум при угле ~30° соответствует расстояниям между атомами кадмия и кислорода второй координационной сферы. Максимум при ~47°–50° соответствует расстояниям Cd–О. Так как образцы 6 в настоящей работе и 1 из работы [8] максимально близки по составу, можно считать, что и структурные группировки при 17°, 30° и 47°–50° в них идентичны.
ЗАКЛЮЧЕНИЕ
Обобщены результаты изучения стеклообразования в системе СdO–В2O3–SiO2. Установлена область стеклообразования в тройной системе СdO–В2O3–SiO2. Показано, что в бинарной системе CdO–В2O3 нельзя получить стекла, содержащие более 85.44% CdO. Образцы кристаллизуются в процессе выработки стекла. Подтверждено присутствие трех максимумов на дифрактограммах стекол 1–6, которые обусловлены наличием повторяющихся межатомных расстояний в структуре ближнего и среднего порядков. Определен химический анализ полученных стекол системы СdO–В2O3–SiO2.
Список литературы
Mori Y., Kuroda L. New Nonlinear Optical Crystal: Cesium Borate // Appl. Phys. Lett. 1993. V. 65. № 21. P. 2614–2615.
Немилов С.В. Научные основы материаловедения стекол / Под ред. Немилова С.В., Никонорова И.В. СПб.; Краснодар: Лан, 2018. 360 с.
International Critical Tables of Numerical Data, Physics, Chemistry and Technology. 1928. V. 4. 503 p.
Subbarao E.C., Hammel F.A. The System Cadmium Oxide-Borie Oxide // J. Electrochem. Soc. 1956. V. 103. № 1. P. 29–33.
Hand W.D., Krogh-Moe J. New Data on the System CdO–B2O3 // J. Am. Ceram. Soc. 1962. V. 45. № 4. P. 197–201.
Hayashi H., Nakayama N., Yoshida M., Kozuka T., Mizuno M., Yamamoto K., Yamamoto T., Noguchi T. Phase Transition under High Pressure (I) Transition of Cadmium Olivine // Rep. Gov. Ind. Res. Inst., Nagoya. 1964. V. 13. № 7. P. 285–290.
Информационная электронная система Sciglass-6.5. Institute of Theoretical Chemistry. Shrewsbury, 2005.
Голубков В.В., Онущенко П.А., Столярова В.Л. О структуре стекол системы PbO–В2O3–SiO2 и CdO–В2O3–SiO2 // Физ. хим. стекла. 2013. Т. 39. № 6. С. 879–890.
Пирютко М.М., Бенедиктова Н.В., Корсак Л.Ф. Усовершенствованный метод определения кремния в виде хинолин-кремне-молибденового комплекса // Стекло и керамика. 1981. № 8. С. 30–31.
Пирютко М.М., Бенедиктова Н.В. Ускоренное титриметрическое определение бора в силикатах // Журн. аналит. химии. 1970. Т. 25. № 1. С. 136–141.
Schwarzenbach G., Flaschka H. Die Komplexometrische Titration. Stuttgart: F. Enke, 1965. 360 p.
Щербов Д.П., Матвеец М.А. Aналитическая химия кадмия М.: Наука, 1973. 254 с.
ГОСТ 6613-86. Сетки проволочные тканые с квадратными ячейками. Технические условия. М.: Стандартинформ, 2006.
Руководство по эксплуатации Яб1.210.077 РЭ. Дифрактометр рентгеновский ДРОН−7. СПб, 2006. 109 с.
Руководство по эксплуатации МДР.01.00.000.РЭ. Дифрактометр рентгеновский Дифрей-401. СПб, 2016. 23 с.
ГОСТ 9077-82. Кварц молотый пылевидный. Общие технические условия. М.: ИПК Издательство стандартов, 2004.
Уманский Я.С., Скаков Ю.А., Иванов А.Н., Расторгуев Л.Н. Кристаллография, рентгенография и электронная микроскопия. М.: Металлургия, 1982. 632 с.
Кржижановская М.Г., Фирсова В.А., Бубнова Р.С. Применение метода Ритвельда для решения задач порошковой дифрактометрии. Санкт-Петербургский университет, 2016. 67 с.
Школьников Е.В. К определению стеклообразующей способности неорганических расплавов // Физ. хим. стекла. 1985. Т. 11. № 4. С. 501–504.
Скрышевский А.Ф. Структурный анализ жидкостей и твердых тел. М.: Высшая школа, 1980. 328 с.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы