

Неорганические материалы, 2022, T. 58, № 12, стр. 1294-1302
Структурная эволюция моноанионных гафний-допированных кластеров олова
Н. А. Борщ 1, *, Н. С. Переславцева 1, С. И. Курганский 2
1 Воронежский государственный технический университет
394006 Воронеж, ул. 20-летия Октября, 84, Россия
2 Воронежский государственный университет
394018 Воронеж, Университетская пл., 1, Россия
* E-mail: n.a.borshch@ya.ru
Поступила в редакцию 09.06.2022
После доработки 20.09.2022
Принята к публикации 21.09.2022
- EDN: TAOKNJ
- DOI: 10.31857/S0002337X22120053
Аннотация
В работе представлены результаты компьютерного эксперимента в рамках теории функционала плотности по расчетам атомной структуры и электронного спектра кластеров ${\text{HfSn}}_{n}^{ - }$ (n = 15–17). На основе правил Уэйда–Мингоса установлены закономерности формирования моноанионных гафний-допированных кластеров олова, позволяющие оптимизировать прогностические исследования по поиску новых наноструктурированных материалов. Сравниваются результаты расчетов с использованием функционалов B3LYP, B3PW91 и PBE в комбинации с базисом SDD. Проведен анализ влияния функционала на результаты оптимизационных расчетов атомной структуры. Предложена оптимальная стратегия компьютерного эксперимента по моделированию пространственной структуры кластеров на основе олова, подтвержденная сопоставлением с известными экспериментальными данными.
ВВЕДЕНИЕ
Проблема поиска новых функциональных материалов – одна из наиболее актуальных в последние десятилетия. Для качественного перехода к новым технологиям в различных сферах производства уже недостаточно традиционных материалов, необходимы радикально новые решения. Одним из наиболее динамично развивающихся направлений является поиск наноструктурированных материалов [1, 2]. Это материалы различной размерности, элементарными строительными блоками для которых служат атомные кластеры [2]. Поскольку свойства атомных кластеров легко варьировать, изменяя число атомов в них и/или атомный состав [3–8], то открываются широкие возможности для конструирования функциональных материалов с заданными свойствами под нужды той или иной производственной задачи.
Наноматериалы на основе олова рассматриваются как перспективные новые материалы. Например, наноклеточные кристаллы на основе олова обладают уникальными термоэлектрическими свойствами [9], одномерные наноструктуры на основе олова (наностержни, нанопроволоки) могут быть использованы для разработки газовых высокочувствительных сенсоров [10], наночастицы олова используются для конструирования новых анодных материалов для литий-ионных аккумуляторов [11]. Очевидно, что работы по синтезу таких материалов невозможны без достоверной информации о структуре и свойствах элементарных блоков, из которых эти материалы конструируются – атомных кластеров.
Экспериментальное исследование пространственной структуры и свойств атомных кластеров в настоящее время технически затруднительно, поэтому особое значение приобретает компьютерный эксперимент. Наиболее эффективным методом в таких экспериментах является теория функционала плотности (DFT), однако существует проблема – часто встречающаяся зависимость результатов расчетов от используемого DFT-функционала [12]. Поэтому важна возможность сравнить результаты расчета с соответствующими экспериментальными данными.
Для достоверного определения пространственной структуры можно комбинировать компьютерный DFT-эксперимент с методом фотоэлектронной спектроскопии [4, 5]. Известно достаточное количество работ, в которых представлены фотоэлектронные спектры различных кластеров, в т.ч. кластеров олова [13]. Сопоставление рассчитанного электронного спектра каждого из наиболее стабильных изомеров кластера с его фотоэлектронным спектром позволяет определить, какие именно структуры были детектированы экспериментально, поскольку профиль электронного спектра значительно зависит от особенностей атомной структуры кластера.
В данной работе представлены результаты оптимизационных расчетов атомной структуры некоторых кластеров ряда ${\text{HfSn}}_{n}^{ - }$, а именно, с n = 15–17. Для этих кластеров известны данные по исследованиям электронного спектра методом фотоэлектронной спектроскопии [13], что позволяет сопоставить результаты компьютерного эксперимента и эксперимента прикладного, тем самым оценив адекватность расчетов. Расчеты проводились в рамках теории функционала плотности с использованием трех функционалов: B3LYP [14, 15], B3PW91 [16] и PBE [17, 18] в комбинации с базисом SDD [19, 20]. Это дало возможность проанализировать влияние выбора DFT-функционала на результаты оптимизационных расчетов атомной структуры, а сопоставление рассчитанных данных с известными экспериментальными [13] позволило предложить оптимальную стратегию компьютерного эксперимента по моделированию пространственной структуры кластеров на основе олова.
МЕТОД РАСЧЕТА
Для каждой оптимизированной структуры получались собственные значения энергии каждой молекулярной орбитали, т.е. энергетический спектр, в котором каждую молекулярную орбиталь можно представить в виде уровня. Теоретические спектры получались после того, как каждый энергетический уровень заменялся гауссовым распределением с полушириной 0.15 эВ и интенсивности всех распределений при каждом значении энергии складывались. Совмещение рассчитанных и экспериментальных спектров по энергетической шкале проводилось по положению верхней заполненной орбитали.
Средняя энергия связи ${{E}_{b}}$ вычислялась по формуле
Для расчетов использовался программный комплекс Gaussian09 [21].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Для кластеров ${\text{HfSn}}_{{{\text{15}}}}^{ - }$ получены шесть стабильных изомеров (рис. 1). Изомеры 15-ARH1, 15-ARH2 и 15-ARH3 можно рассматривать как arachno-многогранники Уэйда–Мингоса [22, 23], которые получаются удалением двух вершин из closo-многогранника с семнадцатью вершинами (см. рис. 1). Изомер 15-NIDO представляет собой nido-многогранник, он получается удалением одной вершины из closo-многогранника с шестнадцатью вершинами. Все эти изомеры можно отнести к дельтаэдрическим структурам, поскольку они представляют собой многогранники с преимущественно треугольными гранями. Атомы олова в этих структурах образуют по четыре или пять связей Sn–Sn. Еще два изомера кластера ${\text{HfSn}}_{{{\text{15}}}}^{ - }$ имеют призматическую структуру, которая может быть описана как сильно искаженная шестиугольная призма с атомом гафния внутри и с тремя дополнительными атомами олова. В изомере 15-PR1 два дополнительных атома олова формируют связи с одним основанием призмы, а один – с противоположным. В изомере 15-PR2 все три дополнительных атома расположены над одним основанием призмы. Большинство атомов олова в этих структурах формируют по три связи Sn–Sn, поэтому их можно отнести к трехсвязным структурам.
Рис. 1.
Атомные структуры и схема формирования стабильных изомеров кластера ${\text{HfSn}}_{{{\text{15}}}}^{ - }$, сопоставление их рассчитанных электронных спектров с экспериментальными [13]: черным цветом показан атом гафния, серым – атомы олова (связи Hf–Sn не показаны для упрощения рисунка).
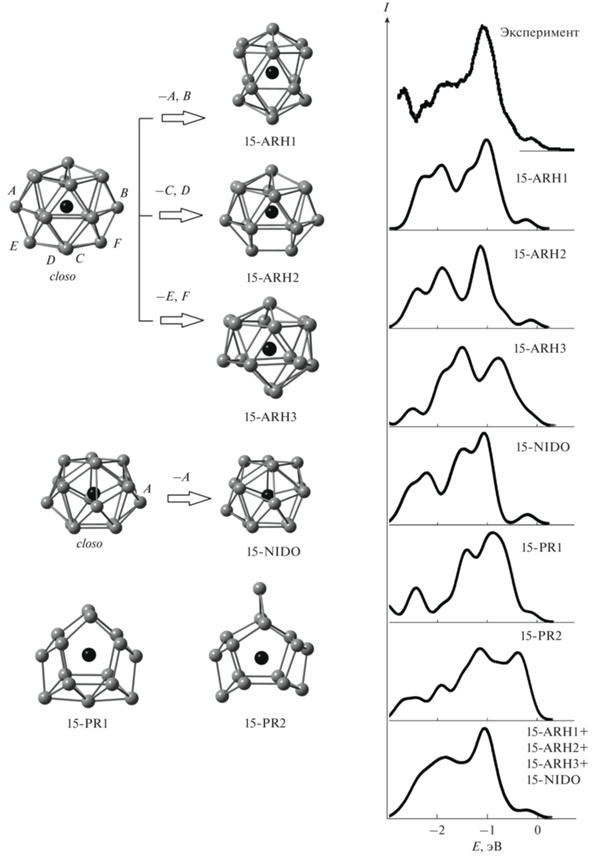
По результатам всех трех расчетов наиболее стабильными изомерами являются изомеры с дельтаэдрической структурой. Средние энергии связи в них близки по результатам B3PW91- и PBE-расчетов (см. табл. 1). Призматические изомеры, согласно этим расчетам, являются заметно менее стабильными, и можно предположить, что они не были детектированы экспериментально. По результатам B3LYP-расчета изомер 15-NIDO является основным, а изомеры 15-ARH1, 15-ARH2 и 15-ARH3 имеют несколько меньшие средние энергии связи. Энергия связи в изомере 15-PR1 при этом сопоставима с энергией связи в arachno-изомерах. Если на основании B3LYP-расчета делать вывод о том, какие структуры могут быть получены экспериментально, то нельзя исключить наблюдение наряду с дельтаэдрическими и призматического изомера 15-PR1.
Таблица 1.
Разности средних энергий связи между основным и остальными изомерами по результатам расчетов с различными функционалами
| Кластер | Изомер | ΔEсв, эВ/атом | ||
|---|---|---|---|---|
| B3LYP | B3PW91 | PBE | ||
| ${\text{HfSn}}_{{{\text{15}}}}^{ - }$ | ARH1 | 0.0096 | 0.0043 | 0.0027 |
| ARH2 | 0.0088 | 0.0006 | 0.0000 | |
| ARH3 | 0.0109 | 0.0052 | 0.0025 | |
| NIDO | 0.0000 | 0.0000 | 0.0031 | |
| PR1 | 0.0114 | 0.0206 | 0.0294 | |
| PR2 | 0.0511 | 0.0800 | 0.0971 | |
| ${\text{HfSn}}_{{{\text{16}}}}^{ - }$ | NIDO | 0.0086 | 0.0172 | 0.0173 |
| ARH | 0.0078 | 0.0209 | 0.0290 | |
| FK | 0.0003 | 0.0000 | 0.0000 | |
| PR1 | 0.0000 | 0.0264 | 0.0447 | |
| PR2 | 0.0097 | 0.0255 | 0.0314 | |
| ${\text{HfSn}}_{{{\text{17}}}}^{ - }$ | ARH | 0.0244 | 0.0028 | 0.0000 |
| NIDO1 | 0.0320 | 0.0041 | 0.0032 | |
| NIDO2 | 0.0229 | 0.0001 | 0.0034 | |
| PR | 0.0000 | 0.0000 | 0.0173 | |
Сопоставление рассчитанных электронных спектров каждого изомера кластера с фотоэлектронным спектром [13] показывает, что рассчитанные спектры каждой из дельтаэдических структур хорошо согласуются с экспериментальным, а профиль их суммарного спектра практически идеально совпадает с профилем фотоэлектронного спектра (рис. 1). Такое совпадение позволяет утверждать, что фотоэлектронный спектр [13] был получен в результате детектирования в эксперименте именно нескольких типов дельтаэдрических изомеров кластера ${\text{HfSn}}_{{{\text{15}}}}^{ - }$. Рассчитанный спектр призматического изомера 15-PR1 также неплохо согласуется с экспериментальным, поэтому нельзя исключить, что такие структуры также могут быть синтезированы.
Стабильные изомеры кластера ${\text{HfSn}}_{{{\text{16}}}}^{ - }$ показаны на рис. 2. Два из них являются эндоэдральными: 16-NIDO1 и 16-NIDO2. Эти изомеры являются nido-многогранниками, которые получаются после удаления одной вершины из closo-многогранника с семнадцатью вершинами. Изомер 16-NIDO1 тоже можно описать как искаженный многогранник Франка–Каспера с атомом гафния в центре. Еще три изомера кластера ${\text{HfSn}}_{{{\text{16}}}}^{ - }$ (16-PR1, 16-PR2 и 16-ARH) можно назвать квазиэндоэдральными – они представляют собой эндоструктуру из пятнадцати атомов олова с дополнительным атомом Sn, связь которого с атомом гафния существенно слабее, чем у остальных. Призматические изомеры 16-PR1 и 16-PR2 получаются добавлением дополнительного атома олова к структуре, аналогичной кластеру 15-PR1. В изомере 16-PR1 дополнительный атом олова формирует две связи Sn–Sn с вершинными атомами, а в изомере 16-PR2 – пять связей с атомами передней (относительно рис. 2) грани. Изомер 16-ARH представляет собой структуру, аналогичную структуре 15-ARH3, с дополнительным атомом олова.
Рис. 2.
Атомные структуры и схема формирования стабильных изомеров кластера ${\text{HfSn}}_{{{\text{16}}}}^{ - }$, сопоставление их рассчитанных электронных спектров с экспериментальными [13]: черным цветом показан атом гафния, серым – атомы олова (связи Hf–Sn не показаны для упрощения рисунка).
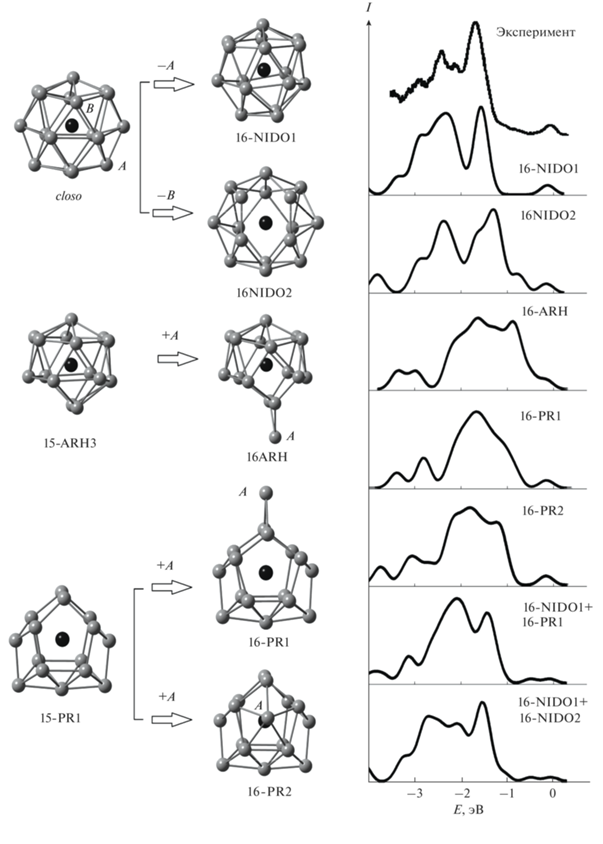
Согласно расчету с использованием B3LYP-функционала, наибольшая средняя энергия связи соответствует изомерам 16-PR1 и 16-NIDO1. В B3PW91- и PBE-расчетах 16-NIDO1 является основным изомером с небольшим энергетическим отрывом от изомера 16-NIDO2 и со значительным – от остальных. Сопоставление рассчитанного электронного спектра изомера 16-NIDO1 с фотоэлектронным [13] показывает их практически полное совпадение. Хорошо согласуется с экспериментальным также спектр изомера 16-NIDO2. Небольшие отличия профиля этого спектра от профиля фотоэлектронного спектра наблюдаются только у потолка валентной полосы. Учитывая, что по результатам всех трех расчетов разница в энергиях связи этого изомера и основных невелика, а в B3PW91- и PBE-расчетах это второй по стабильности изомер, можно предположить, что могут быть синтезированы и 16-NIDO1, и 16-NIDO2 изомеры. Их суммарный спектр также показан на рис. 2, где видно, что он хорошо согласуется с экспериментальным.
Рассчитанные спектры остальных изомеров значительно хуже согласуются с экспериментальным. На рис. 2 показан также суммарный спектр изомеров 16-PR1 и 16-NIDO1, каждый из которых, согласно результатам B3LYP-расчета, равновероятно мог быть детектирован экспериментально. Как видно из рис. 2, суммарный спектр этих изомеров плохо согласуется с экспериментальным. Отличия касаются и энергетического положения особенностей, и их относительных интенсивностей. Поэтому можно заключить, что наблюдение в эксперименте изомера 16-PR1 наряду с изомером 16-NIDO1 все же маловероятно и результаты B3LYP-расчета содержат существенные неточности.
Для кластера ${\text{HfSn}}_{{{\text{17}}}}^{ - }$ получены четыре стабильных изомера (рис. 3). Один из них – изомер 17-NIDO1 – является эндоэдральным. Этот изомер имеет структуру nido-многогранника, который представляет собой closo-многогранник с восемнадцатью вершинами, из которого одна вершина удалена. Остальные изомеры (17-PR, 17-NIDO2, 17-ARH) являются квазиэндоэдральными и представляют собой эндоструктуры из пятнадцати атомов олова, формирующих связи Hf–Sn, с двумя дополнительными атомами, для которых расстояние Hf–Sn существенно больше. Изомер 17-PR построен аналогично изомеру 16-PR и имеет в основе своей структуры призматический кластер 15-PR1. Изомеры 17-NIDO2 и 17-ARH получены добавлением двух дополнительных атомов олова к структурам, идентичным кластерам 15-NIDO1 и 15-ARH1 соответственно.
Рис. 3.
Атомные структуры и схема формирования стабильных изомеров кластера ${\text{HfSn}}_{{{\text{17}}}}^{ - }$, сопоставление их рассчитанных электронных спектров с экспериментальными [13]: черным цветом показан атом гафния, серым – атомы олова (связи Hf–Sn не показаны для упрощения рисунка).
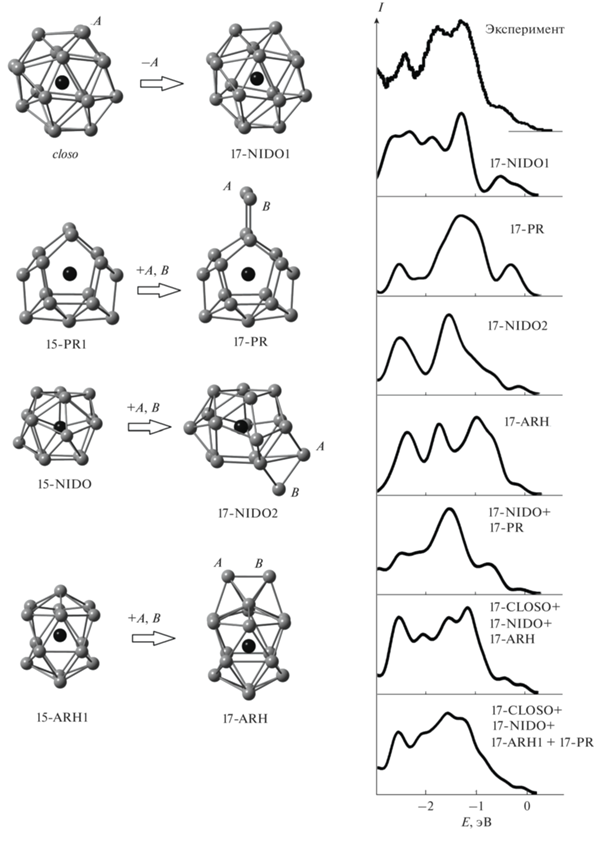
По результатам B3LYP-расчета основным изомером кластера ${\text{HfSn}}_{{{\text{17}}}}^{ - }$ является призматический изомер 17-PR, причем средняя энергия связи в нем существенно больше, чем в остальных. В расчете с использованием B3PW91-функционала изомеры 17-PR и 17-NIDO2 имеют практически равные средние энергии связи и оба могут считаться основными и равновероятно детектируемыми экспериментально. Средние энергии связи для кластеров 17-ARH и 17-NIDO1 лишь немного меньше, чем в основных, поэтому по результатам этого расчета можно предположить, что их наблюдение в эксперименте также возможно. В PBE-расчете средние энергии связи во всех рассматриваемых изомерах, кроме призматического 17-PR, примерно равны, поэтому, основываясь на этом расчете, можно заключить, что все эти структуры экспериментально равновероятны.
Наилучшее соответствие профилей рассчитанного и экспериментального спектров у изомеров 17-NIDO1 и 17-NIDO2 (рис. 3). Рассчитанный электронный спектр призматического изомера 17-PR, который является основным по результатам B3LYP-расчета, имеет общие особенности с фотоэлектронным [13], однако отличия также значительны. Это не позволяет подтвердить возникающее на основе B3LYP-расчета предположение, что призматический изомер является единственным, детектируемым экспериментально. Наилучшее согласие с экспериментом показывают суммарные спектры нескольких изомеров: 17-NIDO1, 17-NIDO2 и 17-ARH, что соответствует результатам PBE-расчета, или всех четырех изомеров, что соответствует результатам B3PW91-расчета. Таким образом, исключать экспериментальное наблюдение призматической структуры нельзя, однако определенно она не является единственно возможной.
Количество валентных электронов в анионных кластерах ${\text{HfSn}}_{n}^{ - }$ не удовлетворяет ни одному из правил Уэйда–Мингоса в 4n-модификации [22], однако находится ровно между значениями 4n + 4 и 4n + 6. Оказалось, что при формировании стабильных изомеров кластеров ${\text{HfSn}}_{n}^{ - }$ работают оба эти правила: часть основных изомеров имеет структуру arachno-многогранников, что характерно для систем с 4n + 6 валентными электронами, а часть – структуру nido-многогранников, как системы с числом валентных электронов 4n + 4. При n ≥ 15 происходит постепенный переход от эндоструктур к квазиэндоструктурам, и для кластеров ${\text{HfSn}}_{{{\text{16}}}}^{ - }$ и ${\text{HfSn}}_{{{\text{17}}}}^{ - }$ наряду с замкнутыми структурами могут быть синтезированы квазиэндоэдральные структуры. При этом в основе этих квазизамкнутых структур – многогранники, подобные изомерам кластера ${\text{HfSn}}_{{{\text{15}}}}^{ - }$, к которым добавляются дополнительные атомы олова. Таким образом, в этих структурах атом гафния инкапсулирован внутри многогранника из пятнадцати атомов олова, а один или два дополнительных атома Sn расположены на существенно большем расстоянии от него, и для них связь Hf–Sn значительно ослаблена.
Результаты оптимизационных расчетов атомной структуры кластеров ${\text{HfSn}}_{n}^{ - }$ значительно зависят от используемого функционала. Наилучшее согласие с экспериментальными данными показывают результаты, полученные с использованием функционала B3PW91. Оптимизация пространственной структуры с применением B3LYP-функционала может приводить к завышению относительной энергии связи трехсвязных структур по сравнению с дельтаэдрическими. Использование PBE-функционала для оптимизационных расчетов, напротив, может приводить к завышению средних энергий связи в дельтаэдрических структурах относительно трехсвязных. Чтобы исключить эти ошибки, наиболее эффективной стратегией поиска основных изомеров кластеров является использование всех трех функционалов, а интерпретация результатов должна проводиться с учетом их особенностей, описанных выше.
ЗАКЛЮЧЕНИЕ
Формирование наиболее стабильных изомеров кластеров ${\text{HfSn}}_{n}^{ - }$ (n = 15–17) может быть описано в рамках 4n-модификации правила Уэйда—Мингоса: часть основных изомеров имеет структуру arachno-многогранников, что характерно для систем с 4n + 6 валентными электронами, а часть – структуру nido-многогранников, как системы с числом валентных электронов 4n + 4. Эндокластеры со структурой arachno- или nido-многогранников могут служить основой для формирования более крупных кластеров.
Сравнение результатов, полученных в расчетах с различными функционалами, показывает, что наилучшее согласие с экспериментальными данными достигается при использовании функционала B3PW91. Применение PBE-функционала может приводить к завышению средних энергий связи в дельтаэдрических структурах относительно трехсвязных, а применение функционала B3LYP, наоборот, к их занижению.
Список литературы
Zhu C., Yang G., Li H., Du D., Lin Y. Electrochemical Sensors and Biosensors Based on Nanomaterials and Nanostructures // Anal. Chem. 2015. V. 87. P. 230–249. https://doi.org/10.1021/ac5039863
Jena P., Sun Q. Super Atomic Clusters: Design Rules and Potential for Building Blocks of Materials // Chem. Rev. 2018. V. 118. № 11. P. 5755–5870. https://doi.org/10.1021/acs.chemrev.7b00524
Shi S.-P., Zhao X.-F., Liu X.-Y. Lei D., Yan M., Jiang G. Structural and Electronic Properties in Titanium-Doped Stannum Clusters: Comparison with Their Anions and Cations // J. Clust. Sci. 2018. V. 29. P. 909–919. https://doi.org/10.1007/s10876-018-1384-4
Борщ Н.А., Курганский С.И. Атомная структура и электронные свойства анионных германий-циркониевых кластеров // Неорган. материалы. 2018. Т. 54. № 1. С. 3–10. https://doi.org/10.7868/S0002337X18010013
Borshch N., Kurganskii S. Geometric Structure, Electron-energy Spectrum, and Growth of Anionic Scandium-Silicon Clusters ${\text{ScSi}}_{n}^{ - }$ (n = 6–20) // J. Appl. Phys. 2014. V. 116. № 12. P. 124302-1–124302-8. https://doi.org/10.1063/1.489652810.1063/1.4896528
Dai W.-S., Yang B., Yan S.-T., Xu H.-G., Xu X.-L., Zheng W.-J. Structural and Electronic Properties of ${\text{LaSi}}_{n}^{{--/0}}$ (n = 2–6) Clusters: Anion Photoelectron Spectroscopy and Density Functional Calculations // J. Phys. Chem. A. 2021. V. 125. № 49. P. 10557–10567. https://doi.org/10.1021/acs.jpca.1c0848710.1021/acs.jpca.1c08487
Liu B., Wang X., Yang J. Comparative Research of Configuration, Stability and Electronic Properties of Cationic and Neutral [AuGen]λ and [Gen+1]λ (n = 1–13, λ = 0, +1) Nanoalloy Clusters // Mater. Today Commun. 2021. V. 26. P. 101989-1–101989-10. https://doi.org/10.1016/j.mtcomm.2020.101989
Zhang Y., Yang J., Cheng L. Probing Structure, Thermochemistry, Electron Affinity and Magnetic Moment of Erbium-Doped Silicon Clusters ErSin (n = 3–10) and Their Anions with Density Functional Theory // J. Clust. Sci. 2018. V. 29. P. 301–311. https://doi.org/10.1007/s00894-017-3566-7
Md. Hasan N., Wahid H., Nayan N., Mohamed Ali M.S. Inorganic Thermoelectric Materials: A Review // Int. J. Energy Res. 2020. V. 44. № 8. P. 6170–6222. https://doi.org/10.1002/er.5313
Abdulsattar M.A., Abed H.H., Jabbar R.H., Almaroof N.M. Effect of Formaldehyde Properties on SnO2 Clusters Gas Sensitivity: A DFT Study // J. Mol. Graph. 2021. V. 102. P. 107791-1–107791-7. https://doi.org/10.1016/j.jmgm.2020.107791
Wang M., Zhang X., He X., Zhu B., Tang H., Wang C. In-situ Grown Flower-Like C@SnO2/Cu2O Nanosheet Clusters on Cu Foam as High Performance Anode for Lithium-Ion Batteries // J. Alloys Compd. 2021. V. 856. P. 158202-1–158202-10. https://doi.org/10.1016/j.jallcom.2020.158202
Борщ Н.А., Курганский С.И. Влияние переходных металлов IIIB-группы на формирование замкнутых германиевых кластеров: компьютерный эксперимент в рамках теории функционала плотности // Конденсированные среды и межфазные границы. 2019. Т. 21. № 2. С. 182–190. https://doi.org/10.17308/kcmf.2019.21/756
Atobe J., Koyasu K., Furuse S., Nakajima A. Anion Photoelectron Spectroscopy of Germanium and Tin Clusters Containing a Transition- or Lanthanide-Metal atom; ${\text{MGe}}_{n}^{ - }$ (n = 8–20) and MSnn− (n = 15–17) (M = Sc–V, Y–Nb, and Lu–Ta) // Phys. Chem. Chem. Phys. 2012. V. 14. P. 9403–9410. https://doi.org/10.1039/C2CP23247B10.1039/C2CP23247B
Becke A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior // Phys. Rev. A. 1988. V. 38. P. 3098–3100. https://doi.org/10.1103/PhysRevA.38.3098
Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density // Phys. Rev. B. 1988. V. 37. P. 785–789. https://doi.org/10.1103/PhysRevB.37.785
Becke A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange // J. Chem. Phys. 1993. V. 98. P. 5648–5652. https://doi.org/10.1063/1.464913
Perdew J.P., Burke K., Ernzerhof M. Generalized Gradient Approximation Made Simple // Phys. Rev. Lett. 1996. V. 77. P. 3865–3868. https://doi.org/10.1103/PhysRevLett.77.3865
Perdew J.P., Burke K., Ernzerhof M. Generalized Gradient Approximation Made Simple // Phys. Rev. Lett. 1997. V. 78. P. 1396(E). https://doi.org/10.1103/PhysRevLett.78.1396
Andrae D., Häußermann U., Dolg M., Stoll H., Preuß H. Energy-Adjusted Ab Initio Pseudopotentials for the 2nd and 3rd Row Transition-Elements // Theor. Chem. Acc. 1990. V. 77. P. 123–141. https://doi.org/10.1007/BF01114537
McLean A.D., Chandler G.S. Contracted Gaussian-Basis Sets for Molecular Calculations. 1. 2nd Row Atoms, Z = 11–18 // J. Chem. Phys. 1980. V. 72. P. 5639–5648. https://doi.org/10.1063/1.438980
Frisch M.J. et al. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013.
Wade K. The Structural Significance of the Number of Skeletal Bonding Electron-Pairs in Carboranes, the Higher Boranes and Borane Anions, and Various Transition-Metal Carbonyl Cluster Compounds // J. Chem. Soc. D. 1971. P. 792–793. https://doi.org/10.1039/C29710000792
Mingos D.A. A General Theory for Cluster and Ring Compounds of the Main Group and Transition Elements // Nature Phys. Sci. 1972. V. 236. P. 99–102. https://doi.org/10.1038/physci236099a0
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы