

Неорганические материалы, 2022, T. 58, № 12, стр. 1279-1286
Влияние температуры на скорость роста нитевидных нанокристаллов полупроводников
В. А. Небольсин 1, *, В. А. Юрьев 1, Н. Свайкат 1, А. Ю. Воробъев 1, А. С. Самофалова 1
1 Воронежский государственный технический университет
394026 Воронеж, Московский пр., 14, Россия
* E-mail: vcmsao13@mail.ru
Поступила в редакцию 08.04.2022
После доработки 20.07.2022
Принята к публикации 09.08.2022
- EDN: BOATMP
- DOI: 10.31857/S0002337X22110124
Аннотация
Исследована зависимость скорости роста нитевидных нанокристаллов (ННК) от температуры. С учетом полученных экспериментальных данных и критического анализа современных представлений о влиянии температуры на кинетику роста ННК показано, что температурные зависимости скорости роста ННК можно предсказать термодинамическим путем, а лимитирующей стадией является кристаллизация на границе кристалл/жидкость.
ВВЕДЕНИЕ
Несмотря на обилие работ и очевидный прогресс в исследованиях полупроводниковых нитевидных микро- и нанокристаллов (ННК), наблюдаемый в последние годы, механизм роста пар → жидкость → кристалл (ПЖК), еще не до конца понят. В частности, ответ на вопрос о стадии, определяющей скорость процесса, является одним из основных в понимании ПЖК-механизма. Однако важные экспериментальные сведения о кинетике роста ННК остаются ограниченными и противоречивыми [1–7]. Известно, что скорость и механизм ПЖК-процесса сильно зависят от условий кристаллизации [2]. При этом температура (Т) является фактором, оказывающим наибольшее влияние на скорость роста ННК ($v$), с помощью которого можно судить и о механизме, и о его лимитирующей стадии. Но в данном важнейшем вопросе имеется много неясностей и разногласий [2, 6].
Цель настоящей работы – выяснить основные причины разногласий в вопросе влияния температуры на скорость роста ННК и показать, что ход зависимости $v$(T) определяется термодинамикой, а не кинетикой.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
ННК Si получали кристаллизацией из газовой фазы в хлоридно-водородной системе с использованием в качестве катализаторов частиц Cu, Au, Ni, Pt и Cu–Al размером от 50 нм до 20 мкм [2, 8]. Температурный диапазон роста ННК составлял от 850 до 1100°С. Мольное отношение компонентов SiCl4 : Н2 поддерживалось в интервале от 0.005 до 0.02. Ростовыми подложками служили пластины Si с ориентацией {111}. Скорость роста ННК определялась по методике “меток времени” [2]. Выращенные кристаллы исследовались методами растровой электронной и сканирующей зондовой микроскопии. Кинетические характеристики ННК Si, Ge, GaAs и др. также анализировались по данным [1–3, 6, 7, 9–11].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
На рис. 1 представлены температурные зависимости скорости роста ННК Si, выращенных с участием различных металлов (М) в интервале температур от 1000 до 1100°С. Видно, что с увеличением t скорость роста ННК понижается. Снижение $v$ наблюдается для разных типов М-катализаторов, разных диаметров ННК и для различных концентраций SiCl4 в газовой фазе. В то же время с повышением t наблюдается усиление радиального роста ННК (рис. 2).
Рис. 1.
Температурные зависимости скорости роста ННК Si cо средним радиусом 500 нм; катализаторы: 1 – Al–Cu, 2 – Cu, 3 – Au, 4 – Pt.
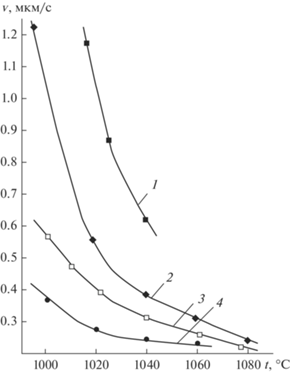
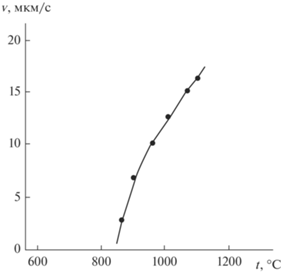
Убывающая температурная зависимость $v$ (рис. 1) не характерна для роста ННК Si и Ge, получаемых при более низких t (<1000°С), для них с повышением t скорость экспоненциально увеличивается (рис. 3а), а энергия активации составляет для Si ∼200 кДж/моль, для Ge ∼130 кДж/моль [1]. В качестве лимитирующих стадий (среди кинетических) здесь можно предположить процессы адсорбции–десорбции и собственно поверхностные реакции [12].
Рис. 3.
Логарифмические зависимости аксиальной скорости роста ННК Si и Ge, выращенных из SiH4 и GeH4 при давлении ${{р}_{{{\text{Si}}{{{\text{H}}}_{4}}}}} = {\text{ }}{{р}_{{{\text{Ge}}{{{\text{H}}}_{4}}}}}$ = 1.33 × 103 Па, от обратной температуры [1] (а); температурные зависимости скорости роста ННК Si для нанопроволок с радиусом r = 1 (1), 2 (2) и 25 нм (3) [7] (б); сравнение экспериментальной (2) и теоретической (1) температурных зависимостей скоростей роста нанопроволок GaAs [6, 9, 10] (черные кружки – экспериментальные данные [10], светлые кружки – результаты расчетов [9]) (в); температурные зависимости скорости роста ННК GaAs, GaP и InAs, полученных методом газофазной эпитаксии из металлорганических соединений [11] (г).
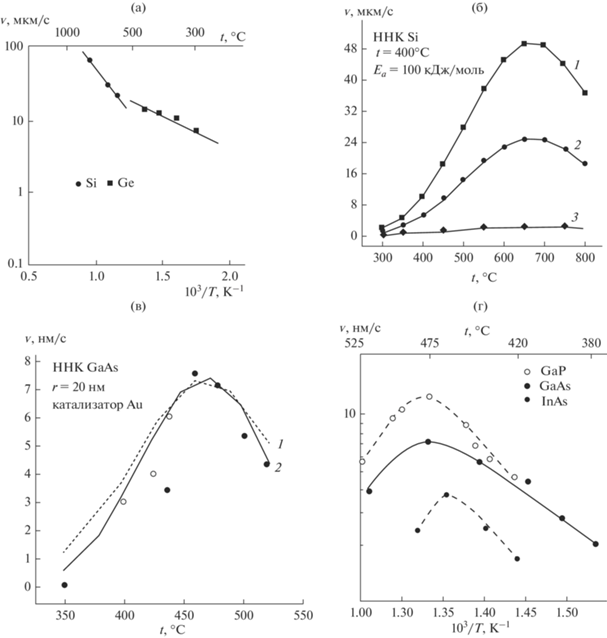
На рис. 3б приведены полученные в работе [6] зависимости $v$ = f(t) для роста ННК Si различных радиусов. Видно, что $v$ увеличивается с ростом t, достигает пиковых значений, а затем уменьшается. Энергия активации, определенная по кривой 1, составляет Еа = 100 кДж/моль. На рис. 3в показаны зависимости $v$ = f(t) для ННК GaAs с r = 20 нм [6], полученные теоретически (кривая 1) в работе [10] и экспериментально (кривая 2) в работе [9]. Для нанопроволок GaAs при t = 480°C Еа = 118 кДж/моль. При этом скорость роста ННК GaAs полярными гранями (111)А и ($\bar {1}\bar {1}\bar {1}$)В различается в пять раз, а для граней {211} и {111} ННК Si ${{v}_{{\left\{ {211} \right\}}}}$ $ \gg $ ${{v}_{{\left\{ {111} \right\}}}}$ [2].
Экспериментальные логарифмические зависимости скорости роста от обратной температуры для ННК GaAs, GaP и InAs, синтезированных методом газофазной эпитаксии с участием частиц Au с радиусом ∼25 нм в интервале температур 350–525°С, приведены на рис. 3г [11]. При низких t (рис. 3г) кривые скорости роста ННК GaAs, GaP и InAs ассимптотически приближаются к предельным кривым, соответствующим типичной энергии активации Еа. Однако при повышении температуры, в области 470–475°С, для ННК GaAs, GaP и InAs наклон кривых существенно уменьшается, а $v$ достигает максимума. При t > 450–475°С скорость роста ННК фактически убывает с температурой.
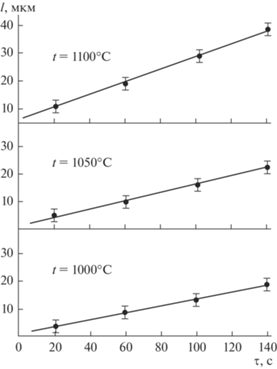
Для нескольких температур исследован характер удлинения ННК Si (l) с течением времени (рис. 4). Как видно из рис. 4, функция l(τ) является линейной с почти постоянным углом наклона.
Рис. 4.
Зависимости удлинения ННК Si диаметром 0.8 мкм от времени выращивания при различных температурах; катализатор Ni.
Для объяснения наблюдающихся различий зависимостей $v$ = f(t) проанализируем имеющиеся представления и модели. Считается, что для экспериментов по химическому паровому осаждению при больших размерах частиц катализатора (r > 1 мкм), высоких ростовых температурах (~1000°С для Si и ~700°С для GaAs) и, следовательно, очень малых длинах диффузионного пробега ${{\bar {\lambda }}_{a}}$ (${{\bar {\lambda }}_{a}} \ll 1$ мкм) адсорбированных атомов (адатомов) характерен адсорбционно-контролируемый рост ННК, определяющий активационную природу зависимости $v$(t) [1, 2]. Картина роста таких нитей [5, 6] показывает, что их скорость лимитируется адсорбционно-десорбционными процессами на поверхности капли как результат прямых соударений атомов парового вещества с жидкой фазой [2]. Следовательно, длина ННК не может быть больше толщины осажденного слоя (l '), если нет десорбции. Но фактически это не так и l/l ' ≈ 7–10 [2].
Появление новых современных источников кристаллизуемого материала позволило существенно снизить ростовые температуры и уменьшить диаметры ННК до 10–100 нм. Поэтому принимается, что прямое поступление атомов из газовой фазы в каплю пренебрежимо мало, а контроль роста ННК обеспечивают диффузионный и адсорбционно-десорбционный вклады с поверхности подложки и боковых стенок кристаллов [5, 6].
Так, для объяснения зависимости, показанной на рис. 3г, авторы [5, 11] прибегают к предположениям, что в высокотемпературной области (при t > 450–475°C на рис. 3г) средняя длина диффузионного пути адатомов на поверхности подложки ${{\bar {\lambda }}_{a}}$ лимитируется десорбцией, поэтому ${{\bar {\lambda }}_{a}}$ уменьшается. В области t < 450°C, где десорбция мала, уменьшение t приводит к возрастанию концентрации адатомов $N_{1}^{c}$ кристаллизуемого материала на поверхности подложки. Из-за возрастающей $N_{1}^{c}$ адатомы не успевают достигнуть движущейся вершины ННК, поскольку их захватывают растущие островки. Следовательно, при низких t величина ${{\bar {\lambda }}_{a}}$ лимитируется адсорбцией на поверхности подложки. Но давайте проанализируем эти процессы детально на примере роста ННК Si.
Пусть растущие ННК Si находятся в атмосфере пара кристаллизуемого вещества при давлении р и температуре T. Тогда в соответствии с кинетической теорией газов первоначально на единицу площади поверхности ростовой подложки с нанокаплями M-катализатора в единицу времени осаждается p/(2mkT)1/2 частиц массы m вещества (здесь k – постоянная Больцмана) [13]. Будем рассматривать адсорбцию лишь на подложке и боковых стенках ННК. Тогда концентрация адатомов равна
где N0 – поверхностная плотность атомов (для (111) Si N0 = 7.84 × 1018 м–2), Eads − энергия активации процесса адсорбции.О том, что термоактивируемая адсорбция (1) не лимитирует рост ННК, свидетельствует зависимость скорости роста от их кристаллографической ориентации на одинаковых по ориентировке подложках [2], т. е. зависимость $v$ от структуры растущей торцевой грани, а также интенсификация образования кристаллических слоев по двумерному механизму на подложке и боковых стенках кристаллов Si с ростом t в широком интервале температур: 500–1000°С [1] и 800–1100°С (рис. 2). При быстрой адсорбции концентрация адатомов на подложке и боковых стенках ННК возрастает, атомы захватываются растущей поверхностью и идет усиливающийся рост по механизму пар → кристалл (наблюдается экспоненциальная зависимость скорости радиального роста ННК от t), хотя ход зависимости $v$(t) для осевого роста ННК в этих температурных диапазонах неоднозначен (рис. 3).
Однако из положения адсорбции под влиянием тепловых колебаний адатом может покинуть поверхность со скоростью десорбции ~$\exp \left( { - {{E}_{{des}}}{\text{/}}kT} \right)$, где Edes − энергия активации процесса десорбции при переходе адатома обратно в паровую фазу. Время жизни адатомов τa на поверхности ростовой подложки
(2)
$1{\text{/}}{{\tau }_{a}} \cong \nu _{a}^{c}{\kern 1pt} \exp \left( { - {{E}_{{des}}}{\text{/}}kT} \right),$(3)
$N_{1}^{c} = p{{\left( {\nu _{a}^{c}} \right)}^{{ - 1}}}{{\left( {2\pi mkT} \right)}^{2}}\exp \left( {{{E}_{{des}}}{\text{/}}kT} \right).$Поскольку в качестве ростовых подложек для роста ННК как правило используются пластины Si{111}, проведем оценку $N_{1}^{c}$ и τa для грани (111) Si в контакте с собственным паром при t = 927°C и р = 1.6 × 10–5 Па. Принимая mSi = 4.76 × 10–27 кг, Edes = 231.8 кДж/моль и $\nu _{a}^{c}$ ≈ 1013 Гц, получим $N_{1}^{c}$ ≈ ≈ 3 × 1016 м–2. Так как поверхностная плотность атомов на грани Si (111) равна 1.54 × 1019 м–2, то доля позиций, заполненных адатомами, составляет ~1.46 × 10–3. При понижении t до 727°C и постоянстве плотности потока доля заполненных мест увеличится до ~1.46 × 10–1. Тогда длительность пребывания адатома на Si-поверхности τa составит ~0.06 с при t = 927°C и ~6 с при t = 727°C. С дальнейшим понижением температуры τa возрастет еще больше.
Адатомы совершают тепловые колебания, которые приводят к перескокам в соседние положения адсорбции, это обеспечивает диффузию по поверхности подложки и, в конечном счете, переход атомов на боковые стенки ННК. Количество перескоков в единицу времени определяется эффективным коэффициентом поверхностной диффузии
где $U_{a}^{c}$ − энергия активации диффузии, а – межатомное расстояние.На основании уравнений (2) и (4) вычислим длину свободного пробега адатома за время его пребывания τa на ростовой Si-подложке
(5)
${{\bar {\lambda }}_{a}} \approx {{\left( {D_{a}^{c}{{\tau }_{a}}} \right)}^{{1/2}}} = a{\kern 1pt} \exp \left[ {\left( {{{E}_{{des}}} - U_{a}^{c}} \right){\text{/}}2kT} \right].$Энергия активации диффузии для атомов Si на грани (111) составляет $U_{a}^{c}$ ≈ 115 кДж/моль, а Edes = = 231 кДж/моль [13]. При t = 727°C среднее расстояние прохождения адатома за время нахождения его на грани (111) Si равно ${{\bar {\lambda }}_{a}}$ ≈ 1.8 × 103а ≈ 8.1 × × 10–7 м (здесь а ≈ 0.45 нм). Так как Edes > $U_{a}^{c}$, то ${{\bar {\lambda }}_{a}}$ зависит от соотношения ${{E}_{{des}}}{\text{/}}kT$ и средняя длина диффузионного пробега адатомов ${{\bar {\lambda }}_{a}}$ должна существенно возрастать с уменьшением t. Следовательно, при десорбционном контроле процесса с понижением температуры скорость роста ННК должна экспоненциально возрастать, что противоречит экспериментальным данным (рис. 1 и 3).
Вероятность поступления адатомов из соседних мест адсорбции на площадку а2 составляет ~$N_{1}^{c}{{a}^{2}}\nu _{a}^{c}{\kern 1pt} \exp \left( { - U_{a}^{c}{\text{/}}kT} \right)$, в то время как вероятность поступления атомов из газовой атмосферы пропорциональна ~$p{{a}^{2}}{\text{/}}{{\left( {2\pi mkT} \right)}^{{1/2}}}$. С учетом (2) соотношение этих вероятностей равно $\exp \left[ {\left( {{{E}_{{des}}} - U_{a}^{c}} \right){\text{/}}2kT} \right]$. Если учесть, что ${{E}_{{des}}} \gg U_{a}^{c}$, то можно считать, что основная доля материала для заполнения мест адсорбции поступает за счет миграции атомов из соседних мест адсорбции, а не путем прямой конденсации из пара. Так, по оценкам [14], для грани Si (111) при t = 727°C отношение указанных выше вероятностей равно ~4 × 106. Из этих оценок следует, что при осаждении Si из пара $\exp \left[ {\left( {{{E}_{{des}}} - U_{a}^{c}} \right){\text{/}}2kT} \right] \gg 1$. Таким образом, нуклеация на поверхности Si-подложки и ННК с ростом t не может уменьшать скорость роста.
Однако поверхностная диффузия на подложке и боковой поверхности ННК также не может контролировать скорость роста кристаллов, поскольку в условиях разреженной атмосферы и больших ${{\bar {\lambda }}_{a}}$ (~3–10 мкм), больших $D_{a}^{c}$ и малых транспортных путей доставки питающего материала в силу малости размеров ННК (диаметр d ~ ~ 10–100 нм и длина l ~ 1 мкм) диффузия, наоборот, протекает очень быстро (l < ${{\bar {\lambda }}_{a}}$). Так, в случае роста ННК GaAs при t = 580°C ${{\bar {\lambda }}_{a}}$ атома Ga на поверхности GaAs ($\bar {1}\bar {1}\bar {1}$) B составляет ∼6 мкм, а на боковой поверхности ННК GaAs {110} по расчетам – от 2–3 до 8–10 мкм [5]. Аналогично, при типичных длинах ННК Si l ~ 1 мкм и типичных коэффициентах поверхностной диффузии (для Si(111) при t = 727°C, по оценкам, $D_{a}^{c}$ ~ 10–11 м2/с) скорость поверхностной диффузии составляет ${{v}_{{DS}}}$ ~ $D_{a}^{c}{\text{/}}l$ ~ 10–5 м/с. Такой величины могли бы достигать в пределе скорости роста ННК Si при лимитировании процесса стадией поверхностной диффузии. Однако наблюдаемые скорости роста ННК (1–10 нм/с) на три–четыре порядка ниже полученных оценочных величин.
Выводы о диффузионном режиме роста ННК также противоречат и наблюдаемым экспоненциальным, т.е. сильным, температурным зависимостям $v$ = f(t) (типичный вид таких зависимостей показан рис. 3а), свидетельствующим об активационном характере процесса (Еа обычно составляет 40–200 кДж/моль). Но известно, что температурная зависимость диффузии слабая, степенная (с показателем степени не более 1.5–2), а энергия активация диффузии обычно не превышает 4–20 кДж/моль [2].
Кроме того, наблюдаемые пиковые зависимости $v$ = f(t) и ln $v$ = f(1/T) c характерной ниспадающей ветвью при более высоких температурах (правые ветви кривых на рис. 3б, 3в и левая ветвь кривых на рис. 3г) не получается объяснить ни в рамках кинетической, включая адсорбционно-десорбционную, ни в рамках диффузионной моделей [1–11].
Вместе с тем, непонятна недооценка авторами многих моделей стадии встраивания атомов вещества в решетку ННК при столь низких температурах ростовых процессов (340–640°C). Учитывая эту стадию, характерный вид кривых ln $v$ = f(1/T) можно было бы интерпретировать следующим образом.
Рассмотрим реакцию кристаллизации вещества А на границе жидкость (L)/кристалл (S), которая приводит к росту ННК: АL ⇔ AS, (ΔH ≶ 0), где ΔH – тепловой эффект (энтальпия) реакции при постоянном давлении. При кристаллизации Si, Ge, GaAs и др. процесс характеризуется отрицательной теплотой реакции (ΔH < 0, т.е. реакция экзотермическая, теплота выделяется). Несмотря на это, в начале температурного диапазона с повышением t скорость роста ННК быстро увеличивается (рис. 3). Данный факт наряду с экспоненциальной зависимостью $v$(Т) в начальной температурной области (рис. 3а (прямые линии), 3б и 3в (левые ветви кривых) и рис. 3г (правые ветви)) и зависимостью скорости роста от ориентации ННК [2] служит сильным доводом в пользу кинетического режима роста с лимитирующей стадией встраивания вещества в решетку кристалла. Кроме того, из линейной функции l = f(τ) (рис. 4) следует, что $v$ = dl/dτ = const и не зависит от концентрации кристаллизуемого вещества при t = const. Это возможно только для поверхностного процесса нулевого порядка по объемной концентрации реагента, поскольку порядок реакций в диффузионной области всегда первый. В этом случае состав капли вблизи границы кристалл/жидкость мало отличается от состава ННК и процесс не требует диффузии.
В кинетической области кривые скорости роста приближаются к кривым, соответствующим характерным для кристаллизации значениям Eа (>100 кДж/моль). Но при повышении t до 650–700°C (рис. 3б) и 450–475°C (рис. 3в, 3г) наклон кривых уменьшается по мере того, как все более существенными становятся термодинамические факторы.
В области высоких t скорость роста ННК фактически убывает с ростом температуры (рис. 3б, 3в (правая ветвь кривых) и рис. 3г (левая ветвь)), поэтому кажется, что процесс перешел в диффузионный режим [5, 6]. На самом деле зависимость $v$ = f(t), характеризующая процесс с ΔH < 0, определяется термодинамическими причинами. С ростом t равновесие процесса кристаллизации смещается в сторону эндотермической реакции растворения кристаллического вещества и скорость роста ННК падает. При этом максимумам на кривых рис. 3б–3г соответствует t, при которой все факторы оказывают на $v$ одинаковое влияние.
Очевидно, в исследованном интервале температур кинетический член $\exp \left( { - {{E}_{а}}{\text{/}}RT} \right)$, где R – газовая постоянная, не вносит заметного вклада в наблюдаемую скорость роста ННК, если она определяется термодинамикой, а не кинетикой процесса. Более того, большая величина энергии активации (100–200 кДж/моль) [6] не может служить признаком кинетического режима для десорбции, поскольку процесс десорбции эндотермичен и ΔH > 0. Убывающую зависимость $v$ = f(t) (рис. 1) нельзя объяснить и снижением выхода Si, так как суммарная теплота реакции из смеси SiCl4 + + H2 положительна (ΔH > 0, реакция эндотермическая) и с увеличением t скорость роста ННК должна возрастать.
Для того чтобы понять возможность влияния термодинамических факторов на рост ННК Si, рассмотрим реакцию на границе кристалл/газ
(6)
${\text{S}}{{{\text{i}}}_{L}} \Leftrightarrow {\text{S}}{{{\text{i}}}_{S}},~\,\,\,\,\Delta H < 0.$Если предположить, что скорость роста ННК определяется объемной диффузией или реакцией на межфазной границе, то, упрощенно, выделим три стадии ПЖК-процесса вблизи равновесия: 1) диффузия Si в жидкой капле к растущей грани ННК; 2) встраивание атомов Si в решетку кристалла (6); 3) диффузия атомарного Si от торцевой грани ННК к поверхности капли.
Скорость первой стадии можно выразить как перенос массы вещества
где ${{v}_{{D1}}}$ – скорость диффузии атомов Si к растущей поверхности, kD1 – коэффициент массопереноса Si, Сsurf и СL – концентрации кристаллизуемого вещества вблизи поверхности капли и на границе с кристаллом.Скорость обратимой реакции кристаллизации (6) ${{v}_{k}}$ выразим как
где kk dir и kk back – константы скорости прямой и обратной реакции, СS – равновесная концентрация атомов на границе с кристаллом.Скорость диффузии атомарного Si от торцевой грани ННК выразится как
При записи выражения (9) предполагалось, что изначально жидкая фаза содержит равновесную концентрацию Si и, следовательно, СL = СS.
Если рост ННК проходит в стационарном режиме, то скорости всех звеньев равны: ${{v}_{{D1}}} = {{v}_{{D2}}} = {{v}_{k}} = v$, где $v$ – наблюдаемая скорость роста ННК. Из уравнений (7)–(9) получаем соотношение для скорости роста ННК
(10)
$v = \left[ {k_{{k\,\,dir}}^{{ - 1}} + k_{{D1}}^{{ - 1}} + {{k}_{{k\,back}}}{{{\left( {{{k}_{{D2}}}{{k}_{{k\,dir}}}} \right)}}^{{ - 1}}}} \right]{{С}_{{surf}}}.$Выражение в скобках показывает, что обратная величина константы скорости роста ННК равна сумме обратных значений констант скоростей стадий процесса. Реакция (6) – реакция первого порядка, поэтому можно записать
где K – константа равновесия реакции кристаллизации. Коэффициенты диффузии kD1 и kD2 в (7) и (9) должны быть одинаковы, т.е. где kD – коэффициент массопереноса. Подставляя (11) и (12) в (10), получим(13)
$v = \left[ {{{k}_{\,}}_{{k\,dir}}^{{ - 1}} + k_{D}^{{ - 1}}\left( {1 + {{K}^{{ - 1}}}} \right)} \right]{{С}_{{surf}}}.$Константа ${{k}_{{k\,dir}}}$ возрастает с температурой по экспоненциальной зависимости, которую можно описать уравнением Аррениуса
Коэффициент kD связан с Т степенной функцией. Поэтому скорость диффузионного процесса в первом приближении можно принять
где b – коэффициент пропорциональности. Температурная зависимость константы K определяется интегральным выражением Вант-Гоффа где m – некоторая постоянная. Подставляя (14), (15) и (16) в (13), получаем(17)
$\begin{gathered} v = \left( {{{{\left( {A{\kern 1pt} \exp \left( { - \frac{{\Delta {{E}_{a}}}}{{RT}}} \right)} \right)}}^{{ - 1}}}} \right. + \\ \left. { + \,\,{{b}^{{ - 1}}}{{T}^{{2/3}}}{{{\left( {1 + m{\kern 1pt} \exp \left( { - \frac{{\Delta H}}{{RT}}} \right)} \right)}}^{{ - 1}}}} \right){{С}_{{surf}}}. \\ \end{gathered} $Из выражения (17) следует, что при Сsurf = const и низких Т решающее влияние на скорость роста ННК оказывает первое слагаемое и процесс подчиняется кинетическим закономерностям. В этой области функция ln$v$ = f(1/T) носит линейный характер (рис. 3а, левая ветвь кривых на рис. 3б, 3в и правая ветвь кривых на рис. 3г). Но с ростом Т для реакции кристаллизации с относительно высокими значениями (по модулю) ΔН < 0 зависимость $v$(Т) будет определяться третьим слагаемым в (17) и подчиняться термодинамическим закономерностям (рис. 1, рис. 3б, 3в (правая ветвь кривых) и рис. 3г (левая ветвь)). Для ростовых систем с ΔН ≥ 0 и Т выше максимальных наблюдаемая скорость роста ННК $v$ ~ Т3/2.
Таким образом, можно констатировать, что причины, лежащие в основе различий представлений о влиянии Т на скорость роста ННК, заключаются в превалирующей роли термодинамического фактора экзотермической реакции кристаллизации, а не кинетики процесса роста.
ЗАКЛЮЧЕНИЕ
Показано, что недооценка влияния термодинамических факторов является основной причиной, определяющей различия результатов изучения влияния Т на скорость роста ННК и противоречивые выводы о лимитирующей стадии ПЖК-процесса. Для обратимых процессов кристаллизации ННК Si, Ge, GaAs и др., у которых теплота реакции отрицательна (ΔН < 0), в области низких Т преобладает кинетический режим с лимитирующей стадией кристаллизации на границе кристалл/жидкость. При повышении Т ход зависимости $v$(T) определяется термодинамическими, а не кинетическими причинами. Интенсифицировать рост ННК в экзотермическом процессе можно, понижая температуру и давление.
Список литературы
Bootsma G.A., Gassen H.J. A Quantitative Study on the Growth of Silicon Whiskers from Silane and Germanium Whiskers From Germane // J. Cryst. Growth. 1971. V. 10. P. 223–234.
Гиваргизов Е.И. Рост нитевидных и пластинчатых кристаллов из пара. М.: Наука, 1977. 304 с.
Weyher J. Some Notes of the Growth Kinetics and Morphology of VLS Si Crystals Grown with Pt and Au as Liquid-Forming Agents // J. Cryst. Growth. 1978. V. 43. P. 235–244.
Lew K.K., Redwing J.M. Growth Characteristics of Silicon Nanowires Synthesized by VLS Growth in Nanoporous Al Templates // J. Cryst. Growth. 2003. V. 254. № 1–2. P. 14–22.
Дубровский В.Г., Сибирев Н.В., Суриc Р.А., Цырлин Г.Э., Устинов В.М., Tchernycheva M., Harmand J.C. О роли поверхностной диффузии адатомов при формировании нанометровых НК // ФТП. 2006. Т. 40. Вып. 9. С. 1103–1110.
Noor Mohammad S. General Theoretical Model for the Vapor-Phase Growth and Growth Rate of Semiconductor Nanowires // J. Vac. Sci. Technol. B. 2010. V. 28. № 2. P. 329–352.
Thombare S.V., Marshall A.F., McIntyre P.C. Kinetics of Ge NW Growth by the Vapor-Solid-Solid Mechanism with a Ni-Based Catalyst // APL Mater. 2013. V. 1. P. 061101.
Небольсин В.А., Щетинин А.А., Даринский Б.М., Попов Е.Е. Кинетика роста НК Si в реакторе с горячими стенками // Изв. вузов. Физика. 1995. Т. 38. № 10. С. 22–27.
Dick K., Deppert K., Karlsson L., Wallenberg L.R., Samuelson L., Seifert W. A New Understanding of Au-Assisted Growth of III–V Semiconductor Nanowires // Adv. Funct. Mater. 2005. V. 15. № 10. P. 1603–1610.
Soci C., Bao X.-Y., Aplin D. P. R., Wang D. A Systematic Study on the Growth of GaAs NWs by MOCV Deposition // Nano Lett. 2008. V. 8. № 12. P. 4275–4282.
Seifert W., Borgstrom M., Deppert K., Dick K.A., Johansson J., Larsson M.W., Martensson T., Skold N., Svensson C.P., Wacaser B.A., Wallenberg L.R., Samuelson L. Growth of One-Dimensional Nanostructures in MOVPE // J. Cryst. Growth. 2004. V. 272. P. 211–220.
Glas F., Dubrovskii V.G. Energetics and Kinetics of Monolayer Formation in Vapor-Liquid-Solid Nanowire Growth // Phys. Rev. Mater. 2020. V. 4. P. 083401.
Современная кристаллография в 4-х томах. Т. 3: Образование кристаллов / Под ред. Чернова А.А. и др. М.: Наука, 1980. 407 с.
Крапухин В.В., Соколов И.А., Кузнецов Г.Д. Физико-химические основы технологии полупроводниковых материалов. М.: Металлургия, 1982. 352 с.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы